

Abstract
Chromatin-organizing factors such as CTCF and cohesins have been implicated in the control of complex viral regulatory programs. We investigated the role of CTCF and cohesins in the control of the switch from latency to the lytic cycle for Kaposi's sarcoma-associated herpesvirus (KSHV). We found that cohesin subunits but not CTCF are required for the repression of KSHV immediate early gene transcription. Depletion of the cohesin subunits Rad21, SMC1, and SMC3 resulted in lytic cycle gene transcription and viral DNA replication. In contrast, depletion of CTCF failed to induce lytic transcription or DNA replication. Chromatin immunoprecipitation with high-throughput sequencing (ChIP-Seq) revealed that cohesins and CTCF bound to several sites within the immediate early control region for ORF50 and to more distal 5′ sites that also regulate the divergently transcribed ORF45-ORF46-ORF47 gene cluster. Rad21 depletion led to a robust increase in ORF45, ORF46, ORF47, and ORF50 transcripts, with similar kinetics to that observed with chemical induction by sodium butyrate. During latency, the chromatin between the ORF45 and ORF50 transcription start sites was enriched in histone H3K4me3, with elevated H3K9ac at the ORF45 promoter and elevated H3K27me3 at the ORF50 promoter. A paused form of RNA polymerase II (Pol II) was loosely associated with the ORF45 promoter region during latency but was converted to an active elongating form upon reactivation induced by Rad21 depletion. Butyrate treatment caused a rapid dissociation of cohesins and loss of CTCF binding at the immediate early gene locus, suggesting that cohesins may be a direct target of butyrate-mediated lytic induction. Our findings implicate cohesins as a major repressor of KSHV lytic gene activation and show that they function coordinately with CTCF to regulate the switch between latent and lytic gene activity.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is the human gammaherpesvirus identified as the causative agent of Kaposi's sarcoma (KS) (9, 19, 50). KSHV has also been implicated as a causative agent of pleural effusion lymphoma (PEL) and multicentric Castleman's disease (MCD) (5, 15, 51, 66). KSHV can be cultured from a latent infection in PEL-derived cell lines, where the viral genome is maintained as a multicopy, chromatin-associated episome with highly restricted gene expression (1, 39, 55, 71). During latent infection, KSHV gene expression is limited to the multicistronic latency transcript, consisting of the LANA (ORF73), vCyclin (ORF72), and vFLIP (ORF71) genes and a downstream promoter driving the transcription of the viral miRNA cluster and an additional open reading frame for the Kaposin (K12) gene (13, 17, 28, 64, 65). Lytic reactivation requires the expression of the immediate early genes, most notably those encoding Rta (ORF50), which is a potent transcriptional activator of most other KSHV lytic cycle genes (22, 69, 84), and ORF45, which can disrupt interferon signaling (85). Complex signaling and epigenetic mechanisms are known to regulate immediate early gene transcription and control the balance between latent and lytic gene expression.
Several chromatin-organizing factors have been shown to bind and regulate KSHV genomes during latent infection. The chromatin insulator protein CTCF colocalizes with cohesins at many sites (56, 62, 78), including the KSHV latency control region (68). CTCF was originally identified as an 11-fingered zinc finger DNA binding protein that bound to CCCTC-like motifs in the c-Myc promoter (36, 40). CTCF has subsequently been shown to bind more extensive DNA sequence elements (2, 35, 60) and has been implicated in many different gene regulatory functions, including chromatin boundary functions, enhancer blocking, DNA looping, epigenetic imprinting, and colocalization with cohesins (54, 59). Cohesins are multiprotein complexes initially identified for their role in sister chromatid cohesion (10, 14, 58). The core components of cohesins are the structural maintenance complex proteins SMC1 and SMC3 and the radiation sensitivity protein Rad21 (SCC1) (26, 52). Structural studies have revealed that SMC1 and SMC3 are heterodimeric ATPases that can form a ring-like structure closed by Rad21. Cohesin rings are thought to either encircle or handcuff sister chromatids to maintain sister chromatid cohesion (23, 41). Several additional factors are known to regulate cohesin loading and unloading on DNA, including Nipped-B (SCC2) and the lysine acetylation of SMC3 by ESCO1 (3, 72, 82). The interaction between CTCF and cohesins is mediated by the cohesin accessory subunit SA-2 (80).
Cohesins have been implicated in several chromosomal functions in addition to sister chromatid cohesion (31, 53, 72, 78). Cohesins were shown to facilitate interactions between transcriptional enhancers and promoters through interactions with Mediator (31). Cohesins have also been implicated in the regulation of RNA polymerase II (RNA Pol) pausing and elongation (18, 33). Naturally occurring mutations in cohesins have been identified as the genetic basis for several human developmental disorders, including Cornelia de Lange and Roberts syndromes (4, 20, 37, 49, 79). These cohesinopathies have a wide spectrum of developmental defects, including facial abnormalities, mental retardation, and increased cancer incidence (48). Mutations have been identified in multiple different cohesin subunits, including the acetylase Esco1, which is the predominant mutation in Roberts syndrome (74). These findings suggest that cohesins and their regulators have broad roles in chromosome biology and gene expression.
In the KSHV genome, CTCF can be detected at ∼17 high-occupancy sites, with some but not all colocalizing with cohesin subunits (34, 68). The most notable of these sites is a cluster of 3 CTCF sites in the latency control region, within the first intron of the LANA transcript. Disrupting mutations of these CTCF sites lead to a loss of episome stability and deregulation of viral gene expression. CTCF binding is also required for cohesin loading at this site. More recent studies have shown that the CTCF-cohesin site in the latency control region is in close physical proximity to the KSHV lytic control region (34). Chromatin conformation capture (3C) revealed a link between the CTCF-cohesin sites at the LANA intron and a region upstream of the immediate early genes for ORF50 and ORF45. The specific target sequence in the lytic control region was not characterized at the molecular level.
In this study, we investigated the chromatin structure and cohesin function at the KSHV lytic control region. We found that depletion of core cohesin subunits, especially Rad21, causes reactivation of latent KSHV immediate early gene expression and viral DNA replication. We show that CTCF and cohesins bind to the region upstream of the divergent promoter for ORF50 and ORF45. We also show that this region contains a bivalent histone modification pattern that is disrupted upon Rad21 depletion. We also show that the histone deacetylase inhibitor sodium butyrate (NaB) leads to a rapid loss of Rad21 and other cohesin subunits from chromatin, suggesting that cohesins play a regulatory role in the transcriptional repression of the KSHV immediate early lytic gene cluster.
MATERIALS AND METHODS
Cells and plasmids.
KSHV-positive PEL cells (BCBL1, BC-1, and BC-3) and both Epstein-Barr virus (EBV)- and KSHV-positive cells (JSC-1) were grown in RPMI medium (Gibco BRL) containing 10% heat-inactivated fetal bovine serum and the antibiotics penicillin and streptomycin (50 U/ml). 293T cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and antibiotics. All cells were cultured at 37°C in a 5% CO2 environment. KSHV lytic activation was induced by the addition of 1 mM sodium butyrate (and/or 20 ng/ml tetradecanoyl phorbol acetate [TPA]) for 8 h or 24 h before analysis as indicated in the figure legends. Where indicated, cells were treated with 13 μM ganciclovir (GCV; Sigma) at the time of short hairpin RNA (shRNA)-expressing lentivirus infection to inhibit KSHV lytic replication.
Generation of shRNA lentiviruses and lentivirus infection.
Lentiviruses were generated by transient transfection of 293T cells by use of a three-plasmid system (a pLKO plasmid expressing shRNA against cellular CTCF, Rad21, SMC1, or SMC3; pMD2.G [envelope plasmid expressing vesicular stomatitis virus glycoprotein]; and psPAX2 [packaging plasmid]). Viral supernatant was harvested at 2 days posttransfection, concentrated, and stored at −20°C until use. For shRNA knockdown experiments, PEL cells were infected with concentrated lentiviruses by spin infection for 90 min at 450 × g and 25°C in the presence of 8 μg/ml of Polybrene (Sigma). After spin infection, cells were seeded at 0.4 × 106/ml for 48 h before puromycin selection. Cells were then harvested at 1 day or 3 days post-puromycin selection, as indicated in the figure legends.
ChIP.
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (12). Briefly, cells were fixed in 1% formaldehyde for 15 min. DNAs were sonicated to 200- to 400-bp DNA fragments on a Diagenode Bioruptor according to the manufacturer's protocol. Quantification of precipitated DNA was determined using SYBR green probe real-time PCR and the Absolute Quantification program (ABI 7900HT Fast real-time PCR system; Applied Biosystems). Antibodies used in the ChIP assay are listed below. Primers for ChIP assays (sequences are available upon request) were designed using Primer Express, version 2.0 (Applied Biosystems). PCR data were normalized to input values that were quantified in parallel for each experiment. IPs were performed in triplicate for each antibody, and the experiments were repeated at least two times.
ChIP-Seq.
ChIP with high-throughput sequencing (ChIP-Seq) was performed as described previously (43). Briefly, 1 × 107 BCBL1 cells were used per assay, with either rabbit anti-cellular SMC1 (Bethyl) or anti-CTCF (Millipore) antibody or control rabbit IgG (Santa Cruz Biotechnology). A Diagenode Bioruptor was used to sonicate genomic DNA into 150- to 300-bp DNA fragments according to the manufacturer's protocol. DNA fragments of ∼150 to 300 bp were isolated by agarose gel purification, ligated to primers, and subjected to Solexa-based sequencing using the manufacturer's recommendations (Illumina, Inc.). Peak-calling analysis of the KSHV genome was performed as described previously (43).
Antibodies.
The following antibodies were used for ChIP assays: anti-IgG (Santa Cruz Biotechnology), anti-CTCF (Millipore), anti-SMC1 (Bethyl), anti-Rad21 (Abcam) anti-trimethyl H3K4 (Millipore), anti-H3K27 (Millipore), anti-acetylated H3K9 (Millipore), RNA polymerase II (Millipore), and RNA polymerase II pS2 (Abcam) and pS5 (Abcam) antibodies. The following mouse monoclonal antibodies were used for ChIP assays: anti-IgG (Santa Cruz Biotechnology) and RNA polymerase II (Millipore) antibodies. Rabbit polyclonal anti-SMC1 (Bethyl) and anti-SMC3 (Bethyl), rat anti-KSHV LANA antibody (Advanced Biotechnologies Inc.), and mouse monoclonal anti-actin (Sigma) and Rad21 (Santa Cruz Biotechnology) antibodies were used for Western blotting. Anti-KSHV ORF45 antibody was provided by Yan Yuan, University of Pennsylvania, and anti-KSHV ORF50 antibody was provided by Erle Robertson, University of Pennsylvania.
Isolation of KSHV genomic DNA and quantification of its copy number.
The intracellular KSHV DNA copy number was determined by quantitative PCR (qPCR) analysis of purified total genomic DNA. Briefly, 106 BCBL1 cells were resuspended in SDS lysis buffer (1% SDS, 20 mM NaCl, 4 mM EDTA, 20 mM Tris, pH 8.0) with proteinase K for at least 6 h at 50°C. The cell lysate was then subjected to phenol-chloroform extraction and ethanol precipitation. Precipitated DNA was then assayed by real-time PCR, using primers for the KSHV ORF50 promoter region (5′-CCC GCC CAG AAA CCA GTA G-3′ and 5′-TGC GGA GTA AGG TTG ACT TTT TAA-3′) and for normalization to the cellular actin DNA signal (5′-GCC ATG GTT GTG CCA TTA CA-3′ and 5′-GGC CAG GTT CTC TTT TTA TTT CTG-3′).
RT-PCR.
Total RNA was isolated using an RNeasy kit (Qiagen) and then further treated with DNase I (New England Biolabs) at 37°C for 30 min. For cDNA synthesis, 2 μg of total RNA was incubated with a 5 μM concentration of random decamers (Ambion), 150 U of Superscript II RNase H− reverse transcriptase (RT; Invitrogen), 1.6 U of RNase inhibitor, a 1 mM concentration of each deoxynucleoside triphosphate (dNTP), and 3.3 mM dithiothreitol (DTT) in a 20-μl reaction mixture according to the manufacturer's protocol (Invitrogen). After heat inactivation at 65°C for 10 min, the sample was diluted with 180 μl distilled water. Diluted RT products were analyzed by real-time PCR with a SYBR green probe, using the ΔCT method (ABI Prism 7900 sequence detection system; Applied Biosystems). The sequences of primers used for RT-PCR assay are available upon request. The level of beta-actin in each sample was used as an internal control for each quantitative PCR. A quantitative PCR assay with RNA but without reverse transcription was conducted to serve as a negative control for each reaction.
PFGE.
BCBL1 cells infected with lentivirus expressing shControl, shRad21, or shCTCF shRNA were resuspended in 1.0% agarose plugs and incubated in lysis buffer (0.2 M EDTA [pH 8.0], 1% SDS, 1 mg/ml proteinase K) at 50°C for 48 h. The agarose plugs were washed twice in Tris-EDTA (TE) buffer (pH 7.5). Pulsed-field gel electrophoresis (PFGE) was performed for 22 h at 14°C, with a linear ramping pulse of 60 to 120 s through 120°C as described previously (Bio-Rad CHEF Mapper). DNA was transferred to nylon membranes by established methods for Southern blotting. The DNA was detected by hybridization with a 32P-labeled probe specific for the KSHV TR region and visualized with a Molecular Dynamics phosphorimager.
RESULTS
shRNA depletion of cohesin subunits results in KSHV reactivation in BCBL1 cells.
To investigate the function of cohesins in regulation of KSHV latency and reactivation, we generated a puromycin-resistant lentivirus expressing shRNAs targeting SMC1, SMC3, and Rad21. BCBL1 cells were infected with lentivirus and selected with puromycin for 5 days postinfection. Western blotting was used to measure the extent of shRNA depletion (Fig. 1A). SMC1 levels were partly depleted by shSMC1 as well as by shSMC3. SMC3 was partially depleted by shSMC3. Rad21 was depleted by shRad21 as well as by shSMC1 and shSMC3. The codependent depletion of cohesin subunits may not be unexpected, since these proteins are known to form a complex that may be unstable in the absence of any one major component. More relevant to KSHV, we observed that KSHV immediate early proteins ORF50 and ORF45 were elevated in BCBL1 cells infected with any of the cohesin-targeting shRNAs but not with the shControl vector (Fig. 1A). Actin levels were not significantly affected by lentivirus infection or shRNA depletion of cohesins (Fig. 1A, bottom panel).
Fig 1.

shRNA knockdown of cohesin subunits results in KSHV lytic reactivation in BCBL1 cells. (A) BCBL1 cells were infected with a lentivirus expressing shControl, shSMC1, shSMC3, or shRAD21 and selected with puromycin for 3 days. Cell extracts were analyzed by Western blotting with antibodies to SMC1, SMC3, Rad21, ORF45, ORF50, and actin, as indicated. (B) PFGE and Southern blot analysis of KSHV genomes in BCBL1 cells infected with shControl, shSMC1, shSMC3, or shRad21. Circular (C), linear (L), and sublinear (SL) KSHV genomes are indicated by arrows. (C) BCBL1 cells were infected with shControl (lane 1), shRad21 (lane 2), or shCTCF (lane 3) and then assayed by Western blotting with antibodies to CTCF, Rad21, ORF45, and actin, as indicated. (D) Same as panel B, except that GCV was added to BCBL1 cells (lanes 2 and 4). Lane 5, BCBL1 cells infected with shCTCF. (E) KSHV mRNA expression was determined by RT-qPCR amplification of ORF50, ORF73, ORF25, and PAN mRNAs and was quantified relative to cellular actin mRNA. BCBL1 cells were treated with shControl, shSMC1, shSMC3, and shRad21 as shown in panels A and B. Error bars represent standard deviations (SD) from the means for three independent qPCRs. (F) KSHV DNA copy number was determined by qPCR with KSHV-specific primers and expressed relative to cellular actin. Copy numbers for KSHV were determined for BCBL1 cells treated with shControl, shSMC1, shSMC3, and shRad21. Error bars represent standard deviations from the means for three independent qPCRs.
To determine if cohesin depletion altered the DNA structure of the KSHV genome, we assayed KSHV DNA by PFGE and Southern blot analysis (Fig. 1B). We found that KSHV genomes were present as an equal mixture of circular and linear forms in BCBL1 cells infected with shControl lentivirus (Fig. 1B, lane 1). In contrast, BCBL1 cells infected with shSMC1, shSMC3, or shRad21 lentivirus showed a large (>10-fold) increase in linear and sublinear forms of the KSHV genome (Fig. 1B, lanes 2 to 4).
We also assayed the effect of CTCF depletion on BCBL1 cells (Fig. 1C and D). CTCF was depleted efficiently (>80%) (Fig. 1C, top panel). However, unlike shRad21, shCTCF did not induce ORF45 expression (Fig. 1C). We also noted that shRNA depletion of CTCF did not induce linear genome formation as analyzed by PFGE (Fig. 1D, lane 5). To determine if the increase in linear genomes induced by shRad21 was due to activation of the viral lytic cycle, we used GCV to inhibit viral DNA polymerase (Fig. 1D, lanes 2 and 4). We found that addition of GCV to shRad21-infected cells prevented the accumulation of linear genomes (Fig. 1D, lane 4), indicating that the amplification of linear genomes reflects viral lytic replication. The ability of cohesin depletion to induce the KSHV lytic cycle was further confirmed by RT-PCR analysis of KSHV lytic genes for ORF50, ORF73, ORF25, and PAN (Fig. 1E). Depletion of SMC1, SMC3, or Rad21 resulted in an ∼10- to 50-fold increase in KSHV lytic mRNA in BCBL1 cells. Additionally, we showed by qPCR that the KSHV DNA copy number increased ∼10- to 100-fold after cohesin depletion (Fig. 1F).
Rad21 depletion causes KSHV reactivation in multiple PEL cell lines.
To investigate whether cohesins regulate KSHV latency in PEL cells other than BCBL1 cells, we assayed the effect of Rad21 depletion in the BC1, BC3, and JSC-1 cell lines (Fig. 2). PEL cells were infected with shControl or shRad21 lentivirus and then assayed by Western blotting to assess Rad21 depletion (Fig. 2A). shRad21 lentivirus infection depleted Rad21 levels to various extents, with BC1 and BC3 cells showing only ∼30% reduction and with more robust reductions in BCBL1 and JSC-1 cells. JSC-1 and BCBL1 cells showed a strong induction of the lytic antigen ORF45, while BC3 showed only a modest activation and BC1 showed little or no induction. To further assess the effects of Rad21 depletion on KSHV reactivation, we analyzed KSHV DNA by PFGE (Fig. 2B). We found that shRad21 lentivirus infection increased linear and sublinear forms of the viral genome in BC3, JSC-1, and BCBL1 cells but had little effect in BC1 cells. qPCR analysis of the KSHV DNA copy number indicated that shRad21 increased the genome copy number in BC3, JSC-1, and BCBL1 cells but not in BC1 cells (Fig. 2C). These findings suggest that depletion of Rad21 induces KSHV lytic replication in multiple different PEL cells. The failure of BC1 cells to reactivate in response to shRad21 lentivirus infection may be explained partly by the poor depletion of Rad21 in these cells (∼50% depleted). However, BC1 cells also failed to respond efficiently to NaB-induced lytic activation (data not shown), suggesting that they have a more restrictive latency than that of other PEL cells tested.
Fig 2.
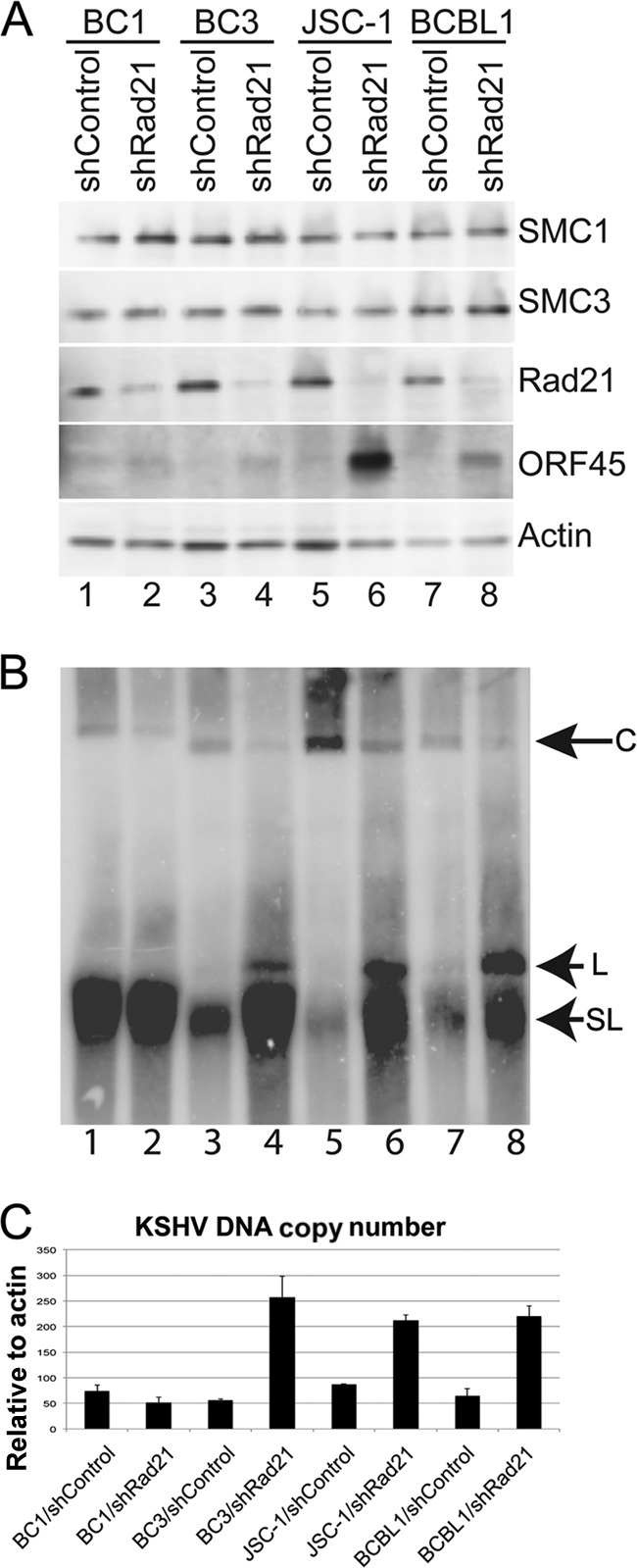
Rad21 depletion induces KSHV lytic reactivation in multiple different KSHV-positive cell lines. (A) KSHV-positive cell lines BC1, BC3, JSC-1, and BCBL1 were infected with shControl or shRad21 lentivirus, selected for 1 day, and then assayed by Western blotting with antibodies to SMC1, SMC3, Rad21, ORF45, and actin, as indicated. (B) PFGE and Southern blot analysis of KSHV genomes in BC1 (lanes 1 and 2), BC3 (lanes 3 and 4), JSC-1 (lanes 5 and 6), and BCBL1 (lanes 7 and 8) cells after infection with shControl (lanes 1, 3, 5, and 7) or shRad21 (lanes 2, 4, 6, and 8). Circular (C), linear (L), and sublinear (SL) genomes are indicated by arrows. (C) DNA copy numbers were determined by qPCR amplification of KSHV DNA and expressed relative to actin.
Cohesins bind at CTCF sites within the immediate early control region.
To generate a high-resolution map of CTCF and cohesin binding sites on the KSHV genome in BCBL1 cells, we performed ChIP-Seq experiments with CTCF and SMC1 antibodies (Fig. 3). ChIP-Seq peaks for CTCF and SMC1 were identified using a Poisson distribution model (modified MACS program) and then normalized to an IgG control (Fig. 3A) (43). As reported previously, most cohesin (e.g., SMC1) peaks overlapped extensively with CTCF peaks (56, 62, 68, 78). However, several CTCF peaks were not co-occupied with cohesins (nucleotides [nt] ∼22,000, 27,000, and 120,000). A major CTCF-cohesin peak was detected within the first intron of the LANA transcript (nt ∼127,400), consistent with our previous findings (32, 34, 68). We also found that CTCF and cohesins bound efficiently to the TR region, which had not been reported previously. Since we previously found that the major CTCF-cohesin peak was capable of forming a 3C-measured DNA loop with the region upstream of the ORF50 promoter, we focused on CTCF and SMC1 binding to this region (Fig. 3B and C). We found that CTCF bound to sites in the ORF45 gene (nt ∼67,755), the ORF46 gene (nt ∼68,744 to 68,763), the ORF49 start site (nt ∼72,294 to 72,312), and the ORF50 termination site (nt ∼74,575 to 74,594). A consensus CTCF binding motif was identified for each of these peak regions (data not shown). SMC1 bound to all of these sites, but with slightly different occupancies. While CTCF bound more extensively at the ORF45-ORF46 peak, SMC1 bound more extensively at the ORF49 and ORF50 regions.
Fig 3.

ChIP-Seq analysis of LANA, CTCF, and SMC1 binding to the KSHV genome in BCBL1 cells. (A) ChIP-Seq binding peaks for LANA (black), CTCF (blue), and SMC1 (red) are shown (obtained using the UCSC browser), spanning the entire KSHV genome with 3 terminal repeats (TRs). (B) Expanded view of ChIP-Seq peaks for the region encompassing the immediate early promoter regions for the ORF45, ORF46, ORF47, ORF48, ORF49, and ORF50 genes (nt 67,452 to 75,890). (C) Schematic diagram of open reading frames, transcripts, and transcription start sites for KSHV immediate early genes in this locus.
Rad21 depletion alters histone and RNA polymerase II posttranslational modifications at the KSHV immediate early control region.
To analyze epigenetic changes induced by Rad21 depletion, we performed a conventional ChIP assay with a series of primers spanning the regions of the lytic immediate early gene cluster (Fig. 4). Conventional ChIP assays confirmed that CTCF bound at the ORF45 (primer B), ORF46 (primer D), and ORF49 (primer K) regions (Fig. 4B, blue bars). The histone modifications H3K4me3, H3K9Ac, and H3K27me3 were examined throughout this region. We found that H3K4me3 was elevated throughout the region between the CTCF sites at positions ∼69,000 (primer D) and ∼72,600 (primer K). H3K9Ac was elevated at the ORF46 region (primer E), while H3K27me3 was notably depleted at this position relative to the surrounding regions. The effect of shRad21 or shCTCF depletion was also measured using ChIP assay across this region (Fig. 4B and C, pink and green bars). We found that CTCF binding was reduced by shCTCF, as expected, but was also reduced by shRad21 (Fig. 4B, top graph). shRad21 depletion also caused a loss of H3K4me3, H3K9Ac, and K3K27me3 at all positions in the lytic control region that were tested. In contrast, shCTCF depletion had little effect on histone modifications in this region.
Fig 4.

Rad21 depletion alters histone and RNA polymerase II modifications at the KSHV immediate early gene cluster. (A) Positions of qPCR primers (A to L) used for ChIP assays. Red vertical arrowheads indicate CTCF-cohesin binding sites identified by ChIP-Seq. (B) CTCF, H3K4me3, H3K9Ac, and H3K27me3 ChIP assay results are shown for shControl (blue), shRad21 (red), and shCTCF (green) and the indicated primers. (C) Same as panel B, but for RNA Pol II, RNA Pol II pS2, RNA Pol II pS5, Rad21, and control mouse IgG (mIgG) ChIP assays. Error bars represent SD from the means for 3 independent PCRs.
To further assess the effect of shRad21 on KSHV lytic transcription control, we assayed RNA Pol II and its phosphorylated CTD isoforms (pS2 and pS5) by ChIP assay (Fig. 4C). In shControl lentivirus-infected cells, we found that RNA Pol II was distributed weakly throughout the immediate early control region, with a minor peak of RNA Pol II pS5 occurring within the ORF46 gene (primer E). In contrast, shRad21 depletion produced a significant increase in RNA Pol II occupancy at the ORF47 locus (primer F) and a general increase in RNA Pol II pS2 and a decrease in pS5 throughout the immediate early gene locus. Depletion of CTCF produced a slight increase in total RNA Pol II occupancy at several primer positions but had no effect on either phospho-isoform of RNA Pol II. Taken together, these findings suggest that Rad21 depletion causes a general loss of histone occupancy and an increase in RNA Pol II pS2 at the lytic control region. Since pS2 corresponds to the elongation-competent form of RNA polymerase, this suggests that Rad21 depletion promotes RNA polymerase loading and elongation throughout the lytic control region.
Rad21 depletion activates immediate early gene transcription similarly to chemical inducing agents.
The effects of shRad21 and shCTCF on transcription of the ORF45 to ORF50 genes within the lytic control region were assayed by RT-qPCR (Fig. 5). Depletion of Rad21 resulted in large increases in ORF45 (primers B, C, and D), ORF47 (primer F), ORF49 (primer K), and ORF50 (primer L) (Fig. 5A, dark gray bars). Depletion of CTCF showed no similar increase in transcription throughout this region (Fig. 5A, light gray bars). Early gene transcription was also measured by chemical induction with NaB for 8 h (Fig. 5B) or with NaB and TPA for 24 h (Fig. 5C). We found that chemical induction for short (8 h) and long (24 h) times produced a pattern of transcription nearly identical to that observed for Rad21 depletion. The effect of Rad21 depletion on the KSHV lytic control region was re-evaluated using a new set of primer pairs optimized for RT-PCR of KSHV mRNAs (17) rather than detection by ChIP primers optimized for viral DNA fragments. The new mRNA-specific primers revealed that KSHV gene transcripts for all viral genes in this locus were activated by Rad21 depletion, with transcripts for ORF45, ORF46, and ORF50 being the most abundant (Fig. 6A). The same Rad21-depleted cells were evaluated by ChIP assay of Rad21, SMC1, and SMC3 binding (Fig. 6B to D). The ChIP assay revealed that Rad21 depletion led to a complete loss of all cohesin subunits from the KSHV lytic control region, suggesting that Rad21 is essential for cohesin binding to this locus. The early induction of ORF45, ORF46, ORF47, and ORF49 is consistent with previous studies reporting these genes to be immediate early genes similar to ORF50 (77, 84). Our findings suggest that these genes are coordinately activated by lytic-inducing agents and that Rad21 plays a key role in regulating transcription of this lytic control region.
Fig 5.
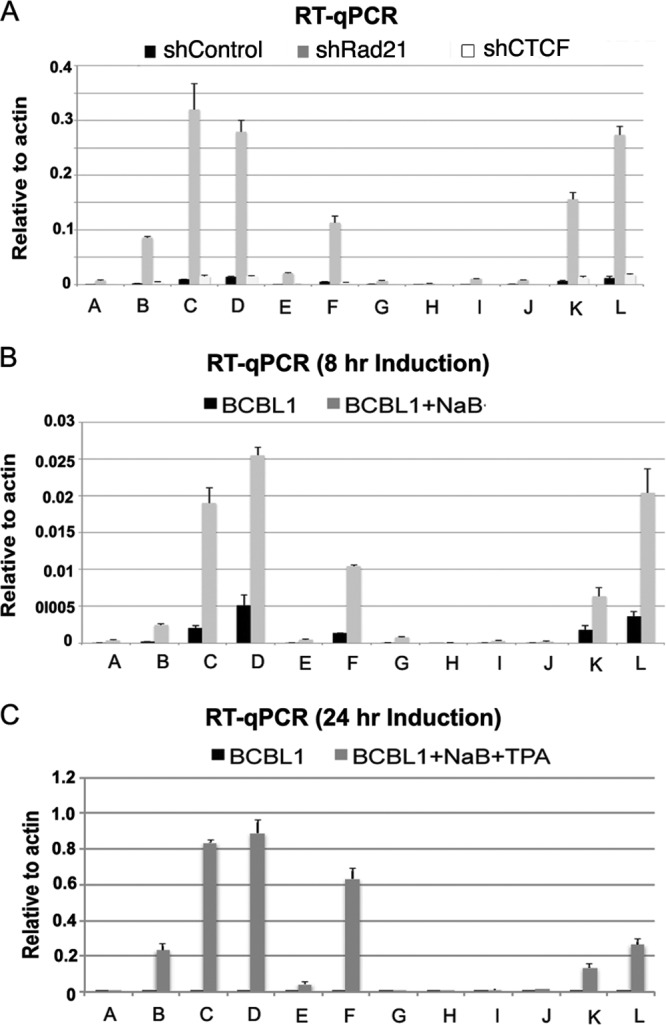
Rad21 depletion activates KSHV immediate early gene transcription similarly to that induced by chemical reactivation. (A) BCBL1 cells infected with shControl (black), shRad21 (dark gray), or shCTCF (light gray) were analyzed for mRNA expression by RT-qPCR, using primers A to L for the KSHV immediate early control region. (B and C) BCBL1 cells were treated with 1 mM sodium butyrate (B) or with NaB plus 20 ng/ml phorbol ester (C) and then assayed for mRNA expression by RT-qPCR as described for panel A.
Fig 6.

Rad21 depletion results in a loss of all cohesin subunit binding to the KSHV lytic locus. (A) RT-PCR analysis with gene-specific primers for ORF45, ORF46, ORF47, ORF48, ORF49, and ORF50, using RNA from BCBL1 cells transduced with shControl (blue) or shRad21 (red). mRNA was quantified relative to that for cellular actin. (B) Rad21 antibody ChIP samples were assayed with primers A to L and with BCBL cells transduced with shControl (blue) or shRad21 (red). (C and D) Same as panel B, but with SMC1 (C) or SMC3 (D) antibody. Error bars represent SD for 3 independent PCRs.
NaB treatment disrupts CTCF-cohesin binding at the lytic control region.
To explore the possibility that NaB acts on cohesin binding to regulatory chromatin, we assayed the effect of NaB treatment on CTCF and cohesin binding in ChIP assays (Fig. 7). BCBL1 cells were treated with or without NaB for 8 h prior to ChIP analysis. We found that both CTCF and cohesin binding was partially disrupted after 8 h of NaB treatment. The most significant losses were observed for cohesin subunits SMC1 and Rad21 at the ORF49 locus (primer K). These findings suggest that loss of cohesin and CTCF binding at the immediate early locus is an early event during KSHV lytic induction by NaB.
Fig 7.
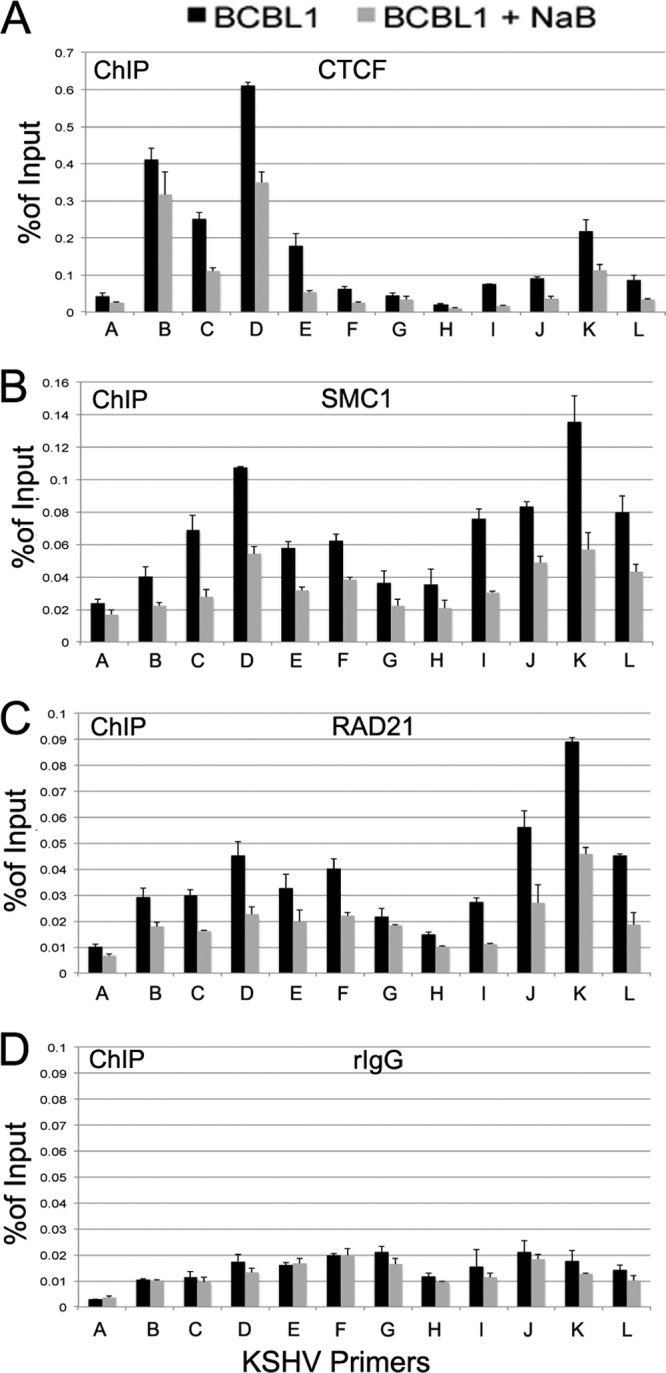
NaB disrupts CTCF and cohesin binding at the lytic control region. BCBL1 cells were left untreated (black bars) or treated with 1 mM NaB for 8 h (gray bars) and then assayed by ChIP assay with antibody to CTCF (A), SMC1 (B), or RAD21 (C) or with control rat IgG (rIgG) (D). ChIP DNAs were assayed with KSHV primers (A to L) targeting the lytic control region and quantified as % of input. Error bars represent SD for 3 independent PCRs.
DISCUSSION
Chromosome structure and organization are thought to play important roles in viral and cellular gene regulation. In this work, we examined the roles of several chromosome structural maintenance proteins in the regulation of KSHV lytic reactivation. We found that cohesins contribute to KSHV latency control by repressing transcription of KSHV immediate early genes. CTCF and cohesins are thought to influence gene expression through the control of histone modification patterns and higher-order DNA conformations (34, 68). In a previous study, we showed that a major CTCF-cohesin binding site located within the latency control region (nt ∼127,450) formed a stable DNA loop with a region upstream of the ORF50 gene promoter, but the limits of 3C analysis could not resolve a more precise interaction site. We reasoned that cohesins are likely to interact with other CTCF-cohesin sites in the KSHV genome, and therefore we generated a high-resolution map of CTCF and cohesin binding sites by ChIP-Seq (Fig. 3). These new data reveal that CTCF and SMC1 co-occupy peaks at positions upstream of the ORF50 gene and within the control regions for the ORF45, ORF47, and ORF50 genes.
Chromatin organization of the lytic control region.
Others have shown that a stem cell-like bivalent pattern of histone modifications is observed at key regions of the KSHV genome, including the ORF50 gene promoter region (73). Our data support the finding that the region upstream of the ORF50 gene promoter is enriched in H3K4me3 and H3K27me3. However, at the ORF46 region, histones were enriched in H3K4me3 and H3K9ac and depleted of H3K27me3. Interestingly, CTCF binding was more robust at the ORF45 and ORF47 sites, while cohesin binding was more robust at the ORF50 transcription initiation site. Histone H3K4me3 formed a broad peak bounded by CTCF sites near the transcription start sites for ORF45 (nt ∼68,763) and ORF50 (nt ∼72,294). RNA Pol II was enriched at the ORF47 region, but no transcription was detected, and RNA Pol II remained in the phospho-S5 form associated with polymerase pausing. We propose that the region between CTCF sites at the ORF45 and ORF50 transcription start sites constitutes a functional chromatin element, which we refer to as the lytic control region (Fig. 8). Depletion of the cohesin subunit Rad21 led to a complete disruption of this chromatin organization, suggesting that cohesins are important for maintaining the histone modification pattern and paused RNA polymerase between the divergently transcribed immediate early genes.
Fig 8.

Model of chromatin organization at the KSHV lytic control region. The lytic control region, encompassing a divergent promoter for ORF50 and ORF48-ORF47-ORF46-ORF45, is shown during transcriptionally inactive latent infection. CTCF (yellow spheres) positions cohesin (pink disks) and inhibits ORF50 gene promoter engagement by RNA Pol II. RNA Pol II remains associated with acetylated histones at the ORF46-ORF47 H3K9Ac peaks in the ORF45 gene promoter region, while a bivalent histone modification (H3K4me3 and H3K27me3) covers the ORF50 gene proximal promoter. CTCF-cohesins mediate interactions between the latent and lytic control regions.
Coordinated control of lytic cycle immediate early genes.
The genomic organization of KSHV suggests that ORF45 to ORF49 may be regulated coordinately with ORF50, since their genes share an upstream promoter regulatory region. Previous studies have shown that the ORF50 and K8 genes can be expressed as a multicistronic immediate early transcript (84) and that the ORF50 gene encodes the primary activator of lytic gene transcription (22, 46, 70, 84). The ORF45 gene was also shown to be an immediate early gene (84) as well as a component of the KSHV tegument (86), and the murine herpesvirus 68 (MHV68) orthologue of ORF45 is required for the immediate early phase of viral replication (29). The ORF46 gene encodes viral uracil deglycosylase, the ORF47 and ORF48 genes encode components of the entry machinery for gL, and the ORF49 gene encodes an orthologue of BRRF1, which has been implicated in EBV lytic gene activation (21, 24, 27). Furthermore, the ORF47-ORF46-ORF45 gene region can be expressed as a multicistronic transcript (77), and the gene cluster is potently activated by Rta, along with autoactivation of the ORF50 gene (6, 8, 63, 76). These observations suggest that the entire locus is coordinately regulated with lytic reactivation signals that activate ORF50 transcription. Our data indicate that several of these genes are coordinately regulated during the immediate early stages of lytic reactivation. We found that sodium butyrate treatment leads to a rapid loss of cohesin binding that correlates temporally with transcription activation of ORF47-ORF46-ORF45 and ORF50. Taken together, these findings support the model that the lytic control region consists of a cohesin-regulated divergent promoter that can initiate transcription of ORF50 as well as ORF47-ORF46-ORF45 in response to sodium butyrate treatment.
Coordinated control of lytic and latent gene expression by cohesins.
Cohesins have been shown to both positively and negatively regulate transcription (11, 47, 61, 67), and more recently, they have been implicated in the control of RNA Pol II pausing (18, 33). The transcriptional effects of cohesins may be related to other biochemical properties, including functions associated with chromatin boundaries, insulators, and DNA looping. The chromatin boundary and insulator functions have been linked closely to CTCF, which co-occupies many of the cohesin sites. Interestingly, we found that depletion of CTCF disrupted cohesin binding but did not result in viral lytic gene activation (Fig. 4). This suggests that cohesins have a more specialized function in transcription repression at the KSHV lytic control region. Cohesin depletion led to an increase in RNA Pol II S2 at the ORF50 promoter region, with a corresponding loss of several histone modifications. CTCF depletion has a much less significant effect on lytic transcription, RNA Pol II recruitment, and histone modifications. CTCF depletion by shRNA may not be as complete as that of Rad21 and other cohesins, and this may partly explain the failure of CTCF depletion to disrupt latency. However, CTCF depletion does lead to reductions of the genome copy number and of LANA expression, consistent with previous findings that CTCF is required for maintaining latent gene expression and for genome maintenance (32, 33, 68). Thus, while CTCF and cohesins may co-occupy many DNA binding sites, they can have functionally separable activities in gene regulation.
Regulation of KSHV lytic control region by cohesins and other factors.
The lytic control region is regulated by numerous cellular factors and signaling pathways, including RBP-jK, C/EBP, octamer binding protein, YY1, SP1, and HIF1α-mediated hypoxia (6–8, 25, 42, 45, 57, 63, 75, 77, 81). Autoactivation of lytic transcripts by Rta has been shown to function through RBP-jK, which binds at several sites in the ORF50 upstream control region. RBP-jK also binds within the latency control region and can be regulated by LANA (30, 38, 44). We note that many RBP-jK sites in the lytic and latency control regions overlap or are in close proximity to the CTCF-cohesin binding sites (45). Although it was not examined in this study, we speculate that RBP-jK may contribute to cohesin function in the structural organization of the KSHV genome. RBP-jK is known to associate with corepressor complexes in the absence of coactivators such as Notch or Rta (16, 83). Thus, CTCF-cohesin may interact with other regulatory factors, including RBP-jK, to maintain bivalent chromatin architecture and histone modifications that can readily respond to environmental stimuli. It is also possible that cohesin depletion has additional indirect and global effects that lead to robust reactivation of KSHV genomes. However, since CTCF-cohesin binds to the ORF50 region (Fig. 3 and 4), and since this binding is lost during early stages of lytic reactivation (Fig. 7), we favor the model that cohesins function as direct repressors of transcription at the KSHV lytic control region.
ACKNOWLEDGMENTS
We acknowledge the Wistar Institute Cancer Center (grant P30 CA10815) and Genomics Core Facility. This work was funded by a grant from the NIH (grant RO1 CA117830) to P.M.L.
We thank Yan Yuan and Erle Robertson at the University of Pennsylvania for the generous supply of KSHV antibodies.
Footnotes
Published ahead of print 27 June 2012
REFERENCES
- 1. Ballestas ME, Kaye KM. 2011. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi's sarcoma-associated herpesvirus latency. Future Microbiol. 6:1399–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barski A, et al. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129:823–837 [DOI] [PubMed] [Google Scholar]
- 3. Beckouet F, et al. 2010. An Smc3 acetylation cycle is essential for establishment of sister chromatid cohesion. Mol. Cell 39:689–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bose T, Gerton JL. 2010. Cohesinopathies, gene expression, and chromatin organization. J. Cell Biol. 189:201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 6. Chang PJ, Boonsiri J, Wang SS, Chen LY, Miller G. 2009. Binding of RBP-Jkappa (CSL) protein to the promoter of the Kaposi's sarcoma-associated herpesvirus ORF47 (gL) gene is a critical but not sufficient determinant of transactivation by ORF50 protein. Virology 398:38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang PJ, et al. 2011. Role of the cellular transcription factor YY1 in the latent-lytic switch of Kaposi's sarcoma-associated herpesvirus. Virology 413:194–204 [DOI] [PubMed] [Google Scholar]
- 8. Chang PJ, Miller G. 2004. Autoregulation of DNA binding and protein stability of Kaposi's sarcoma-associated herpesvirus ORF50 protein. J. Virol. 78:10657–10673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang Y, et al. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 10. Chien R, Zeng W, Ball AR, Yokomori K. 2011. Cohesin: a critical chromatin organizer in mammalian gene regulation. Biochem. Cell Biol. 89:445–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chien R, et al. 2011. Cohesin mediates chromatin interactions that regulate mammalian beta-globin expression. J. Biol. Chem. 286:17870–17878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deng Z, et al. 2002. Telomeric proteins regulate episomal maintenance of Epstein-Barr virus origin of plasmid replication. Mol. Cell 9:493–503 [DOI] [PubMed] [Google Scholar]
- 13. Dittmer D, et al. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 72:8309–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dorsett D. 2011. Cohesin: genomic insights into controlling gene transcription and development. Curr. Opin. Genet. Dev. 21:199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dupin N, et al. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ehebauer M, Hayward P, Martinez-Arias A. 2006. Notch signaling pathway. Sci. STKE 2006:cm7 doi:10.1126/stke.3642006cm7 [DOI] [PubMed] [Google Scholar]
- 17. Fakhari FD, Dittmer DP. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213–6223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fay A, et al. 2011. Cohesin selectively binds and regulates genes with paused RNA polymerase. Curr. Biol. 21:1624–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ganem D. 2006. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu. Rev. Pathol. 1:273–296 [DOI] [PubMed] [Google Scholar]
- 20. Gillis LA, et al. 2004. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am. J. Hum. Genet. 75:610–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonzalez CM, et al. 2006. Identification and characterization of the Orf49 protein of Kaposi's sarcoma-associated herpesvirus. J. Virol. 80:3062–3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gradoville L, et al. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207–6212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haering CH, Farcas AM, Arumugam P, Metson J, Nasmyth K. 2008. The cohesin ring concatenates sister DNA molecules. Nature 454:297–301 [DOI] [PubMed] [Google Scholar]
- 24. Hagemeier SR, Barlow EA, Kleman AA, Kenney SC. 2011. The Epstein-Barr virus BRRF1 protein, Na, induces lytic infection in a TRAF2- and p53-dependent manner. J. Virol. 85:4318–4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haque M, Davis DA, Wang V, Widmer I, Yarchoan R. 2003. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) contains hypoxia response elements: relevance to lytic induction by hypoxia. J. Virol. 77:6761–6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hirano T. 2006. At the heart of the chromosome: SMC proteins in action. Nat. Rev. Mol. Cell Biol. 7:311–322 [DOI] [PubMed] [Google Scholar]
- 27. Hong GK, et al. 2004. The BRRF1 early gene of Epstein-Barr virus encodes a transcription factor that enhances induction of lytic infection by BRLF1. J. Virol. 78:4983–4992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jeong J, Papin J, Dittmer D. 2001. Differential regulation of the overlapping Kaposi's sarcoma-associated herpesvirus vGCR (orf74) and LANA (orf73) promoters. J. Virol. 75:1798–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jia Q, et al. 2005. Murine gammaherpesvirus 68 open reading frame 45 plays an essential role during the immediate-early phase of viral replication. J. Virol. 79:5129–5141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jin Y, et al. 2012. Carboxyl-terminal amino acids 1052 to 1082 of the latency-associated nuclear antigen (LANA) interact with RBP-Jκ and are responsible for LANA-mediated RTA repression. J. Virol. 86:4956–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kagey MH, et al. 2010. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467:430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang H, Lieberman PM. 2009. Cell cycle control of Kaposi's sarcoma-associated herpesvirus latency transcription by CTCF-cohesin interactions. J. Virol. 83:6199–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kang H, Lieberman PM. 2011. Mechanism of glycyrrhizic acid inhibition of Kaposi's sarcoma-associated herpesvirus: disruption of CTCF-cohesin-mediated RNA polymerase II pausing and sister chromatid cohesion. J. Virol. 85:11159–11169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kang H, Wiedmer A, Yuan Y, Robertson E, Lieberman PM. 2011. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog. 7:e1002140 doi:10.1371/journal.ppat.1002140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim TH, et al. 2007. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128:1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Klenova EM, et al. 1993. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol. Cell. Biol. 13:7612–7624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krantz ID, et al. 2004. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 36:631–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lan K, Kuppers DA, Robertson ES. 2005. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 79:3468–3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lieberman PM, Hu J, Renne R. 2007. Maintenance and replication during latency, p 379–402 In Arvin A, et al. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 40. Lobanenkov VV, et al. 1990. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene 5:1743–1753 [PubMed] [Google Scholar]
- 41. Losada A. 2007. Cohesin regulation: fashionable ways to wear a ring. Chromosoma 116:321–329 [DOI] [PubMed] [Google Scholar]
- 42. Lu F, Day L, Gao SJ, Lieberman PM. 2006. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J. Virol. 80:5273–5282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu F, et al. 2012. Identification of host-chromosome binding sites and candidate gene targets for Kaposi's sarcoma-associated herpesvirus LANA. J. Virol. 86:5752–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu J, Verma SC, Cai Q, Robertson ES. 2011. The single RBP-Jkappa site within the LANA promoter is crucial for establishing Kaposi's sarcoma-associated herpesvirus latency during primary infection. J. Virol. 85:6148–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lu J, et al. 2012. The RBP-Jkappa binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog. 8:e1002479 doi:10.1371/journal.ppat.1002479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF50 transactivator, a homolog of the EBV R protein. Virology 252:304–312 [DOI] [PubMed] [Google Scholar]
- 47. Majumder P, Boss JM. 2011. Cohesin regulates MHC class II genes through interactions with MHC class II insulators. J. Immunol. 187:4236–4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mannini L, Menga S, Musio A. 2010. The expanding universe of cohesin functions: a new genome stability caretaker involved in human disease and cancer. Hum. Mutat. 31:623–630 [DOI] [PubMed] [Google Scholar]
- 49. McNairn AJ, Gerton JL. 2008. Cohesinopathies: one ring, many obligations. Mutat. Res. 647:103–111 [DOI] [PubMed] [Google Scholar]
- 50. Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi's sarcoma and its associated herpesvirus. Nat. Rev. Cancer 10:707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moore PS, Chang Y. 2003. Kaposi's sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu. Rev. Microbiol. 57:609–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nasmyth K, Haering CH. 2005. The structure and function of SMC and kleisin complexes. Annu. Rev. Biochem. 74:595–648 [DOI] [PubMed] [Google Scholar]
- 53. Nativio R, et al. 2009. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 5:e1000739 doi:10.1371/journal.pgen.1000739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ohlsson R, Lobanenkov V, Klenova E. 2010. Does CTCF mediate between nuclear organization and gene expression? Bioessays 32:37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pantry SN, Medveczky PG. 2009. Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus replication. Semin. Cancer Biol. 19:153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Parelho V, et al. 2008. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132:422–433 [DOI] [PubMed] [Google Scholar]
- 57. Persson LM, Wilson AC. 2009. Wide-scale use of Notch signaling factor CSL/RBP-Jkappa in RTA-mediated activation of Kaposi's sarcoma-associated herpesvirus lytic genes. J. Virol. 84:1334–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Peters JM, Tedeschi A, Schmitz J. 2008. The cohesin complex and its roles in chromosome biology. Genes Dev. 22:3089–3114 [DOI] [PubMed] [Google Scholar]
- 59. Phillips JE, Corces VG. 2009. CTCF: master weaver of the genome. Cell 137:1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rhee HS, Pugh BF. 2011. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147:1408–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rhodes JM, et al. 2010. Positive regulation of c-Myc by cohesin is direct, and evolutionarily conserved. Dev. Biol. 344:637–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rubio ED, et al. 2008. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. U. S. A. 105:8309–8314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sakakibara S, Ueda K, Chen J, Okuno T, Yamanishi K. 2001. Octamer-binding sequence is a key element for the autoregulation of Kaposi's sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 75:6894–6900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Samols MA, Hu J, Skalsky RL, Renne R. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:9301–9305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72:1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schulz TF. 2006. The pleiotropic effects of Kaposi's sarcoma herpesvirus. J. Pathol. 208:187–198 [DOI] [PubMed] [Google Scholar]
- 67. Seitan VC, et al. 2011. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature 476:467–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stedman W, et al. 2008. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 27:654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sun R, et al. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sun R, et al. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 73:2232–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tempera I, Lieberman PM. 2010. Chromatin organization of gammaherpesvirus latent genomes. Biochim. Biophys. Acta 1799:236–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Terret ME, Sherwood R, Rahman S, Qin J, Jallepalli PV. 2009. Cohesin acetylation speeds the replication fork. Nature 462:231–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Toth Z, et al. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 6:e1001013 doi:10.1371/journal.ppat.1001013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vega H, et al. 2009. Phenotypic variability in 49 cases of ESCO2 mutations, including novel missense and codon deletion in the acetyltransferase domain, correlates with ESCO2 expression and establishes the clinical criteria for Roberts syndrome. J. Med. Genet. 47:30–37 [DOI] [PubMed] [Google Scholar]
- 75. Wang SE, et al. 2003. Role of CCAAT/enhancer-binding protein alpha (C/EBPalpha) in activation of the Kaposi's sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 77:600–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang SE, Wu FY, Yu Y, Hayward GS. 2003. CCAAT/enhancer-binding protein-alpha is induced during the early stages of Kaposi's sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J. Virol. 77:9590–9612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang SS, et al. 2012. Positive and negative regulation in the promoter of the ORF46 gene of Kaposi's sarcoma-associated herpesvirus. Virus Res. 165:157–169 [DOI] [PubMed] [Google Scholar]
- 78. Wendt KS, et al. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451:796–801 [DOI] [PubMed] [Google Scholar]
- 79. Whelan G, et al. 2011. Cohesin acetyltransferase Esco2 is a cell viability factor and is required for cohesion in pericentric heterochromatin. EMBO J. 31:71–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xiao T, Wallace J, Felsenfeld G. 2011. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin-dependent insulation activity. Mol. Cell. Biol. 31:2174–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ye J, Shedd D, Miller G. 2005. An Sp1 response element in the Kaposi's sarcoma-associated herpesvirus open reading frame 50 promoter mediates lytic cycle induction by butyrate. J. Virol. 79:1397–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang J, et al. 2008. Acetylation of Smc3 by EcoI is required for S phase sister chromatid cohesion in both human and yeast. Mol. Cell 31:143–151 [DOI] [PubMed] [Google Scholar]
- 83. Zhou S, Hayward SD. 2001. Nuclear localization of CBF1 is regulated by interactions with the SMRT corepressor complex. Mol. Cell. Biol. 21:6222–6232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhu FX, Cusano T, Yuan Y. 1999. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhu FX, King SM, Smith EJ, Levy DE, Yuan Y. 2002. A Kaposi's sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. U. S. A. 99:5573–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhu FX, Yuan Y. 2003. The ORF45 protein of Kaposi's sarcoma-associated herpesvirus is associated with purified virions. J. Virol. 77:4221–4230 [DOI] [PMC free article] [PubMed] [Google Scholar]