

Abstract
The cellular receptor of foamy viruses (FVs) is unknown. The broad spectrum of permissive cells suggests that the cellular receptor is a molecular structure with almost ubiquitous prevalence. Here, we investigated the ability of heparan sulfate (HS), a glycosaminoglycan (GAG) present on the extracellular matrix of many cells, to bind FV particles and to permit prototype FV (PFV) and feline FV (FFV) entry. Permissivity of different cell lines for FV entry correlated with the amount of heparan sulfate present on the cell surface. The resulting 50% cell culture infectious doses (CCID50s) were distributed over a range of 4 logs, which means that the most susceptible cell line tested (HT1080) was more than 10,000 times more susceptible for PFV infection than the least susceptible cell line (CRL-2242). HS surface expression varied over a range of 2 logs. HS expression and FV susceptibility were positively correlated (P < 0.001). Enzymatic digestion of heparan sulfate on HT1080 cells diminished permissivity for PFV entry by a factor of at least 500. Using fast protein liquid chromatography (FPLC), we demonstrated binding of FV vector particles to a gel filtration column packed with heparin, a molecule structurally related to heparan sulfate, allowing for the purification of infectious particles. Both PFV and FFV infection were inhibited by soluble heparin. Our results show that FVs bind to HS and that this interaction is a pivotal step for viral entry, suggesting that HS is a cellular attachment factor for FVs.
INTRODUCTION
Foamy viruses (FVs) represent the only genus of the retroviral subfamily of Spumaretrovirinae. They are named after a cytopathic effect (CPE) characterized by the formation of multinucleated giant cells and a foamy appearance that they induce in adherent cell culture. However, in contrast to this in vitro feature, FV infections in vivo are not associated with any known pathology. Different FV species have been isolated so far, including simian foamy viruses (SFVs), bovine foamy viruses (BFVs), equine foamy virus (EFV), and feline foamy viruses (FFVs) (34, 37). FVs are not endemic in humans, but zoonotic transmission from monkeys to humans has been described elsewhere (3, 5, 6, 16, 18, 44, 45, 48). The best-studied foamy virus isolate is the prototype FV (PFV), which was obtained from an infected Kenyan individual who probably became infected by transspecies infection with SFV (1).
The cellular receptor (or receptors) that permits FV infection is unknown for any of the FV species. The broad tropism of FVs (25) suggests that ubiquitous membrane-bound macromolecules rather than species- or tissue-specific proteins are likely candidates for cellular receptors that mediate viral attachment or entry. We have previously reported that cell lines with a defect in glucosaminoglycan (GAG) synthesis are less susceptible for PFV infection, indicating that the presence of GAGs may favor PFV infection (41).
One prominent GAG is heparan sulfate (HS), a ubiquitous, highly negatively charged polysaccharide of the extracellular matrix that is involved in many biological processes, including angiogenesis, embryonic development, and tissue repair (4, 19, 23). HS is bound as a proteoglycan together with transmembrane or membrane-anchored proteins, forming the heparan sulfate proteoglycan (HSPG).
The highly negatively charged extracellular environment has selected HS-binding proteins for many viruses in order to be able to reach the cell membrane for viral entry. A review by Liu and Thorp lists 16 animal and human viruses that use HS as a receptor for attachment or entry (24), including human T-cell leukemia virus (HTLV) (20, 31, 46), HIV (11, 26, 29, 38, 50), herpes simplex virus (49), cytomegalovirus (9), dengue virus (8), adeno-associated virus type 2 (43), and respiratory syncytial virus (14).
In this study, we hypothesized that HS plays a role in FV attachment. We studied the binding of PFV to heparin by fast protein liquid chromatography (FPLC) and analyzed the role of HS in cellular infection. We then extended our studies to an FFV isolate in order to generalize our findings to other FV species.
MATERIALS AND METHODS
Cell lines and primary cells.
The following cells were cultivated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS): HT1080 (human fibrosarcoma cells, American Type Culture Collection [ATCC] CCL-121), BHK-21 (baby hamster kidney cells, ATCC CCL-10), CRFK (Crandell-Reese feline kidney cells, ATCC CCL-94), COS-7 (green monkey kidney fibroblast cells, ATCC CCL-1651), HepG2 cells (human hepatocellular carcinoma cells, ATCC HB-8065), MRC-5 (human lung fibroblastoma cells, ATCC CCL-171), SK-N-SH (human neuroblastoma cells, ATCC HTB-11), Sog9 cells (mouse fibroblasts, defect in initiation of GAG assembly so that no surface HS is being produced [2]), and Mouse L (mouse fibroblasts [2]). CHO-K1 (Chinese hamster ovary cells, ATCC PTA-6812) and CRL-2242 (a CHO subclone, deficient in xylosyltransferase I, so that no surface GAGs are being produced [21]; ATCC CRL-2242) cells were cultured in F-12K medium supplemented with 10% FCS. HEK-293T (human embryonic kidney cells, ATCC CRL-11268) cells were cultured in minimal essential medium (MEM), supplemented with 10% FCS. hMSC-Tert cells (telomerase-immortalized human mesenchymal stem cell line) were cultured in MEM with Earle's salts supplemented with 10% FCS.
FV molecular clones.
In this study, we used the replication-incompetent PFV clone MD9 as previously described (17, 40). MD9 encodes an enhanced green fluorescent protein (EGFP) expression cassette. The PFV sequences, including the env gene, are based on the infectious PFV molecular clone pPFV2 (39), which is, except for the long terminal repeat (LTR) regions, identical with its parental clone pHSRV1 (35). The env gene of pPFV-2 was derived from human embryonic lung (HEL) cells infected with PFV (15). The replication-competent FFV molecular clone pChatul-3 originated from FFV serially passaged on CRFK cells (36).
PFV vector packaging.
HEK-293T cells (6 × 106) (13) were seeded into 10-cm dishes. After 24 h, cells were transfected using a polyethylenimine transfection reagent (Polyscience). The transfection mix contained 6 μg of DNA of a 4-plasmid FV vector system (17, 40), consisting of 1.5 μg of the PFV vector plasmid pMD9 and 1.5 μg of each of the three packaging plasmids pcoPG4, pcoPP, and pcoPE, encoding the PFV envelope protein (27, 41). One day after transfection, cellular transcription was boosted by the addition of 10 mM sodium butyrate for 8 h. After 2 days, the supernatant was harvested and passed through an 0.45-μm filter (Millipore) to remove floating cells and cellular debris. The virus suspension was kept frozen at −80°C in aliquots until use.
Determination of the CCID50.
Cells were seeded for 24 h in tissue culture plates before infection with a dilution series of a PFV (MD9) vector preparation. In experiments in which susceptibilities of different cells were compared, aliquots of the same vector preparation were used in order to perform the experiments with identical vector concentrations. In experiments with heparinase treatment, cells were treated with heparinase I or heparinase III (40 U/ml and 5 U/ml, respectively; both from Sigma-Aldrich) at 37°C for 1 h before PFV infection. Cells were incubated with vector preparations for 1 h at 37°C, and then excess vector was washed away. To control for possible protease contamination of the heparinase III used, cells were treated in the presence or absence of a protease inhibitor cocktail (Sigma-Aldrich; 1:100 dilution as recommended by the manufacturer). Cells were analyzed 24 h after infection for EGFP expression by flow cytometry. Applying a nonlinear regression analysis (sigmoidal dose-response curve) using GraphPad Prism software (version 4.0c for Macintosh), the 50% cell culture infectious dose (CCID50), corresponding to the dilution at which half-maximal infection was visible, was determined.
FPLC.
PFV vector suspension was purified using an Äkta Explorer10 FPLC machine with UV detection at 280 nm (GE Healthcare) and a Poros 50-μm HE heparin column (Life Technologies). As a control, a Poros-OH 50-μm column (Life Technologies) with no bound ligand attached to the Poros carrier material was used. The columns were equilibrated with 20 mM sodium phosphate buffer (1 M NaCl in 20 mM sodium phosphate buffer, pH 7). After injection of 1 ml of PFV-containing cell culture supernatant at a flow rate of 3 ml/min, the column was washed by increasing the NaCl concentration to 300 mM. For elution of PFV, 500 mM NaCl was applied. Fractions were collected and analyzed for PFV infectivity by transduction experiments.
Flow cytometry.
For detection of HS and CD138 expression, cells were detached from their culture dishes with EDTA–phosphate-buffered saline (PBS). For HS staining, cells were incubated with the anti-HS mouse antibody F58-10E4 (Seikagaku America) or with a mouse IgM isotype control antibody (BD Biosciences), followed by incubation with a secondary rat antibody anti-mouse IgM labeled with phycoerythrin (PE) (BD Biosciences). For CD138 staining, cells were incubated with a PE-labeled anti-CD138 antibody (BD Biosciences) or a PE-labeled isotype control antibody (BD Biosciences). For detection of FFV Gag, cells were fixed with 4% formalin, permeabilized with 0.5% saponin in PBS, and incubated with a rabbit anti-Gag antibody (36) followed by incubation with a fluorescein isothiocyanate (FITC)-coupled goat anti-rabbit antibody (BD Pharmingen). For detection of EGFP expression, cells were left unstained. Cells were fixed with 2% formalin and analyzed using a FACSCalibur flow cytometer (BD Biosciences, Germany). Cell debris was excluded from analysis by forward scatter (FSC)-side scatter (SSC) gating. EGFP expression was detected in the FL1 channel. Quadrants were set according to autofluorescence detected in untransduced cells (for detection of EGFP) or in cells stained with the isotype control antibody (for detection of HS). Flow cytometry data were analyzed using FlowJo software (Tree Star Inc., OR).
RESULTS
PFV binds to heparin column in FPLC.
We generated replication-incompetent foamy virus vector particles (MD9, described in reference 17) expressing EGFP as a reporter gene. Virus particles were pseudotyped with the envelope protein from PFV. Virus-containing cell culture supernatants were loaded onto a Poros 50HE (heparin) FPLC column. As depicted in Fig. 1, FV particles bound to the column and were eluted with 500 mM NaCl. Fractions of the elution peak, but not from previous wash steps with 300 mM NaCl (data not shown), contained infectious particles as demonstrated by subsequent transduction of HT1080 cell cultures. Figure 1B depicts representative fluorescence microscopic pictures of cell cultures transduced with selected FPLC fractions, and Fig. 1C displays flow cytometric analysis of EGFP expression of transduced HT1080 cell cultures. Titration experiments of PFV vector suspensions before and after FPLC showed that FPLC did not reduce the number of infectious particles (data not shown). In a control experiment, we used an FPLC column packed with the carrier material Poros alone (i.e., without the heparin ligand) and found no binding of foamy virus particles to the column (Fig. 2), demonstrating that the interaction described in Fig. 1 was specific for binding of PFV to heparin. These results demonstrate that heparin-containing columns can be used for affinity purification of PFV or PFV vectors.
Fig 1.

PFV binds to heparin. Replication-incompetent PFV encoding an EGFP expression cassette was loaded on an FPLC column packed with Poros-heparin. The column was washed with 20 mM sodium phosphate buffer, pH 7.0, and the column was eluted with 500 mM NaCl, pH 7.0. FPLC fractions were tested for infectivity by transduction of HT1080 cells. The proportion of transduced cells was visualized by fluorescence microscopy and quantified by flow cytometry detecting EGFP fluorescence. (A) FPLC chromatogram displaying the affinity purification of PFV particles. The blue line represents absorbance at 280 nm detecting protein. The yellow line represents the applied NaCl gradient, and the brown line represents the corresponding conductivity. The x axis displays the fractions collected. (B) Fluorescence microscopy of HT1080 cells incubated with the indicated fractions. (C) Flow cytometric quantification of EGFP-expressing cells of HT1080 cultures transduced with the collected FPLC fractions.
Fig 2.

PFV does not bind to Poros carrier. The experiment was performed as described in the legend to Fig. 1. Instead of using a Poros-heparin column, a column packed with Poros alone was used. (A) FPLC chromatogram. (B) Fluorescence microscopy of HT1080 cells incubated with the indicated FPLC fractions indicated below. (C) Flow cytometric quantification of EGFP-expressing cells of HT1080 cultures transduced with the collected FPLC fractions.
Heparin inhibits PFV and FFV infection.
Binding of PFV to the 50HE column suggests that heparin-like molecules may also play a role in viral attachment or entry. If membrane-bound heparin-like molecules play a role in cellular attachment of PFV, soluble heparin should interact with the virus and inhibit the infection. We therefore performed transduction experiments in the presence of a serial dilution of soluble heparin. As depicted in Fig. 3A, heparin inhibited PFV transduction of HT1080 cells with a 50% effective concentration (EC50) of 29.81 μg/ml. Similarly, heparin inhibited FFV infection with an EC50 of 16.71 μg/ml (Fig. 3B). These results support the idea that heparin-like structures play a role in FV infection.
Fig 3.
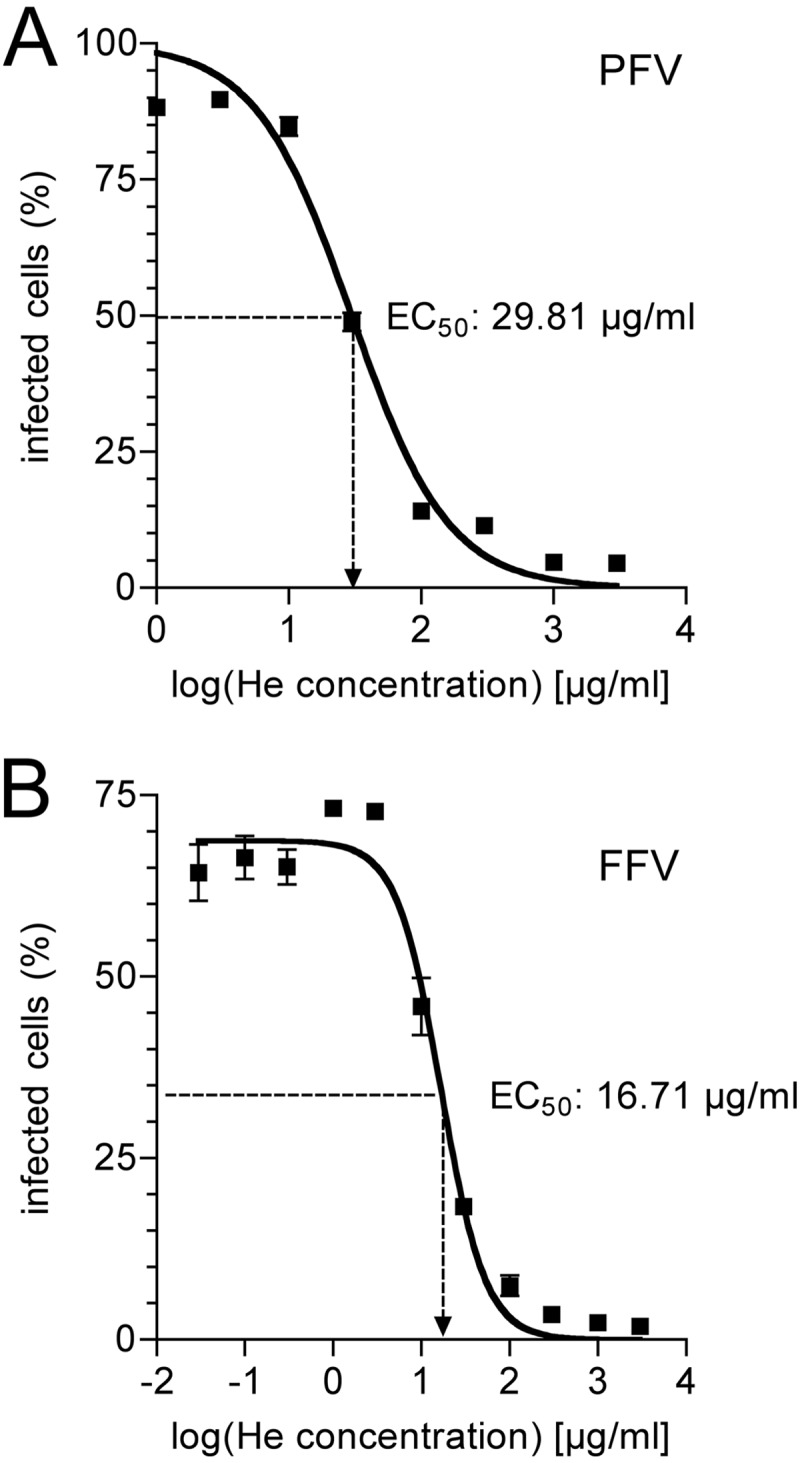
Heparin inhibits PFV and FFV infection. HT1080 cells were incubated with replication-incompetent PFV (MD9) or replication-competent FFV (Chatul-3) in the presence of different concentrations of heparin (He), a GAG closely related to HS. Virus-infected cells were monitored by EGFP expression (MD9) or by immunostaining for FV Gag (FFV) after 24 h by flow cytometry. Using a sigmoidal curve function, the EC50 (the concentration at which the half-maximal inhibitory effect was visible) of heparin was determined by nonlinear regression. (A) HT1080 cells transduced with PFV. (B) HT1080 cells infected with FFV. For both panels, data represent means ± standard deviations from triplicates.
HS-deficient cells show reduced susceptibility.
The structure most closely related to heparin that is present on the cell surface is heparan sulfate (HS). In order to investigate whether cellular HS plays a role in susceptibility to FV infection, we performed transduction experiments with Mouse L cells and their HS-deficient subclone Sog9 (2). Flow cytometric analysis of the Sog9 cells with anti-HS antibodies demonstrated a total lack of HS expression on Sog9 cells, whereas the parental clone Mouse L exhibited HS on the cell surface (Fig. 4A and B). We infected both cell clones with a dilution series of the same replication-incompetent vector preparation and determined the concentration at which 50% of the cells became infected (CCID50). As depicted in Fig. 4C, Sog9 cells were much less susceptible to PFV infection (CCID50, 0.1546) than were Mouse L cells (CCID50, 0.0036), corresponding to a 43-fold-lower susceptibility.
Fig 4.

Cells that lack HS are less susceptible to PFV infection. Mouse L cells and their HS-deficient subclone Sog9 were incubated with a dilution series of the same FV vector suspension, and transduction rate was determined by flow cytometry detecting EGFP expression. (A and B) Flow cytometric determination of surface HS expression. Green line, cells stained with anti-HS antibody; blue line, cells stained with isotype control antibody. (A) Mouse L cells. (B) Sog9 cells. (C) Mouse L and Sog9 cells were incubated with a dilution series of the same vector preparation. EGFP-positive cells were determined after 24 h by flow cytometry. Using a sigmoidal curve function, CCID50 (the vector dilution at which the half-maximal transduction rate was visible) was determined by nonlinear regression.
Susceptibility correlates with HS surface expression.
In order to analyze whether HS determines PFV susceptibility also in other cell types, we quantified HS expression of different cell lines (Fig. 5B) and correlated the amount of surface HS with the susceptibility to FV infection (Fig. 5A). We determined the CCID50 of each cell line by a dilution series similar to the experiments depicted in Fig. 3. The resulting CCID50s were distributed over a range of 4 logs, which means that the most susceptible cell line in the experiment was more than 10,000 times more susceptible to PFV infection than the least susceptible cell line (Fig. 5A). HS surface expression (medians) varied over a range of 2 logs. HS expression and FV susceptibility were positively correlated (P < 0.001).
Fig 5.

The amount of cell surface HS correlates with susceptibility to PFV infection. Relative surface HS expression of different cell lines (CRL-2242, HepG2, 293T, BHK, CRFK-LL, SK-N-SH, MRC, human mesenchymal stem cell line MSC-Tert, COS-7, CHO-K1, HT1080) was determined by the shift of mean fluorescence of cells stained with anti-HS antibody versus cells stained with isotype control antibody. The different cells were incubated with a dilution series of the same vector preparation. EGFP-positive cells were determined after 24 h by flow cytometry. Using a sigmoidal curve function, CCID50 was determined by nonlinear regression. (A) Correlation of relative HS surface expression versus cell-specific CCID50. Data represent means ± standard deviations from triplicates. (B) Histograms of HS expression of the cells from flow cytometric analysis. Cells were stained with anti-HS antibody (blue line) or an isotype-matched control antibody (red line). Numbers indicate geometric mean fluorescence intensity of cells stained with anti-HS (blue numbers) and isotype control (red numbers). (C) Correlation of relative CD138 (syndecan 1) surface expression with cell-specific CCID50. (D) Correlation of surface expression of HS with CD138 (syndecan 1).
In contrast to HS expression, syndecan 1 expression (CD138, a heparan sulfate proteoglycan) negatively correlated with FV susceptibility (Fig. 5C), indicating that HS responsible for FV entry does not require the presence of this particular proteoglycan. In fact, the amounts of cell surface HS and CD138 show a negative correlation for the tested cell lines (Fig. 5D). This analysis was restricted to cell lines of human origin due to the species reactivity of the anti-CD138 antibody used.
Enzymatic degradation of surface HS reduces permissivity.
In order to exclude any cell-type-specific effects independent of HS expression that could bias the previous experiments, we enzymatically digested surface HS and compared the susceptibility of the HS-stripped cells to that of nondigested cells. Treatment of HT1080 cells with heparinase I (Fig. 6A) or III (Fig. 6B) reduced susceptibility by a factor of 530 (heparinase I) or higher (heparinase III; the exact CCID50 of cells treated with heparinase III could not be determined as we were not able to generate a transduction rate of ≥50% in these experiments), demonstrating that HS is a cellular factor involved in PFV infection. To exclude the possibility that the observed effects might have been caused by a potential protease contamination of the heparinases used, we performed additional experiments in the presence of a broad-range protease inhibitor cocktail and found no differences in inhibition of foamy viral transduction (data not shown). As depicted in Fig. 6C, heparinase III treatment markedly reduced heparan sulfate expression on the cells (upper panel) but had no effect on extracellular protein expression (CD138). No differences were seen in the presence or absence of protease inhibitors, indicating that the heparinase used was indeed free of contaminating protease activity.
Fig 6.

Enzymatic removal of cell surface HS reduces susceptibility for PFV infection. HT1080 cells were treated with heparinase I or heparinase III for 1 h and subsequently incubated for 1 h with a dilution series from the same vector preparation. EGFP-positive cells were determined after 24 h by flow cytometry. Using a sigmoidal curve function, CCID50 was determined by nonlinear regression. (A) HT1080 cells treated with (red triangles) or without (green squares) heparinase I. (B) HT1080 cells treated with (red triangles) or without (green squares) heparinase III. (A and B) Data represent means ± standard deviations from triplicates. (C) Flow cytometric analysis of HT1080 cells treated with heparinase III in the presence or absence of a protease inhibitor (PI) cocktail. (Top) Histogram of HS expression (blue line) versus isotype control staining (red line). (Bottom) Histogram of CD138 (syndecan 1) expression (blue line) versus isotype control staining (red line). Numbers indicate geometric mean fluorescence intensity of cells stained with anti-HS or anti-CD138 (blue numbers) and isotype controls (red numbers).
DISCUSSION
Attachment factor or entry receptor?
For many viruses, two different groups of receptor functions have been described: (i) attachment factors that bind the virus to the cell surface and enhance susceptibility but do not trigger viral entry and (ii) entry receptors that trigger a mandatory conformational change in viral entry proteins and whose presence determines whether a cell is susceptible or not. Here, we have shown that FVs bind to HS and that the amount of HS on the cell surface positively correlates with susceptibility. A total lack of HS, however, does not completely abolish FV infection. We therefore suggest that HS is an attachment factor for FVs rather than an entry receptor.
Enveloped viruses enter their target cells by fusion of the viral and cellular membranes, triggered by a conformational change of their envelope proteins (12). Depending on the trigger of this conformational change, pH-dependent and pH-independent mechanisms can be distinguished. A pH-independent conformational change usually results following binding of the virus to a cellular entry receptor. A prominent example for this is HIV (7), but similar mechanisms have been described for other viruses such as paramyxoviruses (32). In contrast, other viruses show a pH-dependent conformational change of their fusion proteins, which is usually being triggered by acidification within the endosome (12). One well-understood example is influenza virus, which binds to the attachment receptor sialic acid (SA) through its envelope protein hemagglutinin (HA) (10). After endosomal uptake and acidification, influenza virus HA undergoes the necessary conformational change to penetrate the endosomal membrane and to trigger membrane fusion (10). Note that the crucial conformational change of influenza virus HA is not triggered by receptor binding but by a change in the pH. In experiments with artificial lipid bilayers, pH-activated HA has been shown to be sufficient to initiate membrane fusion (22). Therefore, influenza viruses do not require entry receptors for membrane fusion but only an attachment factor (SA) in combination with a pH change.
We have previously shown that PFV entry also is dependent on a pH change (30). From what is known for influenza virus entry, it seems plausible that the combination of mere attachment factors and a pH change may also be sufficient for PFV entry. In this scenario, we have identified HS as a major attachment factor involved in PFV infection. Consistent with this, several classes of HSPGs—such as syndecans—are true endocytic receptors that mediate internalization of their ligands into acid endosomes (47), so that HSPG binding would be sufficient to allow viral entry of viruses with pH-dependent fusion proteins.
For some viruses, both pH-dependent (i.e., entry receptor-independent) and pH-independent (i.e., entry receptor-dependent) fusion mechanisms have been described (33). In this regard, our results do not exclude the existence of true entry receptors for FVs. Neither can we exclude that one of these hypothetical FV entry receptors is HS, as we have no data on potential conformational changes of FV Env upon binding to HS that would be sufficient to trigger membrane fusion.
Is HS the only attachment factor for FVs?
While the manuscript was under review, Nasimuzzaman and Persons independently published that heparan sulfate is a receptor molecule for FV entry (28). Their results are in line with our findings with regard to the important role of HS in foamy virus cell entry. However, using serial dilution series of the virus preparations, we were also able to quantify the effects of HS surface expression of foamy viral susceptibility, showing residual FV transduction also in cells that totally lack HS expression, as well as in cells on which HS has been enzymatically removed. We therefore suggest that other attachment factors/receptors may exist that can be used by FVs, alternatively or in addition to HS. However, residual infection despite the absence of the identified receptor molecule has also been observed with influenza virus (42). It may therefore well be that membrane-coated viruses whose fusion proteins become activated by a pH change do not absolutely require the presence of attachment receptors for infection and that both SA and HS are indeed the only attachment factors for these two viruses. In an attempt to identify additional cellular molecules involved in FV entry, we screened the activity of other cellular matrix molecules, such as chondroitin sulfate A, B, and C; hyaluronic acid; keratan sulfate; and N-acetyl-neuraminic acid. None of these diminished FV transduction when added in soluble form to FV preparations before infection (data not shown).
Taken together, our study shows that HS is an attachment factor involved in FV entry and that cellular HS expression positively correlates with cellular susceptibility to FV infection, suggesting that HS is a key factor involved in FV cell entry.
ACKNOWLEDGMENTS
We thank Bianca Fries for excellent technical assistance.
The study was supported by grants from the Deutsche Forschungsgemeinschaft (DFG GRK 1522/1) and the Interdisciplinary Center for Clinical Research Würzburg (IZKF D-23).
Footnotes
Published ahead of print 11 July 2012
REFERENCES
- 1. Achong BG, Mansell PW, Epstein MA, Clifford P. 1971. An unusual virus in cultures from a human nasopharyngeal carcinoma. J. Natl. Cancer Inst. 46:299–307 [PubMed] [Google Scholar]
- 2. Banfield BW, Leduc Y, Esford L, Schubert K, Tufaro F. 1995. Sequential isolation of proteoglycan synthesis mutants by using herpes simplex virus as a selective agent: evidence for a proteoglycan-independent virus entry pathway. J. Virol. 69:3290–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bastone P, Truyen U, Lochelt M. 2003. Potential of zoonotic transmission of non-primate foamy viruses to humans. J. Vet. Med. B Infect. Dis. Vet. Public Health 50:417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bernfield M, et al. 1999. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 68:729–777 [DOI] [PubMed] [Google Scholar]
- 5. Betsem E, Rua R, Tortevoye P, Froment A, Gessain A. 2011. Frequent and recent human acquisition of simian foamy viruses through apes' bites in central Africa. PLoS Pathog. 7:e1002306 doi:10.1371/journal.ppat.1002306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boneva RS, et al. 2007. Clinical and virological characterization of persistent human infection with simian foamy viruses. AIDS Res. Hum. Retroviruses 23:1330–1337 [DOI] [PubMed] [Google Scholar]
- 7. Chan DC, Kim PS. 1998. HIV entry and its inhibition. Cell 93:681–684 [DOI] [PubMed] [Google Scholar]
- 8. Chen Y, et al. 1997. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 3:866–871 [DOI] [PubMed] [Google Scholar]
- 9. Compton T, Nowlin DM, Cooper NR. 1993. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology 193:834–841 [DOI] [PubMed] [Google Scholar]
- 10. Cross KJ, Burleigh LM, Steinhauer DA. 2001. Mechanisms of cell entry by influenza virus. Expert Rev. Mol. Med. 3(21):1–18 [DOI] [PubMed] [Google Scholar]
- 11. de Witte L, et al. 2007. Syndecan-3 is a dendritic cell-specific attachment receptor for HIV-1. Proc. Natl. Acad. Sci. U. S. A. 104:19464–19469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dimitrov DS. 2004. Virus entry: molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2:109–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DuBridge RB, et al. 1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 7:379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feldman SA, Audet S, Beeler JA. 2000. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J. Virol. 74:6442–6447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Flügel RM, Rethwilm A, Maurer B, Darai G. 1987. Nucleotide sequence analysis of the env gene and its flanking regions of the human spumaretrovirus reveals two novel genes. EMBO J. 6:2077–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goldberg TL, et al. 2009. Coinfection of Ugandan red colobus (Procolobus [Piliocolobus] rufomitratus tephrosceles) with novel, divergent delta-, lenti-, and spumaretroviruses. J. Virol. 83:11318–11329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heinkelein M, et al. 2002. Improved primate foamy virus vectors and packaging constructs. J. Virol. 76:3774–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heneine W, Schweizer M, Sandstrom P, Folks T. 2003. Human infection with foamy viruses. Curr. Top. Microbiol. Immunol. 277:181–196 [DOI] [PubMed] [Google Scholar]
- 19. Iozzo RV, San Antonio JD. 2001. Heparan sulfate proteoglycans: heavy hitters in the angiogenesis arena. J. Clin. Invest. 108:349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jones KS, Petrow-Sadowski C, Bertolette DC, Huang Y, Ruscetti FW. 2005. Heparan sulfate proteoglycans mediate attachment and entry of human T-cell leukemia virus type 1 virions into CD4+ T cells. J. Virol. 79:12692–12702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaesberg PR, Ershler WB, Esko JD, Mosher DF. 1989. Chinese hamster ovary cell adhesion to human platelet thrombospondin is dependent on cell surface heparan sulfate proteoglycan. J. Clin. Invest. 83:994–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim CS, Epand RF, Leikina E, Epand RM, Chernomordik LV. 2011. The final conformation of the complete ectodomain of the HA2 subunit of influenza hemagglutinin can by itself drive low pH-dependent fusion. J. Biol. Chem. 286:13226–13234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kjellen L, Lindahl U. 1991. Proteoglycans: structures and interactions. Annu. Rev. Biochem. 60:443–475 [DOI] [PubMed] [Google Scholar]
- 24. Liu J, Thorp SC. 2002. Cell surface heparan sulfate and its roles in assisting viral infections. Med. Res. Rev. 22:1–25 [DOI] [PubMed] [Google Scholar]
- 25. Mergia A, Leung NJ, Blackwell J. 1996. Cell tropism of the simian foamy virus type 1 (SFV-1). J. Med. Primatol. 25:2–7 [DOI] [PubMed] [Google Scholar]
- 26. Mondor I, Ugolini S, Sattentau QJ. 1998. Human immunodeficiency virus type 1 attachment to HeLa CD4 cells is CD4 independent and gp120 dependent and requires cell surface heparans. J. Virol. 72:3623–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Müllers E, et al. 2011. Novel functions of prototype foamy virus Gag glycine-arginine-rich boxes in reverse transcription and particle morphogenesis. J. Virol. 85:1452–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nasimuzzaman M, Persons DA. 2012. Cell membrane-associated heparan sulfate is a receptor for prototype foamy virus in human, monkey, and rodent cells. Mol. Ther. 20:1158–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patel M, et al. 1993. Cell-surface heparan sulfate proteoglycan mediates HIV-1 infection of T-cell lines. AIDS Res. Hum. Retroviruses 9:167–174 [DOI] [PubMed] [Google Scholar]
- 30. Picard-Maureau M, Jarmy G, Berg A, Rethwilm A, Lindemann D. 2003. Foamy virus envelope glycoprotein-mediated entry involves a pH-dependent fusion process. J. Virol. 77:4722–4730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pinon JD, et al. 2003. Human T-cell leukemia virus type 1 envelope glycoprotein gp46 interacts with cell surface heparan sulfate proteoglycans. J. Virol. 77:9922–9930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog. 7:e1002058 doi:10.1371/journal.ppat.1002058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reske A, Pollara G, Krummenacher C, Chain BM, Katz DR. 2007. Understanding HSV-1 entry glycoproteins. Rev. Med. Virol. 17:205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rethwilm A. 2010. Molecular biology of foamy viruses. Med. Microbiol. Immunol. 199:197–207 [DOI] [PubMed] [Google Scholar]
- 35. Rethwilm A, et al. 1990. Infectious DNA of the human spumaretrovirus. Nucleic Acids Res. 18:733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roy J, et al. 2003. Feline foamy virus genome and replication strategy. J. Virol. 77:11324–11331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saib A. 2003. Non-primate foamy viruses. Curr. Top. Microbiol. Immunol. 277:197–211 [DOI] [PubMed] [Google Scholar]
- 38. Saphire AC, Bobardt MD, Zhang Z, David G, Gallay PA. 2001. Syndecans serve as attachment receptors for human immunodeficiency virus type 1 on macrophages. J. Virol. 75:9187–9200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schmidt M, Rethwilm A. 1995. Replicating foamy virus-based vectors directing high level expression of foreign genes. Virology 210:167–178 [DOI] [PubMed] [Google Scholar]
- 40. Stange A, et al. 2005. Characterization of prototype foamy virus gag late assembly domain motifs and their role in particle egress and infectivity. J. Virol. 79:5466–5476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stirnnagel K, et al. 2010. Analysis of prototype foamy virus particle-host cell interaction with autofluorescent retroviral particles. Retrovirology 7:45 doi:10.1186/1742-4690-7-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stray SJ, Cummings RD, Air GM. 2000. Influenza virus infection of desialylated cells. Glycobiology 10:649–658 [DOI] [PubMed] [Google Scholar]
- 43. Summerford C, Bartlett JS, Samulski RJ. 1999. AlphaVbeta5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat. Med. 5:78–82 [DOI] [PubMed] [Google Scholar]
- 44. Switzer WM, et al. 2004. Frequent simian foamy virus infection in persons occupationally exposed to nonhuman primates. J. Virol. 78:2780–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Switzer WM, et al. 2008. Coinfection with HIV-1 and simian foamy virus in West Central Africans. J. Infect. Dis. 197:1389–1393 [DOI] [PubMed] [Google Scholar]
- 46. Tanaka A, et al. 2012. Entry of human T-cell leukemia virus type 1 is augmented by heparin sulfate proteoglycans bearing short heparin-like structures. J. Virol. 86:2959–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wittrup A, et al. 2009. ScFv antibody-induced translocation of cell-surface heparan sulfate proteoglycan to endocytic vesicles: evidence for heparan sulfate epitope specificity and role of both syndecan and glypican. J. Biol. Chem. 284:32959–32967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wolfe ND, et al. 2004. Naturally acquired simian retrovirus infections in central African hunters. Lancet 363:932–937 [DOI] [PubMed] [Google Scholar]
- 49. WuDunn D, Spear PG. 1989. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 63:52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang YJ, et al. 2002. Envelope-dependent, cyclophilin-independent effects of glycosaminoglycans on human immunodeficiency virus type 1 attachment and infection. J. Virol. 76:6332–6343 [DOI] [PMC free article] [PubMed] [Google Scholar]