

Abstract
Breast cancer that recurs as metastatic disease many years after primary tumor resection and adjuvant therapy appears to arise from tumor cells that disseminated early in the course of disease but did not develop into clinically apparent lesions. These long-term surviving, disseminated tumor cells maintain a state of dormancy, but may be triggered to proliferate through largely unknown factors. We now demonstrate that the induction of fibrosis, associated with deposition of type I collagen (Col-I) in the in vivo metastatic microenvironment, induces dormant D2.0R cells to form proliferative metastatic lesions through β1-integrin signaling. In vitro studies using a 3D culture system modeling dormancy demonstrated that Col-I induces quiescent D2.0R cells to proliferate through β1-integrin activation of SRC and FAK, leading to ERK-dependent myosin light chain (MLC) phosphorylation by myosin light chain kinase (MLCK) and actin stress fiber formation. Blocking β1-integrin, Src, ERK or MLCK by shRNA or pharmacologic approaches inhibited Col-I-induced activation of this signaling cascade, cytoskeletal reorganization and proliferation. These findings demonstrate that fibrosis with type I collagen enrichment at the metastatic site may be a critical determinant of cytoskeletal reorganization in dormant tumor cells leading to their transition from dormancy to metastatic growth. Thus, inhibiting Col-I production, its interaction with β1-integrin and downstream signaling of β1-integrin may be important strategies for preventing or treating recurrent metastatic disease.
Keywords: tumor cell dormancy, metastasis, fibrosis, integrin β1 signaling, extracellular matrix
INTRODUCTION
Metastatic disease is the major cause of mortality for breast cancer patients and may occur years or even decades after successful treatment of the primary tumor by surgery and adjuvant therapy. Recent evidence indicates that tumor cell dissemination may be an early event in the disease process (1, 2), yet little is understood about the biologic mechanisms that ensure the survival of a presumably small subset of disseminated tumor cells that lay dormant for many years. Likewise, the critical triggers that regulate the transition of dormant tumor cells into a proliferative state leading to clinical recurrence remain unknown. It has been proposed that dormant tumor cells may exist in a quiescent state for many years or, alternatively, as micrometastases whose cellular proliferation is balanced by apoptosis (3–6) and which can progress to clinical disease once an angiogenic process is activated to support continued tumor growth (7).
The microenvironment has been increasingly recognized as a critical regulator of cancer progression (5, 8–10, 11) and is thought to be a major factor determining survival and growth of disseminated tumor cells at preferential metastatic sites (12). The extracellular matrix (ECM), a key component of the microenvironment, is in immediate contact with tumor cells, regulates aspects of both normal and tumor cell functions and provides a critical source for growth, survival, motility, and angiogenic factors that significantly affect tumor biology and progression. Alterations in the expression of ECM-related genes including fibronectin and type I collagen (Col-I), have been identified in gene expression signatures related to poor prognosis and metastases in breast cancers (13–16). Fibrotic foci with Col-I are often observed in primary tumors and lymph node metastases of breast cancer patients at high risk of recurrence (17, 18). High levels of pro-collagen type I, a marker for Col-I synthesis, have been identified in the serum of patients with recurrent breast cancer (19).
We have recently demonstrated that ECM composition plays a critical role in determining whether solitary dormant tumor cells remain quiescent or begin to actively proliferate using the well-characterized D2.0R/D2A1 mammary cell line model system to study dormant vs. metastatic proliferative growth (20). These cell lines derived from tumors arising from implants of the same D2 hyperplastic alveolar nodule line display distinct metastatic properties (21, 22). Whereas D2A1 cells form metastatic lesions after a few weeks in mice when disseminated to multiple sites by venous injection, D2.0R cells remain dormant for months with occasional formation of metastatic lesions. Using a modified 3D culture system, we demonstrated that the quiescent or proliferative behavior of these cells could be recapitulated in vitro (20, 22) and regulated by the presence of the ECM component fibronectin through activation of β1-integrin (Intβ1). Additionally, we demonstrated that significant alterations in cytoskeletal structure are critical for the dormant-to-proliferative switch requiring the phosphorylation of myosin light chain by myosin light chain kinase (MLCK).
In this study, we demonstrate in vivo that the prior induction of fibrosis with Col-I deposition at a metastatic site results in a dramatic Intβ1-mediated increase in proliferative, metastatic lesions of D2.0R cells, which otherwise remain dormant in normal lungs. Using the in vitro dormancy model, we demonstrate that the proliferative response to Col-I is mediated through Intβ1 and requires Src and focal adhesion kinase (FAK) activation, extracellular signal-regulated kinase (ERK)-dependent activation of MLCK, phosphorylation of MLC, and actin stress fiber formation.
This is the first study to our knowledge that functionally demonstrates that fibrosis with Col-I enrichment at the metastatic site can trigger the dormant-to-proliferative switch through Intβ1 signaling and cytoskeletal reorganization in dormant tumor cells leading to metastatic disease. These results suggest that inhibiting growth-promoting signaling from the ECM through signaling downstream of Intβ1 may be an important strategy for preventing or treating recurrent metastatic disease.
MATERIALS and METHODS
Cell lines and culture
Mouse mammary cancer D2.0R and D2A1 cells (21) (kindly provided by Ann Chambers, London Cancer Center, Ontario) were cultured as previously described in 2-D or 3-D Cultrex® (BME) (Trevigen Inc, Gaithersburg, MD) (20) or in BME mixed with neutralized rat tail collagen I (final concentration 2mg/ml). Cell morphology was imaged by confocal microscopy (LSM-META 510 Zeiss MicroImaging, Inc, Thornwood, NY). ML-7 (Biomol International L.P Plymouth Meeting, PA), W13 and U0126 (Calbiochem, San Diego, CA), and PP1 (Biomol International L.P), were used to inhibit MLCK, calmodulin, ERK and Src activities, respectively. Anti-Intβ1 antibody clone 9EG7 (azide-free) was used to antagonize Intβ1 function and non-specific IgG was used as a control (BD Biosciences, San Jose, CA). Sublines of D2.0R and D2A1 cells were generated to express GFP using the pSICO lentivirus (kindly provided by Tyler Jacks, MIT)
Proliferation assay
2×103 cells were resuspended in 100 μl DMEM low glucose supplemented with 2% FBS +2% BME, or 2% FBS + BME+Col-I respectively, and grown on 96 well plates coated with 50μl BME or BME+ Col-I. Proliferation was measured as described previously (20). Experiments were repeated 3 times with 8 replicates each.
shRNA silencing experiments
Cells were transfected with PLKO-1 plasmids expressing either short hairpin RNAs targeting Src (shSrc) (Clone ID: TRCN0000023596) or scrambled shRNA (Open Biosystems, Huntsville, AL) and cultured in the 3D system 48h later as previously described (20). Additionally, total protein extracts were isolated from the transfected cells in 2D culture at 72 h for western blot analysis. For stable knockdown of Intβ1, cells were transduced with Mission shRNA lentiviral particles targeting either mouse Intβ1 (sh-Intβ1) or with non-target sh-RNA (sh-NT) control (Sigma, St. Louis, MO) and selected with puromycin as per the manufacturer’s protocol.
Western blot
Western blot was carried out as previously described (20). The following primary antibodies were used: goat anti-mouse Intβ1 (0.2μg/ml) (R&D system, Minneapolis, MN); monoclonal antibody against Src (1:1000) (Upstate cell signaling solutions, Temecula, CA); and monoclonal antibody against β-actin (1:40,000) (Sigma, St. Louis, MO).
Animal studies
All mice were treated in accordance with the guidelines of the Animal Care and Use of Laboratory Animals (NIH publication No. 86-23, 1985) under an approved animal protocol and in accordance with the guidelines of the Canadian Council of Animal Care.
Experimental metastasis assays
1×106 D2A1-GFP cells were tail-vein injected into 6–8 week-old female BALB/c-nu/nu athymic mice (Charles Rivers Laboratories). Lungs were removed 2 weeks post-injection, inflated with PBS and analyzed by fluorescent single cell whole organ microscopy (SCOM) imaging (Leica DM IRB) as previously described (20). 100X images of the total external surface of each lung were sequentially captured and analyzed using OpenLab software (20, 23). Lungs were then frozen in OCT (Sakura, Torrance, CA) and immunofluorescence was performed on frozen sections.
Induction of pulmonary fibrosis
Eight week-old female CD1/nu/nu athymic mice received 5×108 PFU of adenoviral vector expressing either active TGFβ1 (Ad-TGFβ1223/225) to induce fibrosis or adenovirus-null vector control (Ad-empty) in 20μl PBS to the lungs as previously described (24). 21 days post-infection, 1×106 D2.0R-GFP cells, D2.0R-GFP+non-target-shRNA or D2.0R-GFP+Intβ1 shRNA cells were tail vein-injected to untreated mice or mice receiving Ad-empty or Ad-TGFβ-1223/225. Lungs were removed 4 weeks post-cell injections, inflated with PBS and frozen in OCT for analysis by SCOM.
Immunofluorescence Staining
Cells were cultured in 8 well chamber glass slides fixed and blocked as previously described (20). The following primary antibodies were used: Armenian hamster monoclonal antibody to Intβ1–(FITC) (Abcam, Cambridge, MA), Rabbit polyclonal to FAK Y397 (Abcam, Cambridge, MA), antibody against di-phosphorylated MLC (25) (kindly provided by Dr. Sabina Jankowska, Eisai London Research Laboratories, London UK), phospho-ERK1/ERK2 (Cell Signaling Technologies, Danvers, MA) and Alexa-Fluor ® 488 Phalloidin (Molecular Probes, Eugene, Oregon). Co-staining for Src Y416 and FAKY397: Cells were blocked and stained with M.O.M kit FMK-2201 (Vector laboratories Inc, Burlingame, CA) and incubated with SrcY416 monoclonal antibody (9A6) (Assay Designs, Ann Arbor, MI). For cell viability staining: Calcein AM staining (Trevigen Inc., Gaithersburg, MD) was utilized according to the manufacturer’s protocol
For Col-I staining, frozen lung sections were prepared as previously described (20) and incubated with goat anti-type I Collagen (1:100) (SouthernBiothec, Birmingham, Alabama).
Statistical analyses
Student’s t-test was used for proliferation assays. Statistical analyses of metastatic lesion measurements by SCOM: The pixel counts of GFP-positive metastatic lung lesions were quantified for the entire surface area of each lung. Lesions with >10 and <1000 pixels represented individual tumor cells; lesions with >1000 pixels represented multi-cellular metastatic lesions. Total tumor burden/lung was represented by the total number of pixels detected on the entire surface of each lung. Average tumor burden/lung was represented as the median size of the metastatic lesions. Single-cell lesions (<1000 pixels) and multi-cellular clusters (>1000 pixels) were quantified and the distribution of the sizes of metastatic lesions (represented by the number of pixels per lesion) per lung was calculated. Statistical analyses of differences in the distribution of tumor burden and metastatic lesion size between groups utilized unpaired Student’s t test for two-group comparisons and one-way analysis of variance (ANOVA) planned comparisons for multiple groups. The contrast comparisons tested the differences between fibrotic lungs vs. normal lungs for each cell line, and between fibrotic lungs in the different cell lines, after subtracting the control baseline value of the normal lungs for each cell line. Both unadjusted and Bonferroni corrected P-values were determined and considered significant if <0.05.
RESULTS
Fibrosis triggers metastatic outgrowth of dormant D2.0R cells
Mice instilled with Ad-TGFβ1 developed fibrosis by 3 weeks with extensive deposition of matrix including Col-I, whereas mice treated with Ad-empty or left untreated did not develop fibrosis (Fig. 1A and Supp-Fig. 1 and 2A). 21 days post-viral transduction, after virus clearance and absence of exogenous viral TGFβ1 expression (24, 26) D2.0R-GFP cells were injected via tail vein into all three mice groups. Lungs were removed 4 weeks later (Supp-Fig. 1) and tumor cells in the lungs were imaged by SCOM (Fig. 1B–D). Since initial experiments demonstrated that growth of D2.OR-GFP cells in the Ad-empty-instilled lungs and untreated lungs was not significantly different (Supp-Fig. 2B–D), subsequent experiments were conducted using either Ad-empty or Ad-TGFβ1-treated mice.
Figure 1. Fibrosis induces the dormant-to-proliferative switch of D2.0R cells in the lung.

A) Left: Lung sections of Ad-empty (no fibrosis) and Ad-TGFβ1 treated (fibrosis) mice stained for Col-I (red). Right: Metastatic outgrowth of D2.0R-GFP cells (green) in fibrotic lungs stained for Col-I (red) (immunofluorescence, confocal microscopy, X63). B) Total D2.0R-GFP tumor cell burden/lung in fibrotic lungs (n=5) compared to non-fibrotic lungs (n=5; ***p ≤0.001). C) Average size of metastatic lesions/lung in fibrotic lungs (n=5) compared to non-fibrotic lungs (n=5; **p ≤0.01). D) Percentage of single cells vs. multicellular proliferative metastatic lesions in non-fibrotic (n=5) and fibrotic lungs (n=5). Insert: SCOM images of D2.0R-GFP lung lesions in mice, x100.
Mice with fibrotic lungs (Ad-TGFβ1) exhibited a significant increase in proliferative metastatic lesions with a marked increase in collagen deposition (Fig. 1A) compared to Ad-empty control mice (Fig. 1B–D). Total tumor burden per lung was 18-fold higher in mice with fibrotic lungs (6.3×106 pixels) compared to mice that received null vector (3.5×105 pixels) (Fig. 1B p ≤ 0.001). The average size of the metastatic lesions was 18-fold higher in mice with fibrotic lungs (2.3×105 pixels) compared to mice receiving Ad-empty (1.3 ×104 pixels) (Fig. 1C; p ≤ 0.01). Multicellular metastatic lesions and individual metastatic cells expressing GFP were quantified on the entire lung surface. Fluorescence density >1000 pixels represented multicellular, proliferative metastatic lesions, whereas foci <1000 pixels indicated individual, dormant metastatic cells (Fig. 1D insert). 79 ± 3.3% of the foci in fibrotic lungs were multicellular lesions, with only 21 ± 3.3% of the foci persisting as single, quiescent cells. In contrast, only 40 ± 10.8% of the lung lesions were multi-cellular in mice without fibrosis (p ≤ 0.01), while 60% ± 10.8% of the lung lesions persisted as single quiescent cells (Fig. 1D).
Although Ad-TGFβ1 vector no longer produces TGFβ1 at the time of cell injections into mice, we wished to determine whether TGFβ1 might be capable of initiating the dormant-to-proliferative switch. D2.0R cells cultured in 3-D with BME remained quiescent and did not proliferate in response to TGFβ1. Interestingly, TGFβ1 slightly increased the proliferation of D2.0R cells, which were induced to proliferate with Col-I. (Supp-Fig. 2E, F). These results strongly suggest that TGFβ1 is not an initiating factor to induce the proliferation of dormant D2.0R cells in vivo, but may augment their proliferative response after induction of the dormant-to-proliferative switch by Col-I.
Col-I triggers the transition from dormancy to proliferation through Intβ1
Given that Col-I is enriched in the fibrotic environment, we explored whether this ECM protein might play a direct role in the dormant-to-proliferative switch. D2.0R and D2A1 cells were either cultured on BME or on BME+Col-I. While D2.0R and D2A1 cells cultured in BME alone displayed a rounded morphology at 24h, both cell lines cultured in BME+Col-I acquired a flattened, spindle morphology within 24h (Supp-Fig. 4A). D2.0R cells in BME remained quiescent during the nine-day assay period, while D2.0R cells cultured in BME + Col-I proliferated within 24h (Fig. 2A; p ≤ 0.0001). Similarly, supplementation of BME with Col-I accelerated the transition from quiescence to proliferation of D2A1 cells (Fig 2A; p ≤ 0.0001).
Figure 2. Col-I induces the transition from quiescence to proliferation of D2.0R and D2A1 cells through Intβ1.
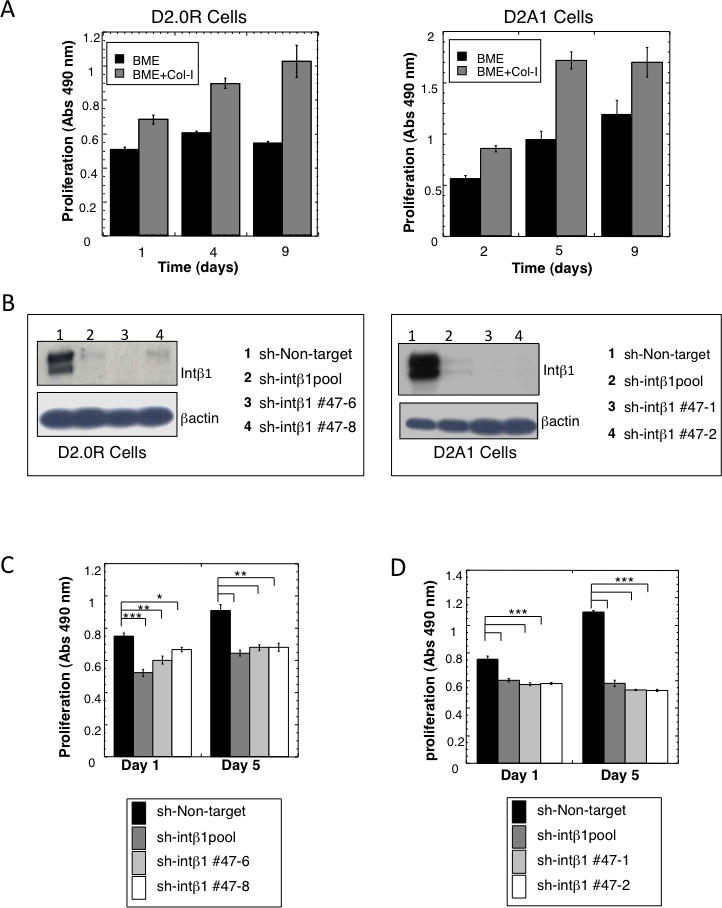
A) Proliferation of D2.0R and D2A1 cells in BME+Col-I, compared to cells grown in BME (mean ± SE, n=8, p ≤0.0001). B) Western blot for Intβ1 expression. Left panel: D2.0R cell lines. Right panel: D2A1 cell lines. C–D) Proliferation of D2.0R (C) and D2A1 cells (D) in BME+ Col-I (mean ± SE; n=8). (*p ≤0.05; **p ≤0.01; ***p ≤0.0001). Representative results of three independent experiments for all data shown.
Since Intβ1 serves as a receptor for Col-I, we examined whether the Col-I induced dormant-to proliferative switch was dependent on Intβ1 expression. sh-Intβ1 or sh-NT control was stably expressed in pooled and selected clones of D2.0R cells (dormant phenotype) and D2A1 (metastatic) cells (Fig. 2B). Both D2.0R and D2A1 cells with reduced Intβ1 expression were significantly less responsive to Col-I induced proliferation (Fig. 2C&D; p ≤ 0.0001) than parental cells or cells with the non-target shRNA vector. As demonstrated for quiescent D2.0R cells cultured on BME, quiescent D2.0R and D2A1 cells with reduced Intβ1 expression remained viable as demonstrated by calcein AM staining while maintaining a rounded morphology when cultured on BME+Col-I (Supp-Fig. 4B).
Intβ1 mediates the switch of solitary dormant D2.0R tumor cells to metastatic growth in vivo
Mice were tail vein-injected with D2.0R-GFP cells expressing shRNA for Intβ1 (clone #47-6,D2.0R-sh-Intβ1), or D2.0R-GFP expressing non-target shRNA (D2.0R-sh-NT). Lungs were removed 4 weeks post-injection and analyzed by SCOM (Fig. 3A–C). Metastatic burden in the lungs was analyzed by ANOVA in relation to both presence or absence of fibrosis and the Intβ1 expression (Fig. 3A). A significant 5-fold difference in the total tumor burden/lung in mice with fibrotic lungs receiving D2.0R sh-non target cells (sh-NT) compared to mice with non-fibrotic lungs was observed (n=8; p=0.018 unadjusted, p=0.05 with Bonferroni correction). Importantly, no difference was observed in total burden between normal and fibrotic lungs receiving D2.0R-sh-Intβ1 cells (sh-Intβ1) (n=10; p=0.89 unadjusted, p=1 with Bonferroni correction), indicating that fibrosis is associated with significant induction of metastatic lesions that could be prevented by the loss of Intβ1 expression in D2.0R cells. Viable single dormant cells were observed in these lungs, suggesting that the reduction in metastatic burden was not due to the lack of cell viability as a result of the reduction in Intβ1 expression, in keeping with the in vitro results above (Supplementary Figure 4B). Comparing the fibrosis effect only, revealed that in fibrotic lungs the total tumor burden per lung of D2.0R-sh-NT cells was 5-fold higher then the total tumor burden/lung of D2.0R cells with low expression of Intβ1 (sh-Intβ1) (Figure 3A). However, this did not reach statistical significance (p=0.086 unadjusted, p=0.26 with Bonferroni correction) due to the higher error variance when the previous two class comparisons were directly compared.
Figure 3. Loss of Intβ1 expression inhibits the fibrosis-induced transition from dormancy to metastatic outgrowth in mice.

A) Fold difference in total tumor burden per lung for each cell line in response to fibrosis (total pixels in fibrotic lungs/total pixels in non-fibrotic lungs). Fibrosis significantly increases total tumor burden/lung of D2.0R-sh–NT cells compared to non-fibrotic lungs (*p=0.018 unadjusted, p=0.05 with Bonferroni correction [BFC]), whereas inhibition of Intβ1(sh-Intβ1) abolishes the response to fibrosis (no significant difference, p=0.89 unadjusted, p=1 with BFC). [sh-NT - sh-Intβ1] represents the comparison between the sh-NT class analysis vs sh- Intβ1 analysis (ns). B) Average size of the metastatic lesions in fibrotic lungs (Ad-TGFβ) from D2.0R-GFP- sh-NT cells and D2.0R-GFP-sh-Intβ1 cells (*p=0.00014 uncorrected; p=0.00042641 with BFC) as well as for D2.0R-GFP-sh-non-target (sh-NT) cells in non-fibrotic lungs (Ad-empty) (p=0.013 unadjusted; p=0.039 with BFC). C) Percentage of single cells vs. proliferative metastatic lesions in non fibrotic vs. fibrotic lungs of D2.0R-GFP-sh-NT or D2.0R-GFP sh-Intβ1 cells. D2.0R-GFP- sh-NT cells had a significantly higher percentage of multi-cellular, proliferative lesions in fibrotic lungs compared to D2.0R-GFP-sh-Intβ1 cells (p=0.001 uncorrected; p= 0.003 with BFC) as well as for D2.0R-GFP- sh-NT cells in non-fibrotic lungs (p=1.75E-05 uncorrected; p= 5.25E-05 with BFC).
The average size of metastatic lesions in the fibrotic lungs was significantly higher for D2.0R-sh-NT lesions (3.1×105 pixels) compared to D2.0R-sh-Intβ1 lesions (812 pixels) (p ≤ 0.05; Fig. 3B). Similarly, the average size of the metastatic lesions of D2.0R-sh-NT cells in the fibrotic lungs was significantly higher compared to D2.0R-sh-NT cell lesions in non-fibrotic lungs (p ≤ 0.001; Fig. 3B). Importantly, as shown in Fig. 3C, the majority of D2.0R-sh-Intβ1 cells persisted as single cells in lungs with or without induced fibrosis (82%± 12.5% and 97% ± 3.3%, respectively, p = n.s.). In contrast, mice with pre-existing lung fibrosis and D2.0R-sh-non target cells had only 7± 4.2% single cell foci and 93± 4.2% proliferative, multicellular foci compared to mice with fibrotic lungs injected with D2.0R-sh-Intβ1 cells exhibiting only 18% ± 12.5% proliferative foci (p ≤ 0.01; Fig. 3C).
Similar results were obtained using metastatic D2A1 cells with reduced Intβ1 expression (Supp-Fig. 5 A–C). Furthermore, the metastatic lesions that developed from D2A1 sh-non target-GFP cells in normal lungs exhibited significant expression of Col-I, whereas regions in the same lungs without D2A1 metastatic lesions did not exhibit significant Col-I expression (Supp-Fig. 5D) further highlighting the association between proliferative growth and Col-I expression for D2A1 cells.
Intβ1 activation by Col-I induces the phosphorylation of Src and FAK
Interactions between focal adhesion kinase (FAK) and Src are known to mediate integrin signaling (27). Within 24h, D2.0R cells on BME+Col-I proliferated with an associated increase in FAKY397 phosphorylation, which co-localized with Intβ1. These changes were absent in cells cultured only on BME (Supp-Figs 6A and 7A). Inhibition of Intβ1 in D2.0R cells cultured on BME+Col-I with either αIntβ1 antibody (αIntβ1Ab) or shRNA led to a marked reduction in both in pFAKY397 and co-localization with Intβ1 compared to controls (Supp-Figs 6B–C & 7A–B). BME+Col-I significantly enhanced phosphorylation and co-localization of pSrcY416 and pFAKY397 in D2.0R cells (Fig. 4A).
Figure 4. Src activation is required for Col-I-induced FAKY397 phosphorylation and the transition from quiescence to proliferation of D2.0R cells.

A, C) Co-immunofluorescence staining for SrcY416, FAKY397 and nuclear staining with DAPI. A) Cells cultured on BME and BME+Col-I for 24h. C) D2.0R cells cultured on BME+Col-I for 24h with nonspecific IgG (100μg/ml) or D2.0R non-target shRNA cells or D2.0R cells treated with anti-Intβ1 antibody (100μg/ml) or pooled D2.0R cells and D2.0R-sh-Intβ1 cells B) Co-immunofluorescence staining for Intβ1, FAKY397 and nuclear staining with DAPI of D2.0R cells cultured on BME+Col-I for 24h treated either with PP1 (10 μM) or sh-Src. D) Proliferation of D2.0R cells in BME or BME+Col-I with or without 10 μM PP1, non-target shRNA, or sh-Src (n=8; mean ±SE; P ≤ 0.0001). Confocal microscopy, x63, white bar = 20um. Representative of three independent experiments.
Inhibition of Src activity in D2.0R cells cultured on BME+Col-I using the inhibitor PP1 or Src shRNA (Supp-Fig. 8) prevented phosphorylation of FAKY397, co-localization with Intβ1 (Fig. 4B) and proliferation (p ≤ 0.0001) (Fig 4D). Inhibition of Intβ1 with αInt β1AB or shRNA significantly reduced p FAKY397 and p SrcY416 and their co-localization compared to controls (Fig. 4C and Supp-Fig 7A–B).
FAK and Src activate the extracellular signal regulated kinase (ERK) leading to proliferation (28). Quiescent D2.0R cells cultured on BME had minimal activated ERK (pERK) (Figure 5A and Supp-Fig. 7A). The increased pERK, actin stress fiber formation and proliferation in D2.0R cells cultured on BME+Col-I (Fig. 5A) were inhibited with αIntβ1AB (Fig. 5B), Intβ1 shRNA (Fig. 5C and Supp-Fig 7A–B), or through inhibition of Src activity with PP1 or Src shRNA (Fig. 5D). Scrambled shRNA had no effect (data not shown).
Figure 5. CoI-I induces actin stress fiber formation and activation of ERK through Intβ1 and Src.

A–D) Co-immunofluorescence staining of D2.0R cells for pERK, F-actin with phalloidin and nuclear staining with DAPI after A) 24h culture on BME or BME +Col-I; B) D2.0R cells on BME+Col-I treated with non-specific IgG (100μg/ml) or anti-Intβ1 antibody (100μg/ml); C) sh-non target and sh-Intβ1; D) 10 μM PP1, or sh-Src. Confocal microscopy x63, white bar = 20 microns. Representative of three independent experiments.
Col-I induced Intβ1 signaling activates ERK leading to phosphorylation of MLC by MLCK and actin stress fiber formation
The dormant-to-proliferative switch of D2.0R cells requires actin stress fiber formation through phosphorylation of MLC (pMLC) by MLCK (20) which can be regulated by ERK (29). D2.0R cells in BME+Col-I demonstrated a marked increase in pMLC and actin stress fibers (Fig. 6A and Supp-Fig. 7A) compared to BME alone. Inhibition of MLCK directly by ML-7 (30) or through inhibition of upstream signaling molecules that regulate MLCK activity (ERK using U-0126 or Ca2+ calmodulin using W13) reduced pMLC, inhibited actin stress fiber formation and proliferation (p ≤ 0.0001) (Fig. 6A–B). Inhibiting Intβ1 function also reduced pMLC and actin stress fiber formation compared to controls (Fig. 6C and Supp-Fig 7A). Inhibiting Src activity with PP1 or shRNA also reduced pMLC, actin stress fiber formation (Fig. 6D) and proliferation.
Figure 6. Col-I mediated signaling through Intβ1 induces phosphorylation of MLC by MLCK leading to proliferation of quiescent cells.

A, C–D) Co-immunofluorescence staining for phospho-MLC, F-actin and nuclear localization with DAPI. of D2.0R cells cultured for 24h on BME or BME+Col-I A) either untreated or treated with ML-7 (5 μM), U-0126 (10μM) or W-13 (10μM). B) Proliferative response to Col-I with or without inhibitors (n=8, mean ±SE; P ≤0.0001). C) D2.0R cells on BME+Col-I with non-specific IgG (100μg/ml), anti-Intβ1 antibody (100μg/ml) and D2.0R cells stably expressing non-target-shRNA and D2.0R sh-Intβ1cells (pooled or clone #47-6). D) D2.0R cells cultured on BME+Col-I (Control) or treated with 10 μM PP1, or sh-Src. Confocal microscopy x63, white bar = 20 microns. Representative results of three independent experiments.
DISCUSSION
Our results indicate that tumor cells already carrying critical genetic alterations can remain dormant or be triggered to proliferate by changes occurring in their microenvironment. Thus, tumor dormancy may be maintained in an initially non-permissive microenvironment, but transition to a proliferative state may result from extrinsic changes within the microenvironment. This study demonstrates in vivo for the first time how changes in the ECM composition produced by stromal cells can initiate the transition of solitary dormant cells to metastatic proliferation. Several gene expression studies have identified ECM-related genes (including Col-I) in prognostic signatures for poor outcome, metastases and tumor recurrence (13, 15). In this study, we demonstrate that an in vivo fibrotic environment that contains high amounts of Col-I, as well as other ECM proteins, may induce the transition from a dormant-to-proliferative state through Intβ1. Mice with fibrotic lungs displayed greatly incrased numbers and sizes of proliferative, metastatic lesions compared to mice without fibrosis.
Intβ1 is the primary receptor that interacts with Col-I. Previous studies have reported Intβ1’s important role in different stages of tumor progression (31–35). We demonstrated that Intβ1 is a key regulator in the switch from dormancy to metastatic growth. Reduced expression of Intβ1 in D2.0R cells disseminated to pre-existing fibrotic lungs resulted in a 5-fold reduction in proliferative metastatic lesions compared to control cells. Similarly, in the context of a normal lung environment, loss of Intβ1 expression in metastatic D2A1 cells resulted in a 9-fold reduction of metastatic lesions and lower tumor burden compared to D2A1-sh-NT cells. Metastatic lesions arising from D2A1 cells in non-fibrotic lungs were associated with significant deposition of Col-I suggesting that the metastatic D2A1 cells are able to induce a Col-I stromal response to stimulate their growth, whereas D2.0R cells require an exogenous fibrotic stimulus to initiate their proliferative response. Previous studies in head and neck carcinoma have demonstrated critical cross-talk between urokinase receptor and Intβ1 in tumor dormancy (36, 37). We have determined that Col-I increases Intα1 mRNA, but not α2 or α7 integrin in D2.0R cells in vitro, suggesting that α1β1 is the most likely candidate Col-I receptor on D2.0R cells (data no shown).
Based upon our previous study, fibronectin may also contribute to the dormant-to-proliferative switch in this in vivo fibrosis model system since fibronectin also activates Intβ1 in D2.0R and D2A1 cells (20). While we do not exclude the influences of other factors in the microenvironment of the fibrotic lungs, such as a possible role of TGFβ in sustaining/enhancing the proliferation of the emerging micrometastasis, our results using the 3D in vitro model system for dormancy (20) demonstrated that a signaling cascade induced by Col-I is sufficient to trigger the dormant–to-proliferative switch of D2.0R cells through activation of Intβ1, Src, FAK, and ERK leading to phosphorylation of MLC by MLCK, and actin stress fiber formation - all critical for the proliferative response.
Our previous study demonstrated the critical role of actin stress fiber formation in the transition from dormancy to proliferative growth. In this study, we have further demonstrated that actin stress fiber formation is regulated through Col-I activation and is dependent upon Intβ1, FAK, Src, and ERK, a key regulator of the cell cycle and MLCK (29), which phosphorylates MLC leading to actin stress fiber formation. Furthermore, we demonstrated that activated ERK is critical for actin stress fiber formation through phosphorylation of MLC by MLCK, required for the transition from quiescence to proliferation.
Our results are consistent with several clinical correlations between enriched stromal Col-I and cancer recurrence and reduced survival. Women with high breast density, associated with enriched stromal Col-I have an increased risk for local recurrence after mastectomy or radiotherapy (38). Fibrotic foci (FF), are found around some invasive ductal carcinomas and their recurring lesions residing at lymph nodes and bones. Notably, patients with FF are at higher risk of developing bone and lymph node metastasis, and disease recurrence (17, 39). Osteosclerotic breast cancer bone metastases exhibit marrow fibrosis and new bone formation (40). Therefore, enriched stromal Col-I in a more fibrotic microenvironment or induced by the residing tumor cells may provide a fertile ‘soil’ for the transition from dormancy to metastatic growth.
In summary, we have demonstrated for the first time to our knowledge, how fibrosis with Col-I enrichment may serve as a critical element in modifying the ‘soil’ of metastatic sites and activating dormant tumor cells to proliferate. These results suggest that inhibiting the interaction between dormant tumor cells and growth-promoting changes in the ECM that signal through Intβ1 may be an important new avenue to prevent disease recurrence.
Supplementary Material
Acknowledgments
We thank Drs. Marry Ann Step, Glenn Merlino and Dr. Peter Blumberg for valuable discussions, Dr. Chand Khanna for use of SCOM equipment, the LRBGE Fluorescence Imaging Core, and Anthony Vieira and Dr. Zi-Yao Liu for technical assistance.
Footnotes
The authors have no conflicts to disclose.
Financial Disclosure: This research was supported by the Intramural Program of NIH, Center for Cancer Research, NCI and the Canadian Institutes of Health Research.
References
- 1.Pantel K, Schlimok G, Braun S, et al. Differential expression of proliferation-associated molecules in individual micrometastatic carcinoma cells. J Natl Cancer Inst. 1993;85:1419–24. doi: 10.1093/jnci/85.17.1419. [DOI] [PubMed] [Google Scholar]
- 2.Braun S, Vogl FD, Naume B, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med. 2005;353:793–802. doi: 10.1056/NEJMoa050434. [DOI] [PubMed] [Google Scholar]
- 3.Townson JL, Chambers AF. Dormancy of solitary metastatic cells. Cell Cycle. 2006;5:1744–50. doi: 10.4161/cc.5.16.2864. [DOI] [PubMed] [Google Scholar]
- 4.Wikman H, Vessella R, Pantel K. Cancer micrometastasis and tumour dormancy. APMIS. 2008;116:754–70. doi: 10.1111/j.1600-0463.2008.01033.x. [DOI] [PubMed] [Google Scholar]
- 5.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–46. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–53. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- 7.Naumov GN, Bender E, Zurakowski D, et al. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst. 2006;98:316–25. doi: 10.1093/jnci/djj068. [DOI] [PubMed] [Google Scholar]
- 8.Fidler IJ. The organ microenvironment and cancer metastasis. Differentiation. 2002;70:498–505. doi: 10.1046/j.1432-0436.2002.700904.x. [DOI] [PubMed] [Google Scholar]
- 9.Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70:537–46. doi: 10.1046/j.1432-0436.2002.700907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu M, Polyak K. Microenvironmental regulation of cancer development. Curr Opin Genet Dev. 2008;18:27–34. doi: 10.1016/j.gde.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schedin P, Elias A. Multistep tumorigenesis and the microenvironment. Breast Cancer Res. 2004;6:93–101. doi: 10.1186/bcr772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massague J. New concepts in tissue-specific metastases. Clin Adv Hematol Oncol. 2003;1:576–7. [PubMed] [Google Scholar]
- 13.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 14.Qiu TH, Chandramouli GV, Hunter KW, Alkharouf NW, Green JE, Liu ET. Global expression profiling identifies signatures of tumor virulence in MMTV-PyMT-transgenic mice: correlation to human disease. Cancer Res. 2004;64:5973–81. doi: 10.1158/0008-5472.CAN-04-0242. [DOI] [PubMed] [Google Scholar]
- 15.Calvo A, Catena R, Noble MS, et al. Identification of VEGF-regulated genes associated with increased lung metastatic potential: functional involvement of tenascin-C in tumor growth and lung metastasis. Oncogene. 2008;27:5373–84. doi: 10.1038/onc.2008.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma XJ, Dahiya S, Richardson E, Erlander M, Sgroi DC. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 2009;11:R7. doi: 10.1186/bcr2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasebe T, Sasaki S, Imoto S, Mukai K, Yokose T, Ochiai A. Prognostic significance of fibrotic focus in invasive ductal carcinoma of the breast: a prospective observational study. Mod Pathol. 2002;15:502–16. doi: 10.1038/modpathol.3880555. [DOI] [PubMed] [Google Scholar]
- 18.Van den Eynden GG, Smid M, Van Laere SJ, et al. Gene expression profiles associated with the presence of a fibrotic focus and the growth pattern in lymph node-negative breast cancer. Clin Cancer Res. 2008;14:2944–52. doi: 10.1158/1078-0432.CCR-07-4397. [DOI] [PubMed] [Google Scholar]
- 19.Jensen BV, Johansen JS, Skovsgaard T, Brandt J, Teisner B. Extracellular matrix building marked by the N-terminal propeptide of procollagen type I reflect aggressiveness of recurrent breast cancer. Int J Cancer. 2002;98:582–9. doi: 10.1002/ijc.10187. [DOI] [PubMed] [Google Scholar]
- 20.Barkan D, Kleinman H, Simmons JL, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008;68:6241–50. doi: 10.1158/0008-5472.CAN-07-6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris VL, Koop S, MacDonald IC, et al. Mammary carcinoma cell lines of high and low metastatic potential differ not in extravasation but in subsequent migration and growth. Clin Exp Metastasis. 1994;12:357–67. doi: 10.1007/BF01755879. [DOI] [PubMed] [Google Scholar]
- 22.Naumov GN, MacDonald IC, Weinmeister PM, et al. Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res. 2002;62:2162–8. [PubMed] [Google Scholar]
- 23.Khanna C, Wan X, Bose S, et al. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med. 2004;10:182–6. doi: 10.1038/nm982. [DOI] [PubMed] [Google Scholar]
- 24.Bonniaud P, Kolb M, Galt T, et al. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol. 2004;173:2099–108. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- 25.Ratcliffe MJ, Smales C, Staddon JM. Dephosphorylation of the catenins p120 and p100 in endothelial cells in response to inflammatory stimuli. Biochem J. 1999;338 (Pt 2):471–8. [PMC free article] [PubMed] [Google Scholar]
- 26.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100:768–76. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gabarra-Niecko V, Schaller MD, Dunty JM. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003;22:359–74. doi: 10.1023/a:1023725029589. [DOI] [PubMed] [Google Scholar]
- 28.Yee KL, Weaver VM, Hammer DA. Integrin-mediated signalling through the MAP-kinase pathway. IET Syst Biol. 2008;2:8–15. doi: 10.1049/iet-syb:20060058. [DOI] [PubMed] [Google Scholar]
- 29.Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–92. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saitoh M, Ishikawa T, Matsushima S, Naka M, Hidaka H. Selective inhibition of catalytic activity of smooth muscle myosin light chain kinase. J Biol Chem. 1987;262:7796–801. [PubMed] [Google Scholar]
- 31.White DE, Kurpios NA, Zuo D, et al. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004;6:159–70. doi: 10.1016/j.ccr.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 32.Paszek MJ, Zahir N, Johnson KR, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 33.Park CC, Zhang H, Pallavicini M, et al. Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 2006;66:1526–35. doi: 10.1158/0008-5472.CAN-05-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kren A, Baeriswyl V, Lehembre F, et al. Increased tumor cell dissemination and cellular senescence in the absence of beta1-integrin function. EMBO J. 2007;26:2832–42. doi: 10.1038/sj.emboj.7601738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imanishi Y, Hu B, Jarzynka MJ, et al. Angiopoietin-2 stimulates breast cancer metastasis through the alpha(5)beta(1) integrin-mediated pathway. Cancer Res. 2007;67:4254–63. doi: 10.1158/0008-5472.CAN-06-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147:89–104. doi: 10.1083/jcb.147.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001;12:863–79. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park CC, Rembert J, Chew K, Moore D, Kerlikowske K. High mammographic breast density is independent predictor of local but not distant recurrence after lumpectomy and radiotherapy for invasive breast cancer. Int J Radiat Oncol Biol Phys. 2009;73:75–9. doi: 10.1016/j.ijrobp.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 39.Koyama T, Hasebe T, Tsuda H, et al. Histological factors associated with initial bone metastasis of invasive ductal carcinoma of the breast. Jpn J Cancer Res. 1999;90:294–300. doi: 10.1111/j.1349-7006.1999.tb00747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamby C, Guldhammer B, Vejborg I, et al. The presence of tumor cells in bone marrow at the time of first recurrence of breast cancer. Cancer. 1987;60:1306–12. doi: 10.1002/1097-0142(19870915)60:6<1306::aid-cncr2820600624>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.