

Abstract
A large number of risk alleles have been identified for multiple sclerosis (MS). However, how genetic variations may affect pathogenesis remains largely unknown for most risk alleles. Through direct sequencing of CD24 promoter region, we identified a cluster of 7 new single nucleotide polymorphisms in the CD24 promoter. A hypermorphic haplotype consisting of 3 SNPs was identified through association studies consisting of 935 control and 764 MS patients (P=0.001, odds ratio 1.3). The variant is also associated with more rapid progression of MS (P=0.016, log rank test). In cells that are heterozygous for the risk allele, chromatin immunoprecipitation revealed that risk allele specifically bind to a transcription factor SP1, which is selectively required for the hypermorphic promoter activity of the variant. In MS patients, the CD24 transcript levels associate with the SP1-binding variant in a dose-dependent manner (P=7x10-4). Our data revealed a potential role for SP1-mediated transcriptional regulation in MS pathogenesis.
Keywords: Multiple sclerosis (MS), SP-binding CD24, promoter, risk alleles, single nucleotide polymorphisms (SNP)
Introduction
MS is the most common neurological disease in young adults with a prevalence of about 0.1% in the Caucasian population. Although epidemiological studies, particularly the high concordance of identical twins, support a major genetic contribution [1], HLA-DRB1*15 remains the only known variant that increase MS risk by about 2-3 fold after several genome wide association and linkage analyses [2]. Additional MS risk alleles have been identified by genome-wide association studies [3-8]. However, it is estimated that no less than 50% of heritability of MS remains unexplained [9,10].
CD24 encodes a small glycosyl-phophatidyl inositol (GPI)-anchored glycoprotein that regulates both adaptive and innate immunity [11-17] and is critical for pathogenesis of experimental autoimmune encephalomyelitis [18,19], the widely used animal model for MS. Moreover, CD24 variations in both coding and non-coding regions have significant impacts on MS [20-25] and other autoimmune diseases [20,26-28]. Because no SNP has been reported in the promoter and intron of CD24 and because the sequence for human CD24 is incomplete, we first identified the full-length CD24 gene and sequenced the CD24 locus of 50 population control samples in order to identify CD24 variants that may be associated with risk and progression of MS. Using a combined genetic and functional analysis, we hereby identify a hypermorphic CD24 variant that are associated with risk and progression of MS potentially through specifically binding to transcriptional factor SP1.
Materials and methods
Human samples
Demographic and clinical information of control and MS patients enrolled are listed in Table 1. The enrollment and study design have been approved by the Institutional Review Boards of the Ohio State University, University of Michigan, and the ACP. As controls, a total of 935 human subjects from the general Caucasian population were obtained from the American Red Cross between 1999 and 2006 using leftover peripheral blood samples, as described previously [20,21]. Sporadic Caucasian MS samples were collected during similar period. Samples used include those described in previous studies [18,19] and those provided by ACP. The clinical diagnosis of MS type and the EDSS score [29] were provided by either Dr. Kottil Rammoham or ACP.
Table 1.
Demography and clinical data of subjects enrolled in the study
| Control | Sporadic MS | |
|---|---|---|
| n = 935 | n = 764 | |
| Women/Men | 614/321 | 559/205 |
| Age (years) (mean ± SD) | 46.7 ± 17.1 | 46.5 ± 11.0 |
| Ethnicity | ||
| Caucasian | 935 | 764 |
| African-American | - | - |
| Hispanic-American | - | - |
| Native American | - | - |
| Asian | - | - |
| Other | - | - |
| Age at onset (years) (mean ± SD) | 31.7 ± 10.0 | |
| EDSS | ||
| EDSS < 6.0 | 317 | |
| EDSS ≥ 6.0 | 182 | |
| Time to EDSS 6.0 (years) | 15.6 ± 10.7 | |
| Disease duration (years) | 14.9 ± 11.3 | |
| Clinical course | ||
| RR/SP/PR/PP/Devics | 520/186/1/55/2 | |
Data are expressed as mean ± standard deviation for age, age at onset, time to Expanded Disability Status Scale (EDSS; only across patients having reached EDSS 6.0) and disease duration. RR, relapsing-remitting; SP, secondary progressive; PR, Progressive Relapsing; PP, primary progressive.
Polymorphism identification
The CD24 locus was amplified by PCR. The PCR reactions were carried out in a 20μl volume containing ~20 ng of genomic DNA, 2.0μl 10X Pfx Amplification Buffer, 2.0μl 10X PCRx Enhancer solution, 0.6μl 10 mM mixture of deoxynucleotide triphosphates (dATP, dCTP, dGTP, and dTTP), 0.5μl 50 mM MgSO4, 10 pmol of each primer, and 0.4 unit of Platinum Pfx DNA polymerase (Life Technologies, Grand Island, NY). After a 10-min initial denaturation step at 95°C, 35 cycles of PCR reaction consisting of 95°C for 30 sec, 55°C for 30 sec, and 68°C for 90 sec were carried out, followed by a 10-min final extension step at 68°C in a thermal cycler (Veriti 96 wells, Life Technologies, Grand Island, NY). Potential polymorphisms in the promoter, exon 1 and intron 1 were discovered by DNA sequencing of PCR products from 264 MS samples (21 additional cases excluded due to sequencing failure), 301 control samples (15 additional samples were excluded due to sequencing failure) and 325 MSGG samples (1 additional sample excluded due to sequencing failure). The variants were identified using Mutation Surveyor 3.01 software (SoftGenetics LLC, State College, PA).
Allelic discrimination
Genomic DNA isolated from peripheral blood leukocytes was used to identify CD24 polymorphisms. PCR amplification was performed with P-534C-F (GTG GCA ATG CAC TTG CTC CAG GAC C) and P-442C-R (GCG AGC CAC ACA CGC CGC GCT GGG). The PCR reactions were carried out in a 20μl volume containing 20 ng of genomic DNA, 2.0μl 10X High fidelity PCR Buffer, Amplification Buffer, 2.0μl 10X PCRx Enhancer solution, 0.6μl 10 mM mixture of deoxynucleotide triphosphates (dATP, dCTP, dGTP, and dTTP), 0.5μl 50 mM MgSO4, 5 pmol of each primer, and 0.75 unit of Platinum Taq DNA polymerase High Fidelity (Life Technologies, Grand Island, NY). After a 5-min initial denaturation step at 95°C, 10 cycles of PCR reaction consisting of 95°C for 30 sec, 65°C for 30 sec, and 68°C for 30 sec and 25 cycles of PCR reaction consisting of 95°C for 30 sec, 60°C for 30 sec, and 68°C for 30 sec were carried out, followed by a 10-min final extension step at 68°C in a thermal cycler (Veriti 96 wells, Life Technologies, Grand Island, NY). The PCR products were electrophoresed on 2.5% agarose gels. Those with a band of 143 bp were scored positive for having at least one allele of P-534C-P-442C.
We tested the sensitivity the allele-specific PCR using 31 samples with known P-534C-P-442C haplotype and found the sensitivity to be 100%. However, among 630 subjects with no possible P-534C-P-246G haplotypes, 6 were found positive, yielding a false positive rate of 0.95%. These false positive samples, however, were all identified by subsequent sequencing (see below). Therefore the false positivity at this step does not affect the final results reported here.
The PCR products were then used in a Taqman Assay to determine the composition of P-492G/C in the PCR product. Samples were amplified in a model 7900-HT sequence system (Life Technologies, Grand Island, NY) using forward and reverse primers (P-492-Taqman-F, CGG AGG CGC GGA CTT T and P-492-Taqman-R, GGC CCA AGT TTC CTT TGT TTC C) along with FAM and VIC dye-labeled probes P-492G-probe-VIC (CTT ACC CCC CAA AAG A) P-492C-probe-FAM (TTA CCC CCG AAA AGA). If the genotype G or CG of P-492 was present, the haplotype CGC will be considered to be present on at least one allele. For those samples with P-492G genotypes, a new PCR reaction was carried out to amplified 1491 bp CD24 promoter, using primer F2 (GTT GGT CTG GAA CTC CTG ACC TCA GGT) and R2 (CCT CTG GGT GAA AGT GGG AA). Sanger sequencing was then used to determine homozygocity or heterozygocity of the CD24CGC allele. This step also identified the low rate (0.95%) of false positive samples in the allele-specific PCR reaction.
CD24 mRNA measurement
CD24 mRNA expression was analyzed using real-time PCR (Applied Biosystems ABI Prism 7500 Sequence Detection System, Life Technologies, Grand Island, NY). The average expression was determined by comparing the Ct (cycle number when a preset signal threshold is reached) of CD24 amplification with that of GAPDH. The primers are: CD24-realtime-F (TTC TCC AAG CAC CCA GCA) and CD24-realtime-R (TGG AAT AAA TCT GCG TGG GTA).
Promoter assay
The human CD24 promoter-luciferase gene vectors (pGL2-CD24) were constructed by inserting the DNA fragments from the proximal promoter region of CD24 into the pGL2 vector. The proximal promoter fragment (from 53 to 614 5’ of the translation starting site) covering six polymorphism sites (P-534, P-492, P-442, P-388, P-366 and P-246) were cloned into pGL2 to create human CD24 promoter luciferase plasmids. The luciferase activity was measured in a Veritas Microplate Luminometer (Turner BioSystems, Sunnyvale, CA) using a Dual Luciferase Assay System (Promega, Madison, WI).
Chromatin immunoprecipitation
ChIP of SP1 were carried out using breast cancer cell line MDA-MD453 according to published procedures [30]. The anti-SP1(Santa Cruz Biotechnology, sc-644) and anti-IgG antibodies (1 μg/mL) were used to pull down chromatin associated with SP1. The amounts of the specific DNA fragment were quantitated by real-time PCR and normalized against the genomic DNA preparation from the same cells. The sequences of ChIP real-time PCR primers are: P-534-F (AGAGATAACCCTGCCCGAG) and P-534-R (CCAAGTTTCCTTTGTTTCCC).
Statistical analysis
In the case-control population study, patients and normal controls were examined for any significant differences in their genotype (allele or haplotype) distributions in each of the CD24 polymorphisms at the population level. First, the Hardy-Weinberg equilibrium assumption was confirmed for each polymorphism the controls. We performed either Chi-square test or Fisher's exact test for each polymorphism by comparing the distribution of the genotypes of the cases to that of the normal controls. Then, using the counts of one of the genotypes as a reference, the ORs for the remaining genotype variants were computed. The associated 95% CIs for the ORs were obtained through a multivariate logistic regression model adjusted by age as a potential confounding factor using SPSS 16.0 software (SPSS Inc., Chicago, IL). The risk of MS associated with each haplotype having frequency >1% was compared with the most common haplotype using logistic regression models with the HAPSTAT software [31,32].
In survival analysis, we estimated the Kaplan-Meier survival curve for patients with each of the genotypes. We calculated the observed survival time of a patient as the time from the first day of symptoms to reaching EDSS 6.0 (in years) or to the day of the last follow-up visit if EDSS 6.0 had not been reached. In the latter situation, the recorded survival time was treated as a censored observation. Association between the estimated survival curves and the underlying genotypes or haplotypes were then assessed using a log-rank test. All analyses were performed by using SPSS 16.0 software (SPSS Inc., Chicago, IL).
Results
A novel haplotype in CD24 promoter associate with risk and progression of MS
We first identified the full-length DNA sequence as it was not available (submitted to NCBI database, accession number FJ226006). The localization of CD24 to 6q21 was confirmed by fluorescence in situ hybridization using the intronic probe (Figure S1). By sequencing the entire CD24 locus of 50 general population controls, we identified 7 new SNPs that are tightly clustered within a 553 base-pair (bp) region in the CD24 promoter (Figure 1). Since no SNP exist the 2743 bp intron and only 4 previously reported SNPs were found in the 2017 bp exon 2 [20,21], the promoter is the most polymorphic region in the CD24 gene.
Figure 1.

Identification of 7 new SNPs in CD24 promoter. A diagram of the SNP in the CD24 gene identified by sequencing of 301 Caucasians. All SNP in exon 2 are the same as previously reported, while all SNPs in the promoter region are new. The consensus sites for transcriptional factors are indicated in the promoter region.
To determine whether the new SNPs are associated with MS risks, we carried out a case/control study by sequencing the CD24 promoter of 264 MS case and 301 race-, gender- and age-matched controls (Table 1) [20,21]. This analysis reveals that none of the SNPs in the promoter region is associated with MS risk (Table S1). However, using sequencing data of 301 control and 264 control Caucasians, we identified 2 haplotypes of CD24 promoters, CCGCCCG or CCGCCTG, that are significantly associated with MS risk (Table 2). To establish a high risk allele that can be easily genotyped, we searched for a minimal haplotype. As shown in Table 2, combination of P-534C, P-492G and P-442C constitutes a haplotype that associates with risks, which we refer as CD24CGC. We therefore developed a PCR-based assay to identify heterozygous and homozygous samples. First, we amplified the samples that contained a P-534C-P-442C haplotype. The positive samples were further typed for the genotype of the P-442 position by Taqman-based approach. The promoter region of the samples that have at least one allele of CD24CGC was completely sequenced to determine whether they are heterozygous or homozygous for the CD24CGC allele. The cohort used consists of a total of 764 independent MS samples and 935 age-, race- and gender-matched Caucasian controls (Table 1). As shown in Table 3, 16.8% MS and 12.3% control samples are CD24CGC/CGC, yielding an OR of 1.57 (P=0.003). The heterozygous CD24CGC also showed a statistical trend (OR 1.21, P=0.08). The MS subjects are Caucasians in the United States and samples are obtained from two sources: the Ohio State University used in our previous study [20,21], and those from the Accelerated Cure Project (ACP, Waltham, MA 02451, USA). The distribution of the CD24CGC genotypes of the MS samples obtained from OSU and ACP are not significantly different (Table S2).
Table 2.
Associated CD24 haplotype: frequencies and OR against controls
| Haplotype | Cases, n = 264 | Controls, n = 301 | |||
|---|---|---|---|---|---|
| P-809/P-534/P-492/P-442/P-388/P-366/P-246 | % | % | OR | 95% CI | P |
| CAGCCCA | 47.5 | 52.2 | 1.00 | - | - |
| CCGCCCG | 24.5 | 19.6 | 1.37 | 1.02-1.85 | 0.038 |
| CAGTCCA | 9.9 | 9.8 | 1.11 | 0.74-1.67 | 0.625 |
| ACCCTCG | 3.3 | 5.2 | 0.69 | 0.37-1.27 | 0.234 |
| CCCCTCG | 3.2 | 4.0 | 0.89 | 0.47-1.69 | 0.722 |
| CCGCCTG | 5.3 | 2.9 | 2.07 | 1.11-3.87 | 0.023 |
| CCGTCCG | 0.0 | 2.6 | - | - | - |
| *XCGCXXX | 33.4 | 23.2 | 1.51 | 1.14-2.00 | 0.004 |
These haplotypes were identified based on a comprehensive analysis with all haplotypes (a total of 28) in the LD block being analyzed jointly.
A series of single haplotype analyses by comparing each haplotype with the control led to the same conclusion of the associated haplotypes qualitatively.
Table 3.
CD24 genotype and MS risk as determined by case/control study*
| Genotypes | Cases | Controls | OR | 95% CI | P |
|---|---|---|---|---|---|
|
| |||||
| n (%) | n (%) | ||||
| CGC/CGC | 126 (16.8) | 105 (12.3) | 1.57 | 1.17-2.11 | 0.003 |
| CGC/X** | 300 (39.9) | 324 (37.9) | 1.21 | 0.98-1.50 | 0.080 |
| X/X | 326 (43.4) | 426 (49.8) | 1.00 | ||
32 cases and 21 controls failed in the CGC haplotyping;
non-CGC allele.
To evaluate possible population stratification, we compared the frequency distribution of more than 1122 SNPs within a 30 megabase region in the X-chromosome using r = 0.8 as linkage threshold for marker selection and P = 0.001 as threshold for deviation from Hardy–Weinberg equilibrium in control group to delete questionable genotyping. The typing was done for a different project and the 216 cases and 236 controls were selected before the CD24 genotypes are known. As detailed in the Table S3, despite the high number of markers used, we failed to detect significant population stratification (λ < 1.0).
To determine whether the promoter variant is associated with more rapid progression of MS, we carried out survival analysis using Expanded Disability Status Scale (EDSS) 6.0 as the endpoint. As shown in Figure 2, while 50% of CD24CGC/CGC patients lost ability for independent walking within 18 years after first MS symptom, those with other genotypes reached the same endpoint in approximately 27 years. The association between CD24 genotype and MS progression is statistically significant (P=0.016).
Figure 2.
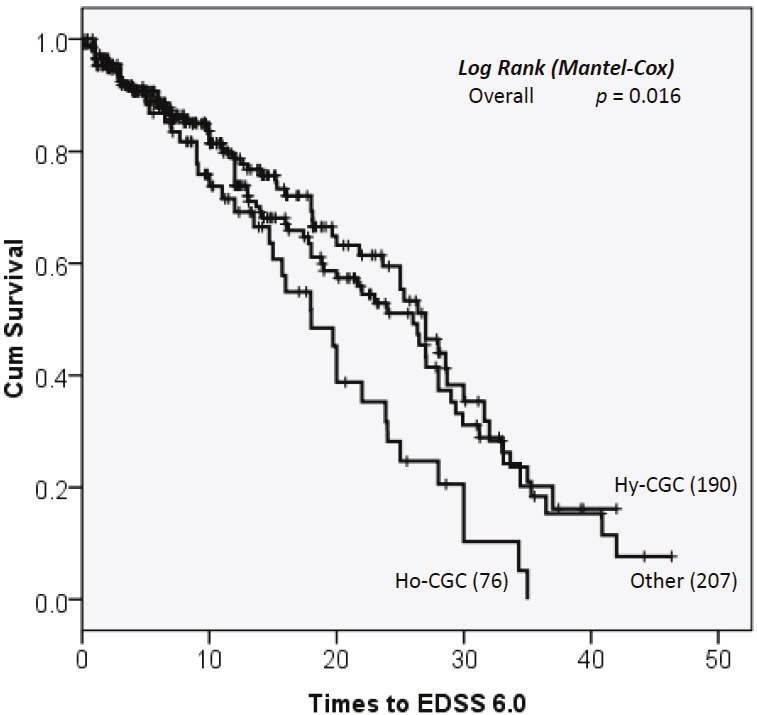
CD24 polymorphism and MS progression. Data shown are Kaplan Meier survival curves depicting the relationship between CD24 genotypes and the time to EDSS6.0. Ho: CD24CGC homozygous; Hy. CD24CGC heterozygous; Other, no CD24CGC allele. P values are calculated by log rank tests.
Hypermorphic promoter activity of the new risk variant
To determine whether the CD24CGC variant affects CD24 mRNA levels in MS patients, we tested CD24 transcript levels in the gender and age-matched MS patients. As shown in Figure 3A, the levels of CD24 mRNA in the peripheral blood leukocytes associate with the gene dosages of the CD24CGC allele (P=7x10-4). To directly demonstrate that the polymorphism dictates promoter activity, we generated luciferase reporters using the CD24 sequence from 3 haplotypes. Compared with the major non-risk haplotypes (AGC and AGT), risk haplotype (CGC) exhibits significantly more activity (Figure 3B). Thus, the risk allele has a hypermorphic CD24 promoter.
Figure 3.
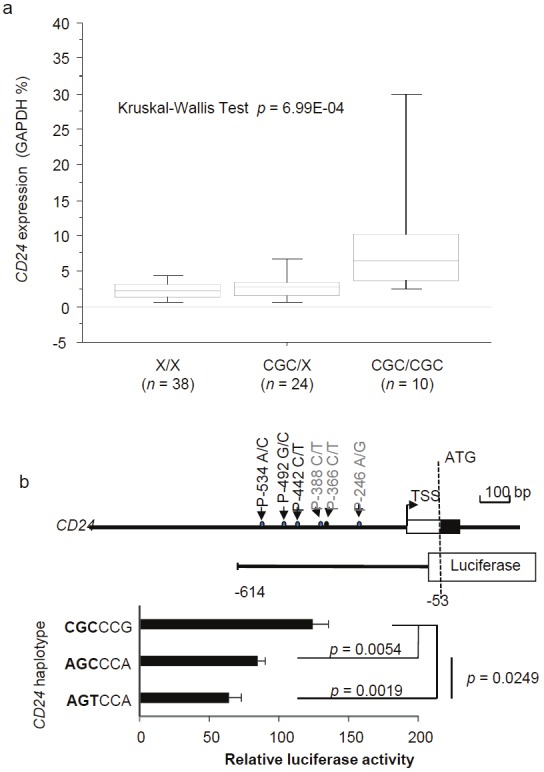
CD24 promoter polymorphism regulates CD24 transcription. A. CD24 transcript levels in PBL of MS patients, as determined by real-time PCR. The CD24 genotypes were indicated in X-axis while the levels of CD24 transcripts were expressed as % of GADPH gene. P values are provided in the panel. B. Promoter activity of the predominant CD24 allele (AGCCCA), risk allele (CGCCCG) and a control minor allele (AGTCCA) that show suggestive protective effect. The top panel shows a diagram 5’ of CD24 gene, highlighting the position of the SNPs and the composition luciferase reporter construct.
CD24 promoter region contains multiple binding sites for GC- or GT-box-binding transcription factors [33], the prototype of which is SP1 (Figure 1). We hypothesized that differential activation of the CD24 gene by SP1 may contribute to differential MS risk. As the first test for the hypothesis, we silenced the SP1 gene using shRNA prior to transfection of the luciferase promoters. As shown in Figure 4A, SP1 silencing by two independent shRNA, but not scrambled shRNA, selectively reduced the hypermorphic CD24 promoter activity without significantly affecting the activity of the ACA promoter. Therefore, SP1 is required only for the enhanced promoter activity of the risk variant.
Figure 4.
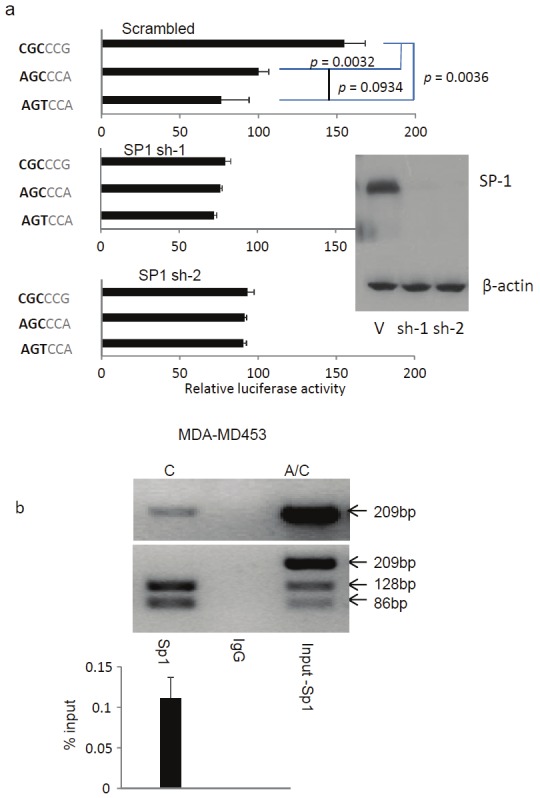
A. An essential role for SP1 in polymorphism CD24 promoter activity. As in b, except that SP1 is silenced by ShRNA prior to luciferase assay. The efficacy of ShRNA is demonstrated by Western blot. B. Endogenous SP1 preferentially interacts with MS risk allele (CD24CGC) in a CD24AGT/CGC cell line, a human breast cancer cell line MDA-MD453. The data shown are PCR-based measurement of SP1-associated CD24 promoter, as revealed by chromatin immunoprecipitation. The top panel shows photograph of undigested PCR products, while the middle panel shows BsrFI digestion of the PCR products. These data show that while the input DNA is heterozygous, essentially all promoters DNA bound to SP1 were of the risk allele, based on P-534A/C-specific RFLP. A quantitative data of the precipitated promoter is shown in the bottom panel. All data in this figure has been repeated three times, error bars are standard derivations of data from independent experiments.
To determine whether different alleles of CD24 locus differentially bind to endogenous SP1, we searched a large panel of human cell lines and found a CD24CGC/AGC cell line. This allowed us to directly compare endogenous interaction between SP1 and promoter of different alleles of CD24 by chromatin immunoprecipitation (ChIP). We carried out a chromatin immunoprecipitation (ChIP) with anti-SP1 mAb and compared the CD24 promoters DNA co-precipitated by the anti-SP1 mAb. As shown in Figure 4B, endogenous SP1 binds strongly to CD24 promoter. Remarkably, when the PCR products were genotyped by RFLP using P-534C-specific restriction enzyme (BsrFI), essentially all SP1-bound CD24 promoter products were found to be derived from the CD24CGC risk allele. In toto, the data in Figure 4 demonstrate that the MS risk allele is hypermorphic because of its stronger interaction with SP1.
Discussion
Taken together, the data presented herein demonstrate that three SNPs within the promoter region constitute an allele with high impact on risk and progression of MS. In addition to the new variant reported herein, multiple CD24 variants increase risk and/or progression of other autoimmune diseases and in all but one MS cohorts [20-23,26-28,34]. Since CD24 is not mapped correctly in human genome until very recently, and since none of the CD24 SNP was included in any GWAS MS studies [3-8], it is not surprising that CD24 has not been implicated in any linkage and GWAS analyses.
The risk alleles increase CD24 expression by encoding a hypermorphic promoter (this study), enhancing mRNA stability [20] and possibly by increasing the efficacy of GPI attachment to CD24 [21]. The strong correlation between expression levels and risks of autoimmune diseases [20-23] suggests an important role for the CD24 gene in the pathogenesis of autoimmune diseases in human. In support of this notion, we reported that mice with targeted mutation of CD24 are resistant to EAE and that CD24 promotes local expansion of autoreactive T cells in the central nervous system [18,19]. In addition, CD24 has been shown to regulate innate response to danger-associated molecular patterns, such as heat-shock proteins and HMGB1 [11,35]. Furthermore, CD24 plays a significant role in T cell homeostatic proliferation [36], a process likely critical for pathogenesis of autoimmune diseases [17].
Recent studies have implicate a role for transcription factor SP1 in the pathogenesis of MS and SLE. Thus, insertional deletion in interferon response factor 5 (IRF5) produced an MS and SLE risk variant with functional SP1 site [37-39]. Moreover, global analysis of gene expression profile in MS patients suggests a potential involvement of SP1-mediated transcription in gender bias [7,39]. By showing specific binding of endogenous SP1 to a specific MS risk allele in heterozygous cells, our data provide the most compelling evidence that CD24-SP1 interaction may regulate MS pathogenesis.
In conclusion, a CD24 promoter variant consisting of 3 novel SNPs in the promoter region is hereby shown to be associated with risk and progression of MS. The risk allele specifically interacts with SP1, a transcription factor that has been implicated in susceptibility to MS and other autoimmune diseases. These data implicate a critical role for SP1-mediated CD24 transcription in the pathogenesis of MS.
Acknowledgements
We thank Drs. David Ginsburg and Tatiana Foroud for critical reading of the manuscript. Ms Nikki Guinther from the Ohio State University, Ms Hollie Schmidt and Ms Sara Loud from Accelerated Cure Projects for assistance in DNA sample and clinical information, Ms Darla Kroft for editorial and secretarial assistance. This study is supported by grants from National Multiple Sclerosis Society and National Institute of Health. The authors have no conflict of financial interest.
This work is supported by grants from National Multiple Sclerosis society and National Institutes of Health.
Abbreviations
- ACP
accelerated cure project
- ChIP
chromatin immunoprecipitation
- EDSS
Expanded Disability Status Scale (EDSS)
- MS
multiple sclerosis
- SNP
single nucleotide polymorphism
Supporting Information
References
- 1.Ebers GC, Sadovnick AD, Risch NJ. A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature. 1995;377:150–151. doi: 10.1038/377150a0. [DOI] [PubMed] [Google Scholar]
- 2.Ramagopalan SV, Ebers GC. Genes for multiple sclerosis. Lancet. 2008;371:283–285. doi: 10.1016/S0140-6736(08)60145-2. [DOI] [PubMed] [Google Scholar]
- 3.Bahlo M, Booth DR, Broadley SA, Brown MA, Foote SJ, Griffiths LR, Kilpatrick TJ, Lechner-Scott J, Moscato P, Perreau VM, Rubio JP, Scott RJ, Stankovich J, Stewart GJ, Taylor BV, Wiley J, Brown MA, Booth DR, Clarke G, Cox MB, Csurhes PA, Danoy P, Drysdale K, Field J, Foote SJ, Greer JM, Griffiths LR, Guru P, Hadler J, McMorran BJ, Jensen CJ, Johnson LJ, McCallum R, Merriman M, Merriman T, Pryce K, Scott RJ, Stewart GJ, Tajouri L, Wilkins EJ, Rubio JP, Bahlo M, Brown MA, Browning BL, Browning SR, Perera D, Rubio JP, Stankovich J, Broadley S, Butzkueven H, Carroll WM, Chapman C, Kermode AG, Marriott M, Mason D, Heard RN, Pender MP, Slee M, Tubridy N, Lechner-Scott J, Taylor BV, Willoughby E, Kilpatrick TJ. Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat Genet. 2009;41:824–828. doi: 10.1038/ng.396. [DOI] [PubMed] [Google Scholar]
- 4.Aulchenko YS, Hoppenbrouwers IA, Ramagopalan SV, Broer L, Jafari N, Hillert J, Link J, Lundstrom W, Greiner E, Dessa Sadovnick A, Goossens D, Van Broeckhoven C, Del-Favero J, Ebers GC, Oostra BA, van Duijn CM, Hintzen RQ. Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nat Genet. 2008;40:1402–1403. doi: 10.1038/ng.251. [DOI] [PubMed] [Google Scholar]
- 5.Baranzini SE, Wang J, Gibson RA, Galwey N, Naegelin Y, Barkhof F, Radue EW, Lindberg RL, Uitdehaag BM, Johnson MR, Angelakopoulou A, Hall L, Richardson JC, Prinjha RK, Gass A, Geurts JJ, Kragt J, Sombekke M, Vrenken H, Qualley P, Lincoln RR, Gomez R, Caillier SJ, George MF, Mousavi H, Guerrero R, Okuda DT, Cree BA, Green AJ, Waubant E, Goodin DS, Pelletier D, Matthews PM, Hauser SL, Kappos L, Polman CH, Oksenberg JR. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum Mol Genet. 2009;18:767–778. doi: 10.1093/hmg/ddn388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ, Pericak-Vance MA, Gregory SG, Rioux JD, McCauley JL, Haines JL, Barcellos LF, Cree B, Oksenberg JR, Hauser SL. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 7.Sanna S, Pitzalis M, Zoledziewska M, Zara I, Sidore C, Murru R, Whalen MB, Busonero F, Maschio A, Costa G, Melis MC, Deidda F, Poddie F, Morelli L, Farina G, Li Y, Dei M, Lai S, Mulas A, Cuccuru G, Porcu E, Liang L, Zavattari P, Moi L, Deriu E, Urru MF, Bajorek M, Satta MA, Cocco E, Ferrigno P, Sotgiu S, Pugliatti M, Traccis S, Angius A, Melis M, Rosati G, Abecasis GR, Uda M, Marrosu MG, Schlessinger D, Cucca F. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat Genet. 2010;42:495–497. doi: 10.1038/ng.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Strange A, Saarela J, Bellenguez C, Fontaine B, Gillman M, Hemmer B, Gwilliam R, Zipp F, Jayakumar A, Martin R, Leslie S, Hawkins S, Giannoulatou E, D'Alfonso S, Blackburn H, Boneschi FM, Liddle J, Harbo HF, Perez ML, Spurkland A, Waller MJ, Mycko MP, Ricketts M, Comabella M, Hammond N, Kockum I, McCann OT, Ban M, Whittaker P, Kemppinen A, Weston P, Hawkins C, Widaa S, Zajicek J, Dronov S, Robertson N, Bumpstead SJ, Barcellos LF, Ravindrarajah R, Abraham R, Alfredsson L, Ardlie K, Aubin C, Baker A, Baker K, Baranzini SE, Bergamaschi L, Bergamaschi R, Bernstein A, Berthele A, Boggild M, Bradfield JP, Brassat D, Broadley SA, Buck D, Butzkueven H, Capra R, Carroll WM, Cavalla P, Celius EG, Cepok S, Chiavacci R, Clerget-Darpoux F, Clysters K, Comi G, Cossburn M, Cournu-Rebeix I, Cox MB, Cozen W, Cree BA, Cross AH, Cusi D, Daly MJ, Davis E, de Bakker PI, Debouverie M, D'Hooghe MB, Dixon K, Dobosi R, Dubois B, Ellinghaus D, Elovaara I, Esposito F, Fontenille C, Foote S, Franke A, Galimberti D, Ghezzi A, Glessner J, Gomez R, Gout O, Graham C, Grant SF, Guerini FR, Hakonarson H, Hall P, Hamsten A, Hartung HP, Heard RN, Heath S, Hobart J, Hoshi M, Infante-Duarte C, Ingram G, Ingram W, Islam T, Jagodic M, Kabesch M, Kermode AG, Kilpatrick TJ, Kim C, Klopp N, Koivisto K, Larsson M, Lathrop M, Lechner-Scott JS, Leone MA, Leppa V, Liljedahl U, Bomfim IL, Lincoln RR, Link J, Liu J, Lorentzen AR, Lupoli S, Macciardi F, Mack T, Marriott M, Martinelli V, Mason D, McCauley JL, Mentch F, Mero IL, Mihalova T, Montalban X, Mottershead J, Myhr KM, Naldi P, Ollier W, Page A, Palotie A, Pelletier J, Piccio L, Pickersgill T, Piehl F, Pobywajlo S, Quach HL, Ramsay PP, Reunanen M, Reynolds R, Rioux JD, Rodegher M, Roesner S, Rubio JP, Ruckert IM, Salvetti M, Salvi E, Santaniello A, Schaefer CA, Schreiber S, Schulze C, Scott RJ, Sellebjerg F, Selmaj KW, Sexton D, Shen L, Simms-Acuna B, Skidmore S, Sleiman PM, Smestad C, Sorensen PS, Sondergaard HB, Stankovich J, Strange RC, Sulonen AM, Sundqvist E, Syvanen AC, Taddeo F, Taylor B, Blackwell JM, Tienari P, Bramon E, Tourbah A, Brown MA, Tronczynska E, Casas JP, Tubridy N, Corvin A, Vickery J, Jankowski J, Villoslada P, Markus HS, Wang K, Mathew CG, Wason J, Palmer CN, Wichmann HE, Plomin R, Willoughby E, Rautanen A, Winkelmann J, Wittig M, Trembath RC, Yaouanq J, Viswanathan AC, Zhang H, Wood NW, Zuvich R, Deloukas P, Langford C, Duncanson A, Oksenberg JR, Pericak-Vance MA, Haines JL, Olsson T, Hillert J, Ivinson AJ, De Jager PL, Peltonen L, Stewart GJ, Hafler DA, Hauser SL, McVean G, Donnelly P, Compston A. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A, Caillier SJ, Ban M, Goris A, Barcellos LF, Lincoln R, McCauley JL, Sawcer SJ, Compston DA, Dubois B, Hauser SL, Garcia-Blanco MA, Pericak-Vance MA, Haines JL. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 10.Oksenberg JR, Baranzini SE. Multiple sclerosis genetics--is the glass half full, or half empty? Nat Rev Neurol. 2010;6:429–437. doi: 10.1038/nrneurol.2010.91. [DOI] [PubMed] [Google Scholar]
- 11.Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec-10 Selectively Repress Tissue Damage-Induced Immune Responses. Science. 2009;323:1722–1725. doi: 10.1126/science.1168988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Chen GY, Zheng P. CD24-Siglec G/10 discriminates danger- from pathogen-associated molecular patterns. Trends Immunol. 2009;30:557–561. doi: 10.1016/j.it.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Jones B, Aruffo A, Sullivan KM, Linsley PS, Janeway CA Jr. Heat-stable antigen is a costimulatory molecule for CD4 T cell growth. J Exp Med. 1992;175:437–445. doi: 10.1084/jem.175.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Wenger RH, Zhao M, Nielsen PJ. Distinct costimulatory molecules are required for the induction of effector and memory cytotoxic T lymphocytes. J Exp Med. 1997;185:251–262. doi: 10.1084/jem.185.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, Zhou Q, Zheng P, Liu Y. CD28-independent induction of T helper cells and immunoglobulin class switches requires costimulation by the heat-stable antigen. J Exp Med. 1998;187:1151–1156. doi: 10.1084/jem.187.7.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang X, Zheng P, Tang J, Liu Y. CD24: from A to Z. Cell Mol Immunol. 2010;7:100–103. doi: 10.1038/cmi.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Zheng P. CD24: a genetic checkpoint in T cell homeostasis and autoimmune diseases. Trends Immunol. 2007;28:315–320. doi: 10.1016/j.it.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Bai XF, Li O, Zhou Q, Zhang H, Joshi PS, Zheng X, Liu Y, Wang Y, Zheng P. CD24 Controls Expansion and Persistence of Autoreactive T Cells in the Central Nervous System during Experimental Autoimmune Encephalomyelitis. J Exp Med. 2004;200:447–458. doi: 10.1084/jem.20040131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai XF, Liu JQ, Liu X, Guo Y, Cox K, Wen J, Zheng P, Liu Y. The heat-stable antigen determines pathogenicity of self-reactive T cells in experimental autoimmune encephalomyelitis. J Clin Invest. 2000;105:1227–1232. doi: 10.1172/JCI9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Lin S, Rammohan KW, Liu Z, Liu JQ, Liu RH, Guinther N, Lima J, Zhou Q, Wang T, Zheng X, Birmingham DJ, Rovin BH, Hebert LA, Wu Y, Lynn DJ, Cooke G, Yu CY, Zheng P, Liu Y. A dinucleotide deletion in CD24 confers protection against autoimmune diseases. PLoS Genet. 2007;3:e49. doi: 10.1371/journal.pgen.0030049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Q, Rammohan K, Lin S, Robinson N, Li O, Liu X, Bai XF, Yin L, Scarberry B, Du P, You M, Guan K, Zheng P, Liu Y. CD24 is a genetic modifier for risk and progression of multiple sclerosis. Proc Natl Acad Sci USA. 2003;100:15041–15046. doi: 10.1073/pnas.2533866100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otaegui D, Saenz A, Camano P, Blazquez L, Goicoechea M, Ruiz-Martinez J, Olaskoaga J, Emparanza JA, Lopez de Munain A. CD24 V/V is an allele associated with the risk of developing multiple sclerosis in the Spanish population. Mult Scler. 2006;12:511–514. doi: 10.1191/135248506ms1314sr. [DOI] [PubMed] [Google Scholar]
- 23.Ronaghi M, Vallian S, Etemadifar M. CD24 gene polymorphism is associated with the disease progression and susceptibility to multiple sclerosis in the Iranian population. Psychiatry Res. 2009;170:271–272. doi: 10.1016/j.psychres.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez SJ, Rojas JI, Redal MA, Patrucco L, Correale J, Argibay PF, Cristiano E. CD24 as a genetic modifier of disease progression in multiple sclerosis in Argentinean patients. J Neurol Sci. 2011;307:18–21. doi: 10.1016/j.jns.2011.05.032. [DOI] [PubMed] [Google Scholar]
- 25.Kollaee A, Ghaffarpor M, Pourmahmoudian H, Shahbazi M, Zamani M. Investigation of CD24 and Its expression in iranian relapsing-remitting multiple sclerosis. Int J Neurosci. 2011;121:684–690. doi: 10.3109/00207454.2011.610529. [DOI] [PubMed] [Google Scholar]
- 26.Piotrowski P, Lianeri M, Wudarski M, Lacki JK, Jagodzinski PP. CD24 Ala57Val gene polymorphism and the risk of systemic lupus erythematosus. Tissue Antigens. 2010;75:696–700. doi: 10.1111/j.1399-0039.2010.01447.x. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez E, Abelson AK, Sabio JM, Gonzalez-Gay MA, Ortego-Centeno N, Jimenez-Alonso J, de Ramon E, Sanchez-Roman J, Lopez-Nevot MA, Gunnarsson I, Svenungsson E, Sturfelt G, Truedsson L, Jonsen A, Gonzalez-Escribano MF, Witte T, Alarcon-Riquelme ME, Martin J. Association of a CD24 gene polymorphism with susceptibility to systemic lupus erythematosus. Arthritis Rheum. 2007;56:3080–3086. doi: 10.1002/art.22871. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez E, Fernandez-Gutierrez B, Gonzalez-Gay MA, Balsa A, Garcia A, Rodriguez L, Pascual-Salcedo D, Gonzalez-Escribano MF, Martin J. Investigating the role of CD24 gene polymorphisms in rheumatoid arthritis. Ann Rheum Dis. 2008;67:1197–1198. doi: 10.1136/ard.2007.084475. [DOI] [PubMed] [Google Scholar]
- 29.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33:1444–1452. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 30.Im H, Grass JA, Johnson KD, Boyer ME, Wu J, Bresnick EH. Measurement of protein-DNA interactions in vivo by chromatin immunoprecipitation. Methods Mol Biol. 2004;284:129–146. doi: 10.1385/1-59259-816-1:129. [DOI] [PubMed] [Google Scholar]
- 31.Lin DY, Zeng D, Millikan R. Maximum likelihood estimation of haplotype effects and haplotype-environment interactions in association studies. Genet Epidemiol. 2005;29:299–312. doi: 10.1002/gepi.20098. [DOI] [PubMed] [Google Scholar]
- 32.Zeng D, Lin DY, Avery CL, North KE, Bray MS. Efficient semiparametric estimation of haplotype-disease associations in case-cohort and nested case-control studies. Biostatistics. 2006;7:486–502. doi: 10.1093/biostatistics/kxj021. [DOI] [PubMed] [Google Scholar]
- 33.Philipsen S, Suske G. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res. 1999;27:2991–3000. doi: 10.1093/nar/27.15.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goris A, Maranian M, Walton A, Yeo TW, Ban M, Gray J, Dubois B, Compston A, Sawcer S. CD24 Ala/Val polymorphism and multiple sclerosis. J Neuroimmunol. 2006 doi: 10.1016/j.jneuroim.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Chen GY, Chen X, King S, Cavassani KA, Cheng J, Zheng X, Cao H, Yu H, Qu J, Fang D, Wu W, Bai XF, Liu JQ, Woodiga SA, Chen C, Sun L, Hogaboam CM, Kunkel SL, Zheng P, Liu Y. Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nat Biotechnol. 2011;29:428–435. doi: 10.1038/nbt.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ou L, Zheng P, Liu Y. CD24 is essential for proliferation of T cells in lymphopenic host. J Exp Med. 2004;200:1083–1089. doi: 10.1084/jem.20040779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kristjansdottir G, Sandling JK, Bonetti A, Roos IM, Milani L, Wang C, Gustafsdottir SM, Sigurdsson S, Lundmark A, Tienari PJ, Koivisto K, Elovaara I, Pirttila T, Reunanen M, Peltonen L, Saarela J, Hillert J, Olsson T, Landegren U, Alcina A, Fernandez O, Leyva L, Guerrero M, Lucas M, Izquierdo G, Matesanz F, Syvanen AC. Interferon regulatory factor 5 (IRF5) gene variants are associated with multiple sclerosis in three distinct populations. J Med Genet. 2008;45:362–369. doi: 10.1136/jmg.2007.055012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menon R, Di Dario M, Cordiglieri C, Musio S, La Mantia L, Milanese C, Di Stefano AL, Crabbio M, Franciotta D, Bergamaschi R, Pedotti R, Medico E, Farina C. Gender-based blood transcriptomes and interactomes in multiple sclerosis: Involvement of SP1 dependent gene transcription. J Autoimmun. 2011 doi: 10.1016/j.jaut.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 39.Sigurdsson S, Goring HH, Kristjansdottir G, Milani L, Nordmark G, Sandling JK, Eloranta ML, Feng D, Sangster-Guity N, Gunnarsson I, Svenungsson E, Sturfelt G, Jonsen A, Truedsson L, Barnes BJ, Alm G, Ronnblom L, Syvanen AC. Comprehensive evaluation of the genetic variants of interferon regulatory factor 5 (IRF5) reveals a novel 5 bp length polymorphism as strong risk factor for systemic lupus erythematosus. Hum Mol Genet. 2008;17:872–881. doi: 10.1093/hmg/ddm359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.