

Abstract
Acute promyelocytic leukemia (APL) is driven by a chromosomal translocation whose product, the PML/retinoic acid (RA) receptor α (RARA) fusion protein, affects both nuclear receptor signaling and PML body assembly. Dissection of APL pathogenesis has led to the rediscovery of PML bodies and revealed their role in cell senescence, disease pathogenesis, and responsiveness to treatment. APL is remarkable because of the fortuitous identification of two clinically effective therapies, RA and arsenic, both of which degrade PML/RARA oncoprotein and, together, cure APL. Analysis of arsenic-induced PML or PML/RARA degradation has implicated oxidative stress in the biogenesis of nuclear bodies and SUMO in their degradation.
APL, a malignancy driven by PML/RARA
Leukemias are a type of cancer caused by the malignant proliferation of bone marrow–derived cells that invade the bloodstream, distance organs, and induce loss of normal bone marrow. These dreaded diseases actually represent a wide spectrum ranging from relatively indolent conditions, which rarely shorten survival, to high-malignancy ones for which there are few therapeutic options to date. Leukemias are usually classified as acute or chronic and lymphoid or myeloid, depending on the phenotype of the malignant cell. Leukemias share certain definitive features: (a) deficient formation of blood cells, anemia, and hemorrhages mainly caused by the loss of platelets (thrombopenia) and infections related to myeloid and lymphoid deficiencies; and (b) tumor mass, with high levels of abnormal leukemia cells in the blood, lymphoid organs, or other organs. Some acute leukemias have benefited from treatments with inhibitors of DNA replication, such as DNA cross-linkers, and topoisomerase or nucleotide synthesis inhibitors. These have allowed the overwhelming majority of children with acute lymphoblastic leukemia to be definitively cured. Unfortunately, many of the other acute or chronic leukemias still have a dismal prognosis.
Cancer is linked to the accumulation of genetic or epigenetic events that enable unrestrained proliferation. In leukemias, these events have progressively been discovered, starting with the first chromosomal translocations disclosed in the 1960’s up to more recent full genome sequencing or epigenetic cartographies (Ley et al., 2008; Figueroa et al., 2010). This has resulted in a molecular classification of unprecedented precision. For example, acute myeloid leukemias are all linked by their phenotypic characters but really constitute a mosaic of genetically diverse diseases, each very rare (Look, 1997; Döhner et al., 2010). Critically, molecular features are more accurate than morphology at predicting evolution and response to therapy. For example, a given kinase inhibitor induces remission and prolonged survival only in those cases in which that specific kinase is mutated (Sawyers, 2008).
Acute promyelocytic leukemia (APL) is a rare condition (100 new cases/year in France) though extremely malignant because of its very rapid spontaneous evolution and occurrence of sudden hemorrhages mainly caused by coagulation disorders (Hillestad, 1957; Warrell et al., 1993). Indeed, in addition to diminished platelet counts, APL cells contain enzymes and proteins that, when liberated in the bloodstream, activate the coagulation cascade (Tallman and Kwaan, 1992). APL is associated with specific chromosomal translocations that always involve the retinoic acid (RA) receptor α (RARA) gene on chromosome 17 to create a variety of X-RARA fusions (Piazza et al., 2001). The most common one is the t(15,17) translocation encoding the PML/RARA fusion (Rowley et al., 1977; de Thé et al., 1990, 1991), which is associated with >98% of APL cases. The second most common translocation t(11,17) encodes PLZF/RARA and is associated clinically with RA-resistant APLs (Chen et al., 1993; Licht et al., 1995). In APLs driven by the t(15,17) translocations, PML/RARA is most often the only driving genetic alteration; epigenetic changes appear limited (Welch et al., 2008; Figueroa et al., 2010; Martens et al., 2010; Wartman et al., 2011). Indeed, full genome sequencing has demonstrated rare and inconsistent additional mutations (Welch et al., 2008). It is most likely that these facilitate disease progression, rather than initiation, as well known for FLT3 activation or Myc amplification (Sohal et al., 2003; Jones et al., 2010). It is thus the view of the authors that APL represents a monogenic cancer primarily, if not exclusively, driven by PML/RARA. In that respect, PML/RARA (Fig. 1 a) initiates a typical APL when expressed in transgenic mice (Brown et al., 1997; Grisolano et al., 1997). In that setting, APL development requires a few months, and penetrance is not complete. The latter may either suggest that, in that setting, genetic/epigenetic changes besides PML/RARA expression are required for progression to full transformation or that the cells of origin in mice somehow differ from those in humans. Another possibility is that additional genetic or epigenetic changes are required for PML/RARA posttranslational modifications that significantly affect PML/RARA function in vivo (Zhu et al., 2005; Nasr et al., 2008).
Figure 1.
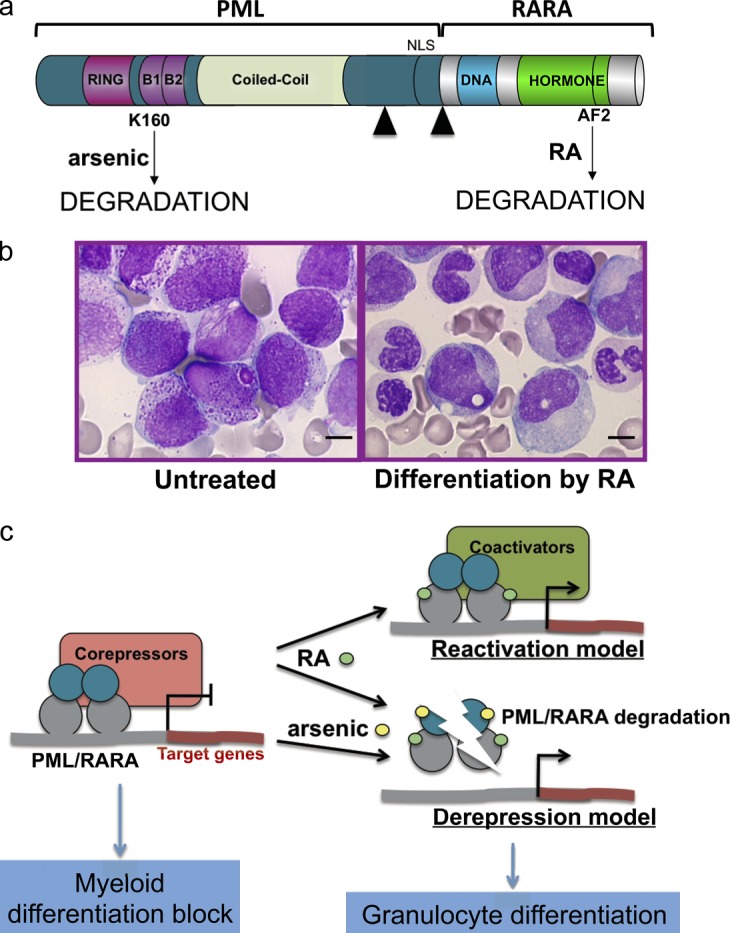
PML/RARA, RA, arsenic, and APL. (a) The PML/RARA fusion protein with the main functional domains of PML and RARA. Note that RA targets the RARA moiety, and arsenic targets the PML moiety, both leading to PML/RARA degradation. Black arrowheads denote the different fusion points. DNA, DNA-binding domain; AF2, activating function 2. (b) RA treatment in vivo elicits the differentiation of leukemia cells (reproduced from Zhu et al. [2002] with permission from Nature Publishing Group). Bars, 5 µm. (c) Current models of RA or arsenic action on transcriptional control and differentiation through gene activation (top) or derepression via degradation (bottom).
RARA is an RA nuclear receptor that acts as a hormone-dependent transcriptional switch. RA has been implicated in many biological responses, notably differentiation of multiple progenitors as well as myeloid cells. PML is the organizer of nuclear domains known as PML nuclear bodies (NBs), as detailed in the next section. Like many other malignancy-associated fusion proteins, PML/RARA has a dual dominant-negative activity on signaling by each of its constitutive partners (Melnick and Licht, 1999). Accordingly, PML/RARA is a potent repressor of nuclear hormone receptor signaling, but it also disrupts PML NBs.
Two therapeutic agents, RA and arsenic trioxide, induce differentiation of promyelocytes in vivo and clinical remission of APL patients (Fig. 1 b). However, although RA induces only transient remissions (Warrell et al., 1993), arsenic by itself cures ≤70% of APL patients (Mathews et al., 2010; Ghavamzadeh et al., 2011). Reactivation of PML/RARA-repressed transcription by RA was logically proposed to explain the induction of differentiation and clearance of APL, exemplifying a remarkable model of both differentiation- and transcription-targeted cancer therapy (Fig. 1 c). Arsenic also induces some differentiation, albeit delayed relative to the effect of RA. Clearly, the transcriptional reactivation model cannot account for the arsenic effect, which does not significantly alter nuclear receptor signaling (Ablain and de Thé, 2011). As we discuss later, both RA and arsenic degrade PML/RARA (Fig. 1 a and see Fig. 3). Thus, abrogation of PML/RARA-mediated repression was proposed to explain arsenic-triggered differentiation (Fig. 1 c). Recent genetic and pharmacological evidence have demonstrated that RA-induced differentiation and clearance of APL may be uncoupled (Nasr et al., 2008; Ablain and de Thé, 2011). For example, in PLZF/RARA-driven APLs (in which PLZF alterations likely substitute for the absence of PML disruption), leukemic cells differentiate upon exposure to RA but fail to clear, suggesting that in PML/RARA-driven APL, restoring PML NBs may contribute to APL regression. Preclinical studies performed in several APL murine models have demonstrated that RA and arsenic dramatically synergize for clearing APL (Lallemand-Breitenbach et al., 1999; Rego et al., 2000). This has led to clinical trials (frontline RA/arsenic combination) that have resulted in the cure of virtually all patients (Shen et al., 2004; Estey et al., 2006; Hu et al., 2009; Tallman and Altman, 2009; de Thé and Chen, 2010). Moreover, a recent clinical meta-analysis has demonstrated that the combination of arsenic and RA in APL patients increased the complete remission rates, shortened the time required to reach remission, and also increased the disease-free survival, perfectly in line with the findings of mouse models (Lallemand-Breitenbach et al., 2005; Wang et al., 2011). Hence, APL is the first example of an oncogene-targeted cancer cure (Quignon et al., 1997; Wang and Chen, 2008; Tallman and Altman, 2009; de Thé and Chen, 2010).
Figure 3.

PML or PML/RARA degradation by arsenic trioxide. Arsenic enhances multi- and/or polysumoylation by SUMO1/2/3 on K65 and K160, but not K490, as demonstrated by Western blotting after short treatment of CHO cells stably expressing PML (top, middle lane); PML proteins are degraded after longer exposure to arsenic (top, right lane). The ubiquitin E3 ligase RFN4 comprises four SIMs and is thus recruited onto dimerized, mesh-associated, hypersumoylated PML to induce its polyubiquitination (poly-Ub). PML is finally degraded by the proteasome machinery, which is also recruited onto NBs. The yellow arrow depicts the events occurring in the first hour after arsenic, and the green one shows subsequent degradation (corresponding to polyubiquitination).
APL research revives mysterious nuclear organelles
The first description of NBs as electron-dense shells with electron-light cores was made in spontaneous tumors of Papilloma virus–infected rabbits (de Thé et al., 1960). These domains were later identified by immunofluorescence analysis using autoimmune sera from patients with primary biliary cirrhosis (Ascoli and Maul, 1991). At the time that the first NB-associated autoantigen, SP100, was cloned (Szostecki et al., 1990), a monoclonal antibody against crude nuclear matrix was shown to recognize nuclear domains also containing SP100 (Stuurman et al., 1992a). This monoclonal antibody actually recognizes PML (Dyck et al., 1994; Koken et al., 1994). In electron microscopy, PML constitutes the outer shell of the sphere (Fig. 2 a, left) and, as formally demonstrated using pml−/− cells, is the organizer of these domains, into which it recruits SP100 and multiple other proteins (Ishov et al., 1999; Lallemand-Breitenbach et al., 2001; Lallemand-Breitenbach and de Thé, 2010).
Figure 2.

PML and NBs. (a) PML NBs in normal and APL cells. From left to right: (normal cells) immunofluorescence and electron microscopy views in CHO cells stably expressing PML. (left) PML is both diffusely distributed in the nucleoplasm and aggregated in NBs. The red arrow points to an individual body, analyzed by electron microscopy. Bar, 1 µm. (right) anti-PML antibodies, labeled with gold particles, show PML distribution in the electron-dense NB shell. Bar, 0.5 µm. (APL cells) NB disruption into micropunctuations by PML/RARA expression (left) and reformation upon arsenic treatment (right). (normal cells) Arsenic controls NB aggregation in non-APL cells. CHO cells stably expressing PML were treated (right) or not treated (left) with 1 µM As2O3 for 1 h. Note the disappearance of the diffuse nuclear staining and increased size of NBs. (b) PML structure and SUMO-induced modifications. Interactions of partner proteins with SUMO–SIM are shown as dotted lines. PML primarily interacts with its partners through K160 sumoylation (1); NB association of sumoylated partners is secured by subsequent SIM–SUMO reciprocal interactions between partners and possibly PML SIM (2). (c) PML cross-linking by disulfide bounds underlies formation of the matrix-associated shell, and polarized SUMO–SIM interactions recruit partner proteins within NBs. PML multimers recruit Ubc9, leading to sumoylation of PML and possibly its partners. S, sulfur. (d) PML targeting to the insoluble nuclear matrix (NM) after arsenic exposure. Reproduced from Lallemand-Breitenbach et al. (2001). Molecular masses are given in kilodaltons. N, nucleoplasm.
That PML/RARA expression disrupts PML NBs (Fig. 2 a, middle; Daniel et al., 1993) indicated that the (elusive) function of NBs might be blocked by PML/RARA, possibly contributing to leukemogenesis. Indeed, this proposal was later substantiated by establishing formal links between PML NBs and self-renewal of normal or cancer stem cells (Ito et al., 2008; Regad et al., 2009). Strikingly, treatment with RA allowed reformation of normal NBs (Daniel et al., 1993; Dyck et al., 1994; Koken et al., 1994; Weis et al., 1994), which was later explained by the fact that PML/RARA was degraded under the effect of RA (Fig. 1 c; Raelson et al., 1996; Zhu et al., 1999) and that PML proteins produced from the nonrearranged PML allele reformed PML NBs. Thus, the NB status mirrored cell malignancy, suggesting that NB reformation may contribute to the RA therapeutic effect. These results have drawn a considerable amount of interest in these domains.
Arsenic also elicits PML/RARA degradation and NB reformation (Fig. 2 a, middle; Zhu et al., 1997) but acts via the PML rather than the RARA moiety of PML/RARA (Fig. 1 a). Indeed, similar to RA, which degrades RARA, arsenic degrades normal PML and, thus, PML/RARA oncoprotein (Fig. 3). This unexpected convergence between two clinically active agents, discovered by chance, strongly reinforced the idea that PML/RARA degradation, and hence PML NB reformation, contributed to clinical remissions (Quignon et al., 1997; de Thé and Chen, 2010; Ablain et al., 2011). PML is normally distributed between NBs and the rest of the nucleoplasm (Fig. 2 a, right). Although PML is more readily detectable by immunofluorescence when associated with NBs, its diffuse, nonmatrix-associated fraction is often actually the most abundant (Fig. 2 d). A pioneering study had demonstrated that arsenic has the unique property of changing this equilibrium by promoting PML targeting onto NBs and nuclear matrix association, before the onset of degradation (Fig. 2 a [right] and d; Zhu et al., 1997). Thus, arsenic became an invaluable tool to approach both APL pathogenesis and NB assembly (Zhu et al., 2002).
PML encages other proteins.
PML forms a shell surrounding the core of NBs and, thus, creates a partition within the nucleoplasm (Bernardi and Pandolfi, 2007; Lallemand-Breitenbach and de Thé, 2010; Lang et al., 2010). PML NBs have the ability to recruit an ever-growing number of proteins, the most extensively studied of which being SP100 and DAXX. Only few partners (such as SP100) are constantly NB associated, whereas most are massively recruited in response to stress. These multiple partner proteins, which all accumulate within the core (Lang et al., 2010), have nothing in common except that they all can undergo sumoylation. Only the PML shell, but not its binding partners, is associated with the nuclear matrix (unpublished data). Consistent with this association, NBs are relatively stable, with its binding partners moving to and from the nucleoplasm (Eskiw et al., 2003; Weidtkamp-Peters et al., 2008). PML is generally considered to be the sole organizer of NBs by a seeding effect. However, a recent study has proposed that SP100 is also important in PML NB formation (Negorev et al., 2010). PML and SP100 appear to stabilize each other (unpublished data; Negorev et al., 2009), so that it is possible that absence of SP100 results in very low levels of PML that preclude self-association into NBs. Similarly, the fact that some NB-associated proteins, such as EIF4e, can form speckles in the absence of PML has led to the proposal that EIF4e domains may preexist and nucleate PML NBs (Cohen et al., 2001). Because PML bodies are primarily defined by the presence of PML, the existence of speckled distribution of other NB-associated proteins (such as EIF4e, Sp100, and PLZF) in PML absence likely denotes the ability of partners to self-aggregate and thus nucleate distinct and specific domains.
Initially, PML NBs were not reported to harbor DNA or RNA within their core (Stuurman et al., 1992a; Boisvert et al., 2000). Yet, nascent messenger RNAs have been found within some NBs (LaMorte et al., 1998). This may reflect different NB makeups according to cell types and/or that NBs may represent a somehow heterogeneous group of domains. Like most other nuclear domains, NBs localize within the interchromatin space, in between chromosomal territories (Matera, 1999). Statistical associations between NBs and transcription sites suggest that neighboring chromatin plays a role in NB localization within the nucleus (Tsukamoto et al., 2000; Shiels et al., 2001; Eskiw et al., 2004). Curiously, association between NBs and specific loci was found for both activated and repressed genes.
Several types of PML bodies.
PML is expressed as splice variants with the same N terminus, but distinct C terminus, sequences (Jensen et al., 2001). PML-I has a nuclear export signal, a nucleolar-targeting signal (primarily active in senescent cells), and an exonuclease III domain of unknown function (Condemine et al., 2007). Although PML-I appears to be the most strongly expressed in nontransformed cells or normal tissues, this has not been extensively investigated in primary cells. All isoforms interact via their coiled coil and hence colocalize in NBs. Yet, when expressed individually in pml−/− cells, isoforms yield NB formation but with distinct morphologies (Condemine et al., 2006), suggesting that their C termini interact with specific cellular components. Thus, each isoform may have particular functions.
Apart from isoform expression, the cellular context appears to exert a great effect. For example, PML aggregates in threads in Ras-transformed cells (Stuurman et al., 1992a), whereas human embryonic stem cells exhibit short threads that seem to join to unidentified structures (Butler et al., 2009). Similarly, PML association with different compartments can occur in response to a variety of stresses, such as telomere shortening, DNA damage, aggregation of misfolded proteins, deregulation of ribosome biogenesis, senescence, and proteasome inhibition (Kamei, 1997; Mattsson et al., 2001; Bernardi et al., 2004; Condemine et al., 2007). In contrast to classical NBs, those generated by alternative lengthening of telomeres or in response to DNA damage contain specific DNA sequences (Draskovic et al., 2009; Chung et al., 2011). Furthermore, significant proximity between NBs and the genome of incoming viruses has been noted, often resulting in viral transcriptional silencing (Everett et al., 2007).
The SUMO connection
SUMOs are ubiquitin-like peptides that conjugate to lysine (K) residues of many proteins, changing their subcellular localization or ability to interact with their partners (Hay, 2005). SUMO can also bind to a short hydrophobic sequence, the SUMO-interacting motif (SIM). PML can be sumoylated on three different K residues and contains a SIM domain (Fig. 2 b; Boddy et al., 1996; Müller et al., 1998; Lallemand-Breitenbach et al., 2001). Nuclear matrix– and NB-associated PML is massively sumoylated (Jeanne et al., 2010), and sumoylation on the critical K160 residue (Fig. 2 b) is the key factor that controls recruitment of most partner proteins (Ishov et al., 1999; Lallemand-Breitenbach et al., 2001). It is currently not known whether conjugation of K160 by the SUMO1 or SUMO2 isoforms recruits similar or different partners.
Conversely, most PML-binding proteins contain a SIM, which is essential for targeting onto NBs (Lin et al., 2006; Cho et al., 2009). Thus, the interaction initiating partner recruitment into NBs is likely to occur between PML-bound SUMO and the partner’s SIM (Fig. 2 b). Importantly, a SIM is also required for sumoylation of most, if not all, NB-associated proteins analyzed to date (Lin et al., 2006; Cho et al., 2009). This may reflect the role of SIMs in anchoring SUMO-conjugating enzyme UBC9 onto its targets (Meulmeester et al., 2008), but it also suggests that sumoylation of partners might occur in situ within NBs, supporting then their transient sequestration through multiple intermolecular SUMO–SIM associations within the NB core (Fig. 2 c and Fig. 4). Although PML’s SIM is not required for partner recruitment into NBs (unpublished data), interaction of sumoylated partners with PML’s own SIM may further contribute to their stable association within NBs.
Figure 4.

PML NB partners and NB-triggered biological responses. (a) PML oxidation elicits PML NB formation. Enzymes, such as SUMO E2 Ubc9, SUMO E3 SMC5/6 subunits, ubiquitin E3 RNF4, the acetyltransferase CBP, or the kinase HIPK2, are recruited onto NBs. Concentrating enzymes and their substrates supports posttranslational modifications of partner proteins, such as SP100, the structural maintenance or chromosome (SMC) complex involved in alternative lengthening telomeres, DAXX, or p53. These modifications affect activity and stability of PML partners and/or lead to their sequestration within NBs, converging into quiescence (stem cells) or a senescence program. (b) Proposed links between PML NB reformation and APL response to treatment. PML/RARA disrupts NBs, which could be reflected in aberrant self-renewal. RA or arsenic, by degrading PML/RARA, allows reformation of PML NBs, as do reactive oxygen species (ROS) and possibly anthracyclins. (c) In therapy-resistant APLs, as a result of mutations in PML/RARA, therapies fail to restore NBs.
Could similar SUMO–SIM interactions also account for PML multimerization and NB assembly as previously suggested (Müller et al., 1998; Shen et al., 2006)? This commonly accepted proposal has limitations because (a) SUMO–SIM interactions are weak and unlikely to confer matrix properties to PML, and (b) PML mutants lacking SIM still yield NBs (unpublished data), as do PML mutants that cannot be sumoylated (Lallemand-Breitenbach et al., 2001). Recent studies have demonstrated that nuclear matrix–associated PML is primarily constituted of disulfide-bound PML homodimers (Fig. 2 c; Jeanne et al., 2010; Zhang et al., 2010), providing a plausible biochemical basis for such association. Nevertheless, specific bodies, such as those encapsulating telomeres, nucleoli, aggregated misfolded proteins, or sites of DNA repair, may result from PML recruitment onto preexisting aggregates of sumoylated partner proteins, notably those bound to telomeres or DNA breaks (Potts and Yu, 2007; Chung et al., 2011). In these specific cases, partner–SUMO and PML–SIM interactions may be critical to nucleate NB assembly.
PML oxidation and arsenic-induced PML/RARA degradation
Disulfide-mediated PML homodimerization was discovered in studies aimed at understanding the biochemical basis of arsenic-induced PML transfer to the nuclear matrix (Fig. 2 d). Arsenic induces oxidative stress (Kawata et al., 2007), which results in PML cross-linking by disulfide bonds, leading to matrix association and NB formation (Jeanne et al., 2010). In addition, a PML cysteine directly binds to arsenic (Jeanne et al., 2010; Zhang et al., 2010). These findings indicate that PML multimerization and aggregation of the mesh forming the outer shell of NBs is primary achieved through the combination of noncovalent (coiled coil) and covalent (disulfide) interactions (Fig. 2 c). Interestingly, the role of long coiled coils has been established for other matrix proteins, such as lamins, and the nuclear matrix comprises large numbers of disulfide-bound proteins (Stuurman et al., 1992b; Pekovic et al., 2011). Control of PML aggregation by redox stress likely explains why, in vivo, NBs have repeatedly been associated with inflammation or neoplastic transformation (Koken et al., 1995). Although these models derive from few studies and thus await independent confirmation, some previous studies had demonstrated that the protein recognized by 5E10 (later shown to be PML) could undergo oxidation within the nuclear matrix (Stuurman et al., 1992b), whereas cysteine starvation (and hence oxidative stress) profoundly affects NB biogenesis (Kamei, 1997).
Nuclear matrix aggregated, disulfide-bound PML is massively sumoylated (Jeanne et al., 2010). PML can directly bind the SUMO E2 ligase UBC9 via its RING finger. Thus, PML multimerization could allow hypersumoylation in trans of the aggregated PML mesh (Fig. 2 c), although some PML sumoylation may also occur before targeting to the matrix (Jeanne et al., 2010). Direct arsenic binding onto PML was also proposed to support UBC9 anchoring, which facilitates PML sumoylation (Zhang et al., 2010), whereas inhibition of SUMO proteases may further enhance PML hypersumoylation.
Arsenic induces both PML and PML/RARA degradation. The molecular mechanisms involved have been extensively reviewed elsewhere (de Thé and Chen, 2010; Lallemand-Breitenbach et al., 2012). In the same manner as for partner protein recruitment, arsenic-elicited PML degradation is initiated by sumoylation of K160 (Fig. 3; Lallemand-Breitenbach et al., 2001). The arsenic-induced, disulfide-linked PML hypersumoylated mesh recruits then a SUMO-dependent ubiquitin ligase, RNF4 (Häkli et al., 2005), which enforces PML polyubiquitination. This allows recruitment of the proteasome and, ultimately, PML degradation (Lallemand-Breitenbach et al., 2008; Tatham et al., 2008). The original evidence for this SUMO-initiated degradation pathway (Lallemand-Breitenbach et al., 2001) was largely ignored because, at the time, SUMOs were believed to compete with and inhibit ubiquitin-initiated proteolysis (Desterro et al., 1998).
Relevance of this model to clinical APL response to arsenic is supported by several recent observations. Cells transformed by PML/RARA point mutants, which cannot bind arsenic and therefore fail to degrade, do not undergo arsenic-induced differentiation ex vivo (Jeanne et al., 2010). Vitamin E derivatives that poison mitochondria and induce reactive oxygen species elicit long-term remissions in murine models of APL (dos Santos et al., 2012). Similarly, nonarsenic oxidants, such as Paraquat, elicit both NB reformation and APL regression in PML/RARA-driven disease only (Jeanne et al., 2010). Finally, mutations adjacent to the arsenic binding site in PML/RARA were found in two arsenic-resistant patients (Fig. 4 c; Goto et al., 2011). In retrospect, reactive oxygen species–induced PML/RARA degradation may explain why anthracyclins, which induce massive production of reactive oxygen species, show some efficacy in APL, alone or in combination with RA (Warrell et al., 1993).
PML NBs as general stress sensors
PML expression is dramatically enhanced by interferons, p53, and Wingless integration site signaling (Stadler et al., 1995: Shtutman et al., 2002; de Stanchina et al., 2004). Such transcriptional control must be taken into account when trying to solve the PML puzzle and may be part of a general response to stress (Dellaire and Bazett-Jones, 2004). Beside oxidative stress, other signals may be important for NB morphogenesis, at least in part through PML posttranslational modifications (such as phosphorylation, acetylation, and sumoylation). PML can be phosphorylated by ATRX, HIPK2, CHK2, or CK2. CHK2 phosphorylation is required for apoptosis (Yang et al., 2002), and CK2-mediated phosphorylation of PML SIM domain results in PML degradation (Scaglioni et al., 2006). PML SIM phosphorylation may modulate its affinity for SUMO-conjugated RNF4, as reported for other SUMO–SIM couples (Hecker et al., 2006; Stehmeier and Muller, 2009).
What happens downstream of stress-induced PML NB formation remains debated. Arsenic regulates partitioning of repressors, such as DAXX, between chromatin and NBs (Lehembre et al., 2001; Lin et al., 2006). Thus, PML oxidation may indirectly regulate transcription of DAXX-bound promoters (Fig. 4 a). PML may also promote sumoylation of partner proteins. In this respect, PML and sumoylation of DAXX are required for interferon-triggered B cell apoptosis (Muromoto et al., 2006). Sumoylation of partners within NBs could facilitate their subsequent phosphorylation or acetylation by locally concentrating sumoylated enzymes and substrates, as suggested for p53 activation by the HIPK2 kinase and Creb-binding protein (CBP) acetylase (Fig. 4 a; Sung et al., 2011). Finally, it is possible that, in the same manner as PML, some partner proteins are ultimately degraded within NBs through the RNF4 pathway (Fig. 4 a). Collectively, NBs appear as domains assembled by oxidative stress and sumoylation, whose assembly controls stress-induced posttranslational modifications of a wide variety of proteins.
The mysterious senescence connection
PML overexpression induces cell senescence, growth arrest, or apoptosis (Quignon et al., 1998). Conversely, pml−/− cells resist several apoptotic signals and do not undergo senescence in response to Ras signaling (Ferbeyre et al., 2000; Pearson et al., 2000). PML is clearly required for some activities of p53 (Gottifredi and Prives, 2001), but the exact mechanisms involved remain debated. PML has been proposed to facilitate p53 acetylation or phosphorylation by CBP or HIPK2, respectively (Pearson et al., 2000; Hofmann et al., 2002), or to sequester MDM2 (Fig. 4 a; Bernardi et al., 2004). Interestingly, although all PML splice variants recruit p53 into NBs, only one of them activates p53 and induces senescence (Bischof et al., 2002). Yet, p53 is not the sole actor in PML-triggered senescence. Indeed, PML can also interact with the E2F–retinoblastoma pathway, although mechanistic details remain imperfectly understood (Vernier et al., 2011). Finally, PML could play a role in the formation of senescence-associated heterochromatin foci, in which heterochromatin proteins (such as HIRA, HP1, and ASF1) transit via NBs before establishment of heterochromatin foci and senescence (Zhang et al., 2005; Ye et al., 2007). In any case, this prosenescent role of PML NBs likely explains why they are frequently observed in early tumor cells and much less as tumors progress (Koken et al., 1995; Gurrieri et al., 2004a). For example, in advanced lung cancers, CK2 activation was proposed to account for PML loss, a process believed to counteract PML-mediated growth suppression and hence favor tumor progression (Scaglioni et al., 2006).
Reintegrating PML NBs in APL pathogenesis and response to therapy
The now well-established role of PML in cell senescence and modulation of p53 signaling sheds new light on the involvement of NB disruption by PML/RARA in APL pathogenesis. Several studies have found that PML/RARA expression impedes p53 signaling and blocks the effects of p53 activation on cell death or clonogenic growth (Pearson et al., 2000; Insinga et al., 2004). Recent studies have unraveled p53 key function in controlling “stemness” or self-renewal (Zheng et al., 2008; Cicalese et al., 2009; Zhao et al., 2010), mediated in part by enforcing G0 arrest of stem cells (Liu et al., 2009). Importantly, very similar findings were reported for PML (Ito et al., 2008; Regad et al., 2009). It is therefore possible that PML directly contributes to this specific function of p53. Thus, NB disruption by PML/RARA could foster proliferation and aberrant stem cell self-renewal. In this respect, it is of note that attempts to induce APL in vivo through deregulation of RARA signaling have been largely unsuccessful (Kogan et al., 2000; Matsushita et al., 2006; Sternsdorf et al., 2006). Importantly, recent studies have demonstrated that the PML coiled-coil dimerization domain is essential for RARA to induce APL, most likely because it allows disruption of PML NBs (Occhionorelli et al., 2011).
The PML relationship with cell senescence and stem cell self-renewal is critical, not only for understanding APL initiation but also the basis of the response to therapy (Fig. 4 b). Indeed, NB reformation in response to RA or arsenic treatments tightly correlates with this response (Daniel et al., 1993; Zhu et al., 1997). Thus, NB reformation could precipitate apoptosis or senescence (Fig. 4 b). In fact, the exquisite treatment response is highly specific to PML/RARA (vs. PLZF/RARA)-driven disease (Nasr et al., 2008), suggesting that NB reformation upon treatment may be a critical determinant of long-term response. In this respect, some differences in the ex vivo sensitivity to RA of murine PML/RARA APLs generated on a pml−/− background have been reported (Rego et al., 2001), a mutation in the nonrearranged PML gene was observed in a treatment-resistant patient (Gurrieri et al., 2004b), and recent data are consistent with an important role of NB reformation in APL eradication in vivo (unpublished data). Collectively, NB reformation upon PML/RARA degradation may be a critical determinant of APL responsiveness to therapy.
Conclusions
Over the past 20 yr, APL has not only been a clinical success story but also incredibly productive for basic science, providing an inspiring example of translational research. The two active drugs were identified largely by chance and shown to represent targeted therapies a posteriori (Quignon et al., 1997; Zhu et al., 2001; Lallemand-Breitenbach et al., 2012). Modeling APL pathogenesis and basis for treatment response in the mouse allowed an unprecedented molecular and cellular understanding of this malignancy (Lallemand-Breitenbach et al., 2005; Nardella et al., 2011). Finally, the APL model highlighted the importance of proteolysis and provided the proof of concept that this could be manipulated for therapeutic purposes (Ablain et al., 2011).
This review specifically highlighted the rising importance of the role played by PML and NBs in APL pathogenesis and therapy. Some of the most decisive insights into NBs biogenesis were derived from the analysis of arsenic action in APL, illustrating the power of chemical biology. The role of reactive oxygen species in mediating PML multimerization and NB biogenesis unravels a striking link between a subcellular domain and a physicochemical parameter, suggesting that PML may act as a redox sensor, a proposal consistent with resistance of pml−/− mice to multiple stress signals.
Acknowledgments
This review is dedicated to the memory of Gerd Maul, an energetic and creative colleague from the Wistar Institute (Philadelphia, PA) who passed away in August 2010 after devoting over 20 yr of his life to PML bodies.
We apologize to our friends and colleagues whose primary work could not be cited because of space restrictions. We warmly thank all members of the laboratory for discussions and J.C. Gluckman for critical review of the manuscript.
The laboratory is supported by Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Université Paris-Diderot, Institut Universitaire de France, Ligue Contre le Cancer, Institut National du Cancer, Association pour la Recherche contre le Cancer (Griffuel Award to H. de Thé), Canceropôle Ile de France, and the European Research Council (STEMAPL advanced grant to H. de Thé).
Footnotes
Abbreviations used in this paper:
- APL
- acute promyelocytic leukemia
- CBP
- Creb-binding protein
- NB
- nuclear body
- RA
- retinoic acid
- RARA
- RA receptor α
- SIM
- SUMO-interacting motif
References
- Ablain J., de Thé H. 2011. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood. 117:5795–5802 10.1182/blood-2011-02-329367 [DOI] [PubMed] [Google Scholar]
- Ablain J., Nasr R., Bazarbachi A., de Thé H. 2011. The drug-induced degradation of oncoproteins: an unexpected achilles’ heel of cancer cells? Cancer Discov. 1:117–127 10.1158/2159-8290.CD-11-0087 [DOI] [PubMed] [Google Scholar]
- Ascoli C.A., Maul G.G. 1991. Identification of a novel nuclear domain. J. Cell Biol. 112:785–795 10.1083/jcb.112.5.785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R., Pandolfi P.P. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8:1006–1016 10.1038/nrm2277 [DOI] [PubMed] [Google Scholar]
- Bernardi R., Scaglioni P.P., Bergmann S., Horn H.F., Vousden K.H., Pandolfi P.P. 2004. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat. Cell Biol. 6:665–672 10.1038/ncb1147 [DOI] [PubMed] [Google Scholar]
- Bischof O., Kirsh O., Pearson M., Itahana K., Pelicci P.G., Dejean A. 2002. Deconstructing PML-induced premature senescence. EMBO J. 21:3358–3369 10.1093/emboj/cdf341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy M.N., Howe K., Etkin L.D., Solomon E., Freemont P.S. 1996. PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene. 13:971–982 [PubMed] [Google Scholar]
- Boisvert F.M., Hendzel M.J., Bazett-Jones D.P. 2000. Promyelocytic leukemia (PML) nuclear bodies are protein structures that do not accumulate RNA. J. Cell Biol. 148:283–292 10.1083/jcb.148.2.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D., Kogan S., Lagasse E., Weissman I., Alcalay M., Pelicci P.G., Atwater S., Bishop J.M. 1997. A PMLRARalpha transgene initiates murine acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 94:2551–2556 10.1073/pnas.94.6.2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J.T., Hall L.L., Smith K.P., Lawrence J.B. 2009. Changing nuclear landscape and unique PML structures during early epigenetic transitions of human embryonic stem cells. J. Cell. Biochem. 107:609–621 10.1002/jcb.22183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Brand N.J., Chen A., Chen S.J., Tong J.H., Wang Z.Y., Waxman S., Zelent A. 1993. Fusion between a novel Krüppel-like zinc finger gene and the retinoic acid receptor-alpha locus due to a variant t(11;17) translocation associated with acute promyelocytic leukaemia. EMBO J. 12:1161–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho G., Lim Y., Golden J.A. 2009. SUMO interaction motifs in Sizn1 are required for promyelocytic leukemia protein nuclear body localization and for transcriptional activation. J. Biol. Chem. 284:19592–19600 10.1074/jbc.M109.010181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung I., Leonhardt H., Rippe K. 2011. De novo assembly of a PML nuclear subcompartment occurs through multiple pathways and induces telomere elongation. J. Cell Sci. 124:3603–3618 10.1242/jcs.084681 [DOI] [PubMed] [Google Scholar]
- Cicalese A., Bonizzi G., Pasi C.E., Faretta M., Ronzoni S., Giulini B., Brisken C., Minucci S., Di Fiore P.P., Pelicci P.G. 2009. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 138:1083–1095 10.1016/j.cell.2009.06.048 [DOI] [PubMed] [Google Scholar]
- Cohen N., Sharma M., Kentsis A., Perez J.M., Strudwick S., Borden K.L. 2001. PML RING suppresses oncogenic transformation by reducing the affinity of eIF4E for mRNA. EMBO J. 20:4547–4559 10.1093/emboj/20.16.4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condemine W., Takahashi Y., Zhu J., Puvion-Dutilleul F., Guegan S., Janin A., de Thé H. 2006. Characterization of endogenous human promyelocytic leukemia isoforms. Cancer Res. 66:6192–6198 10.1158/0008-5472.CAN-05-3792 [DOI] [PubMed] [Google Scholar]
- Condemine W., Takahashi Y., Le Bras M., de Thé H. 2007. A nucleolar targeting signal in PML-I addresses PML to nucleolar caps in stressed or senescent cells. J. Cell Sci. 120:3219–3227 10.1242/jcs.007492 [DOI] [PubMed] [Google Scholar]
- Daniel M.-T., Koken M., Romagné O., Barbey S., Bazarbachi A., Stadler M., Guillemin M.-C., Degos L., Chomienne C., de Thé H. 1993. PML protein expression in hematopoietic and acute promyelocytic leukemia cells. Blood. 82:1858–1867 [PubMed] [Google Scholar]
- Dellaire G., Bazett-Jones D.P. 2004. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays. 26:963–977 10.1002/bies.20089 [DOI] [PubMed] [Google Scholar]
- de Stanchina E., Querido E., Narita M., Davuluri R.V., Pandolfi P.P., Ferbeyre G., Lowe S.W. 2004. PML is a direct p53 target that modulates p53 effector functions. Mol. Cell. 13:523–535 10.1016/S1097-2765(04)00062-0 [DOI] [PubMed] [Google Scholar]
- Desterro J.M.P., Rodriguez M.S., Hay R.T. 1998. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell. 2:233–239 10.1016/S1097-2765(00)80133-1 [DOI] [PubMed] [Google Scholar]
- de Thé G., Rivière M., Bernhard W. 1960. Examen au microscope électronique de la tumeur VX2 du lapin domestique dérivée du papillome de Shope. Bull. Cancer. 47:570–584 [PubMed] [Google Scholar]
- de Thé H., Chen Z. 2010. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat. Rev. Cancer. 10:775–783 10.1038/nrc2943 [DOI] [PubMed] [Google Scholar]
- de Thé H., Chomienne C., Lanotte M., Degos L., Dejean A. 1990. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature. 347:558–561 10.1038/347558a0 [DOI] [PubMed] [Google Scholar]
- de Thé H., Lavau C., Marchio A., Chomienne C., Degos L., Dejean A. 1991. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 66:675–684 10.1016/0092-8674(91)90113-D [DOI] [PubMed] [Google Scholar]
- Döhner H., Estey E.H., Amadori S., Appelbaum F.R., Büchner T., Burnett A.K., Dombret H., Fenaux P., Grimwade D., Larson R.A., et al. 2010. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 115:453–474 10.1182/blood-2009-07-235358 [DOI] [PubMed] [Google Scholar]
- dos Santos G.A., Abreu e Lima R.S., Pestana C.R., Lima A.S., Scheucher P.S., Thomé C.H., Gimenes-Teixeira H.L., Santana-Lemos B.A., Lucena-Araujo A.R., Rodrigues F.P., et al. 2012. (+)α-Tocopheryl succinate inhibits the mitochondrial respiratory chain complex I and is as effective as arsenic trioxide or ATRA against acute promyelocytic leukemia in vivo. Leukemia. 26:451–460 10.1038/leu.2011.216 [DOI] [PubMed] [Google Scholar]
- Draskovic I., Arnoult N., Steiner V., Bacchetti S., Lomonte P., Londoño-Vallejo A. 2009. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc. Natl. Acad. Sci. USA. 106:15726–15731 10.1073/pnas.0907689106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyck J.A., Maul G.G., Miller W.H., Jr., Chen J.D., Kakizuka A., Evans R.M. 1994. A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell. 76:333–343 10.1016/0092-8674(94)90340-9 [DOI] [PubMed] [Google Scholar]
- Eskiw C.H., Dellaire G., Mymryk J.S., Bazett-Jones D.P. 2003. Size, position and dynamic behavior of PML nuclear bodies following cell stress as a paradigm for supramolecular trafficking and assembly. J. Cell Sci. 116:4455–4466 10.1242/jcs.00758 [DOI] [PubMed] [Google Scholar]
- Eskiw C.H., Dellaire G., Bazett-Jones D.P. 2004. Chromatin contributes to structural integrity of promyelocytic leukemia bodies through a SUMO-1-independent mechanism. J. Biol. Chem. 279:9577–9585 10.1074/jbc.M312580200 [DOI] [PubMed] [Google Scholar]
- Estey E., Garcia-Manero G., Ferrajoli A., Faderl S., Verstovsek S., Jones D., Kantarjian H. 2006. Use of all-trans retinoic acid plus arsenic trioxide as an alternative to chemotherapy in untreated acute promyelocytic leukemia. Blood. 107:3469–3473 10.1182/blood-2005-10-4006 [DOI] [PubMed] [Google Scholar]
- Everett R.D., Murray J., Orr A., Preston C.M. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 81:10991–11004 10.1128/JVI.00705-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferbeyre G., de Stanchina E., Querido E., Baptiste N., Prives C., Lowe S.W. 2000. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 14:2015–2027 [PMC free article] [PubMed] [Google Scholar]
- Figueroa M.E., Lugthart S., Li Y., Erpelinck-Verschueren C., Deng X., Christos P.J., Schifano E., Booth J., van Putten W., Skrabanek L., et al. 2010. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 17:13–27 10.1016/j.ccr.2009.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavamzadeh A., Alimoghaddam K., Rostami S., Ghaffari S.H., Jahani M., Iravani M., Mousavi S.A., Bahar B., Jalili M. 2011. Phase II study of single-agent arsenic trioxide for the front-line therapy of acute promyelocytic leukemia. J. Clin. Oncol. 29:2753–2757 10.1200/JCO.2010.32.2107 [DOI] [PubMed] [Google Scholar]
- Goto E., Tomita A., Hayakawa F., Atsumi A., Kiyoi H., Naoe T. 2011. Missense mutations in PML-RARA are critical for the lack of responsiveness to arsenic trioxide treatment. Blood. 118:1600–1609 10.1182/blood-2011-01-329433 [DOI] [PubMed] [Google Scholar]
- Gottifredi V., Prives C. 2001. P53 and PML: new partners in tumor suppression. Trends Cell Biol. 11:184–187 10.1016/S0962-8924(01)01983-3 [DOI] [PubMed] [Google Scholar]
- Grisolano J.L., Wesselschmidt R.L., Pelicci P.G., Ley T.J. 1997. Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood. 89:376–387 [PubMed] [Google Scholar]
- Gurrieri C., Capodieci P., Bernardi R., Scaglioni P.P., Nafa K., Rush L.J., Verbel D.A., Cordon-Cardo C., Pandolfi P.P. 2004a. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J. Natl. Cancer Inst. 96:269–279 10.1093/jnci/djh043 [DOI] [PubMed] [Google Scholar]
- Gurrieri C., Nafa K., Merghoub T., Bernardi R., Capodieci P., Biondi A., Nimer S., Douer D., Cordon-Cardo C., Gallagher R., Pandolfi P.P. 2004b. Mutations of the PML tumor suppressor gene in acute promyelocytic leukemia. Blood. 103:2358–2362 10.1182/blood-2003-07-2200 [DOI] [PubMed] [Google Scholar]
- Häkli M., Karvonen U., Jänne O.A., Palvimo J.J. 2005. SUMO-1 promotes association of SNURF (RNF4) with PML nuclear bodies. Exp. Cell Res. 304:224–233 10.1016/j.yexcr.2004.10.029 [DOI] [PubMed] [Google Scholar]
- Hay R.T. 2005. SUMO: a history of modification. Mol. Cell. 18:1–12 10.1016/j.molcel.2005.03.012 [DOI] [PubMed] [Google Scholar]
- Hecker C.M., Rabiller M., Haglund K., Bayer P., Dikic I. 2006. Specification of SUMO1- and SUMO2-interacting motifs. J. Biol. Chem. 281:16117–16127 10.1074/jbc.M512757200 [DOI] [PubMed] [Google Scholar]
- Hillestad L.K. 1957. Acute promyelocytic leukemia. Acta. Med. Scand. 159:189–194 10.1111/j.0954-6820.1957.tb00124.x [DOI] [PubMed] [Google Scholar]
- Hofmann T.G., Möller A., Sirma H., Zentgraf H., Taya Y., Dröge W., Will H., Schmitz M.L. 2002. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 4:1–10 10.1038/ncb715 [DOI] [PubMed] [Google Scholar]
- Hu J., Liu Y.F., Wu C.F., Xu F., Shen Z.X., Zhu Y.M., Li J.M., Tang W., Zhao W.L., Wu W., et al. 2009. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 106:3342–3347 10.1073/pnas.0813280106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insinga A., Monestiroli S., Ronzoni S., Carbone R., Pearson M., Pruneri G., Viale G., Appella E., Pelicci P., Minucci S. 2004. Impairment of p53 acetylation, stability and function by an oncogenic transcription factor. EMBO J. 23:1144–1154 10.1038/sj.emboj.7600109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishov A.M., Sotnikov A.G., Negorev D., Vladimirova O.V., Neff N., Kamitani T., Yeh E.T., Strauss J.F., III, Maul G.G. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein Daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147:221–234 10.1083/jcb.147.2.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Bernardi R., Morotti A., Matsuoka S., Saglio G., Ikeda Y., Rosenblatt J., Avigan D.E., Teruya-Feldstein J., Pandolfi P.P. 2008. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 453:1072–1078 10.1038/nature07016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanne M., Lallemand-Breitenbach V., Ferhi O., Koken M., Le Bras M., Duffort S., Peres L., Berthier C., Soilihi H., Raught B., de Thé H. 2010. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 18:88–98 10.1016/j.ccr.2010.06.003 [DOI] [PubMed] [Google Scholar]
- Jensen K., Shiels C., Freemont P.S. 2001. PML protein isoforms and the RBCC/TRIM motif. Oncogene. 20:7223–7233 10.1038/sj.onc.1204765 [DOI] [PubMed] [Google Scholar]
- Jones L., Wei G., Sevcikova S., Phan V., Jain S., Shieh A., Wong J.C., Li M., Dubansky J., Maunakea M.L., et al. 2010. Gain of MYC underlies recurrent trisomy of the MYC chromosome in acute promyelocytic leukemia. J. Exp. Med. 207:2581–2594 10.1084/jem.20091071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei H. 1997. Cystine starvation induces reversible large-body formation from nuclear bodies in T24 cells. Exp. Cell Res. 237:207–216 10.1006/excr.1997.3790 [DOI] [PubMed] [Google Scholar]
- Kawata K., Yokoo H., Shimazaki R., Okabe S. 2007. Classification of heavy-metal toxicity by human DNA microarray analysis. Environ. Sci. Technol. 41:3769–3774 10.1021/es062717d [DOI] [PubMed] [Google Scholar]
- Kogan S.C., Hong S.H., Shultz D.B., Privalsky M.L., Bishop J.M. 2000. Leukemia initiated by PMLRARalpha: the PML domain plays a critical role while retinoic acid-mediated transactivation is dispensable. Blood. 95:1541–1550 [PubMed] [Google Scholar]
- Koken M.H.M., Puvion-Dutilleul F., Guillemin M.C., Viron A., Linares-Cruz G., Stuurman N., de Jong L., Szostecki C., Calvo F., Chomienne C., et al. 1994. The t(15;17) translocation alters a nuclear body in a retinoic acid-reversible fashion. EMBO J. 13:1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koken M.H.M., Linares-Cruz G., Quignon F., Viron A., Chelbi-Alix M.K., Sobczak-Thépot J., Juhlin L., Degos L., Calvo F., de Thé H. 1995. The PML growth-suppressor has an altered expression in human oncogenesis. Oncogene. 10:1315–1324 [PubMed] [Google Scholar]
- Lallemand-Breitenbach V., de Thé H. 2010. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2:a000661 10.1101/cshperspect.a000661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V., Guillemin M.-C., Janin A., Daniel M.-T., Degos L., Kogan S.C., Bishop J.M., de Thé H. 1999. Retinoic acid and arsenic synergize to eradicate leukemic cells in a mouse model of acute promyelocytic leukemia. J. Exp. Med. 189:1043–1052 10.1084/jem.189.7.1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V., Zhu J., Puvion F., Koken M., Honoré N., Doubeikovsky A., Duprez E., Pandolfi P.P., Puvion E., Freemont P., de Thé H. 2001. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor α degradation. J. Exp. Med. 193:1361–1371 10.1084/jem.193.12.1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V., Zhu J., Kogan S., Chen Z., de Thé H. 2005. Opinion: how patients have benefited from mouse models of acute promyelocytic leukaemia. Nat. Rev. Cancer. 5:821–827 10.1038/nrc1719 [DOI] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V., Jeanne M., Benhenda S., Nasr R., Lei M., Peres L., Zhou J., Zhu J., Raught B., de Thé H. 2008. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 10:547–555 10.1038/ncb1717 [DOI] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V., Zhu J., Chen Z., de Thé H. 2012. Curing APL through PML/RARA degradation by As2O3. Trends Mol. Med. 18:36–42 10.1016/j.molmed.2011.10.001 [DOI] [PubMed] [Google Scholar]
- LaMorte V.J., Dyck J.A., Ochs R.L., Evans R.M. 1998. Localization of nascent RNA and CREB binding protein with the PML-containing nuclear body. Proc. Natl. Acad. Sci. USA. 95:4991–4996 10.1073/pnas.95.9.4991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang M., Jegou T., Chung I., Richter K., Münch S., Udvarhelyi A., Cremer C., Hemmerich P., Engelhardt J., Hell S.W., Rippe K. 2010. Three-dimensional organization of promyelocytic leukemia nuclear bodies. J. Cell Sci. 123:392–400 10.1242/jcs.053496 [DOI] [PubMed] [Google Scholar]
- Lehembre F., Müller S., Pandolfi P.P., Dejean A. 2001. Regulation of Pax3 transcriptional activity by SUMO-1-modified PML. Oncogene. 20:1–9 10.1038/sj.onc.1204063 [DOI] [PubMed] [Google Scholar]
- Ley T.J., Mardis E.R., Ding L., Fulton B., McLellan M.D., Chen K., Dooling D., Dunford-Shore B.H., McGrath S., Hickenbotham M., et al. 2008. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 456:66–72 10.1038/nature07485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht J.D., Chomienne C., Goy A., Chen A., Scott A.A., Head D.R., Michaux J.L., Wu Y., DeBlasio A., Miller W.H., Jr., et al. 1995. Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood. 85:1083–1094 [PubMed] [Google Scholar]
- Lin D.Y., Huang Y.S., Jeng J.C., Kuo H.Y., Chang C.C., Chao T.T., Ho C.C., Chen Y.C., Lin T.P., Fang H.I., et al. 2006. Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell. 24:341–354 10.1016/j.molcel.2006.10.019 [DOI] [PubMed] [Google Scholar]
- Liu Y., Elf S.E., Miyata Y., Sashida G., Liu Y., Huang G., Di Giandomenico S., Lee J.M., Deblasio A., Menendez S., et al. 2009. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 4:37–48 10.1016/j.stem.2008.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Look A.T. 1997. Oncogenic transcription factors in the human acute leukemias. Science. 278:1059–1064 10.1126/science.278.5340.1059 [DOI] [PubMed] [Google Scholar]
- Martens J.H., Brinkman A.B., Simmer F., Francoijs K.J., Nebbioso A., Ferrara F., Altucci L., Stunnenberg H.G. 2010. PML-RARalpha/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell. 17:173–185 10.1016/j.ccr.2009.12.042 [DOI] [PubMed] [Google Scholar]
- Matera A.G. 1999. Nuclear bodies: multifaceted subdomains of the interchromatin space. Trends Cell Biol. 9:302–309 10.1016/S0962-8924(99)01606-2 [DOI] [PubMed] [Google Scholar]
- Mathews V., George B., Chendamarai E., Lakshmi K.M., Desire S., Balasubramanian P., Viswabandya A., Thirugnanam R., Abraham A., Shaji R.V., et al. 2010. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: long-term follow-up data. J. Clin. Oncol. 28:3866–3871 10.1200/JCO.2010.28.5031 [DOI] [PubMed] [Google Scholar]
- Matsushita H., Scaglioni P.P., Bhaumik M., Rego E.M., Cai L.F., Majid S.M., Miyachi H., Kakizuka A., Miller W.H., Jr., Pandolfi P.P. 2006. In vivo analysis of the role of aberrant histone deacetylase recruitment and RARα blockade in the pathogenesis of acute promyelocytic leukemia. J. Exp. Med. 203:821–828 10.1084/jem.20050616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson K., Pokrovskaja K., Kiss C., Klein G., Szekely L. 2001. Proteins associated with the promyelocytic leukemia gene product (PML)-containing nuclear body move to the nucleolus upon inhibition of proteasome-dependent protein degradation. Proc. Natl. Acad. Sci. USA. 98:1012–1017 10.1073/pnas.031566998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick A., Licht J.D. 1999. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 93:3167–3215 [PubMed] [Google Scholar]
- Meulmeester E., Kunze M., Hsiao H.H., Urlaub H., Melchior F. 2008. Mechanism and consequences for paralog-specific sumoylation of ubiquitin-specific protease 25. Mol. Cell. 30:610–619 10.1016/j.molcel.2008.03.021 [DOI] [PubMed] [Google Scholar]
- Müller S., Matunis M.J., Dejean A. 1998. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J. 17:61–70 10.1093/emboj/17.1.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muromoto R., Ishida M., Sugiyama K., Sekine Y., Oritani K., Shimoda K., Matsuda T. 2006. Sumoylation of Daxx regulates IFN-induced growth suppression of B lymphocytes and the hormone receptor-mediated transactivation. J. Immunol. 177:1160–1170 [DOI] [PubMed] [Google Scholar]
- Nardella C., Lunardi A., Patnaik A., Cantley L.C., Pandolfi P.P. 2011. The APL paradigm and the “co-clinical trial” project. Cancer Discov. 1:108–116 10.1158/2159-8290.CD-11-0061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr R., Guillemin M.C., Ferhi O., Soilihi H., Peres L., Berthier C., Rousselot P., Robledo-Sarmiento M., Lallemand-Breitenbach V., Gourmel B., et al. 2008. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 14:1333–1342 10.1038/nm.1891 [DOI] [PubMed] [Google Scholar]
- Negorev D.G., Vladimirova O.V., Maul G.G. 2009. Differential functions of interferon-upregulated Sp100 isoforms: herpes simplex virus type 1 promoter-based immediate-early gene suppression and PML protection from ICP0-mediated degradation. J. Virol. 83:5168–5180 10.1128/JVI.02083-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negorev D.G., Vladimirova O.V., Kossenkov A.V., Nikonova E.V., Demarest R.M., Capobianco A.J., Showe M.K., Rauscher F.J., III, Showe L.C., Maul G.G. 2010. Sp100 as a potent tumor suppressor: accelerated senescence and rapid malignant transformation of human fibroblasts through modulation of an embryonic stem cell program. Cancer Res. 70:9991–10001 10.1158/0008-5472.CAN-10-1483 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Occhionorelli M., Santoro F., Pallavicini I., Gruszka A., Moretti S., Bossi D., Viale A., Shing D., Ronzoni S., Muradore I., et al. 2011. The self-association coiled-coil domain of PML is sufficient for the oncogenic conversion of the retinoic acid receptor (RAR) alpha. Leukemia. 25:814–820 10.1038/leu.2011.18 [DOI] [PubMed] [Google Scholar]
- Pearson M., Carbone R., Sebastiani C., Cioce M., Fagioli M., Saito S., Higashimoto Y., Appella E., Minucci S., Pandolfi P.P., Pelicci P.G. 2000. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 406:207–210 10.1038/35018127 [DOI] [PubMed] [Google Scholar]
- Pekovic V., Gibbs-Seymour I., Markiewicz E., Alzoghaibi F., Benham A.M., Edwards R., Wenhert M., von Zglinicki T., Hutchison C.J. 2011. Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell. 10:1067–1079 10.1111/j.1474-9726.2011.00750.x [DOI] [PubMed] [Google Scholar]
- Piazza F., Gurrieri C., Pandolfi P.P. 2001. The theory of APL. Oncogene. 20:7216–7222 10.1038/sj.onc.1204855 [DOI] [PubMed] [Google Scholar]
- Potts P.R., Yu H. 2007. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 14:581–590 10.1038/nsmb1259 [DOI] [PubMed] [Google Scholar]
- Quignon F., Chen Z., de Thé H. 1997. Retinoic acid and arsenic: towards oncogene-targeted treatments of acute promyelocytic leukaemia. Biochim. Biophys. Acta. 1333:M53–M61 [DOI] [PubMed] [Google Scholar]
- Quignon F., De Bels F., Koken M., Feunteun J., Ameisen J.-C., de Thé H. 1998. PML induces a novel caspase-independent death process. Nat. Genet. 20:259–265 10.1038/3068 [DOI] [PubMed] [Google Scholar]
- Raelson J.V., Nervi C., Rosenauer A., Benedetti L., Monczak Y., Pearson M., Pelicci P.G., Miller W.H., Jr. 1996. The PML/RAR alpha oncoprotein is a direct molecular target of retinoic acid in acute promyelocytic leukemia cells. Blood. 88:2826–2832 [PubMed] [Google Scholar]
- Regad T., Bellodi C., Nicotera P., Salomoni P. 2009. The tumor suppressor Pml regulates cell fate in the developing neocortex. Nat. Neurosci. 12:132–140 10.1038/nn.2251 [DOI] [PubMed] [Google Scholar]
- Rego E.M., He L.Z., Warrell R.P., Jr., Wang Z.G., Pandolfi P.P. 2000. Retinoic acid (RA) and As2O3 treatment in transgenic models of acute promyelocytic leukemia (APL) unravel the distinct nature of the leukemogenic process induced by the PML-RARalpha and PLZF-RARalpha oncoproteins. Proc. Natl. Acad. Sci. USA. 97:10173–10178 10.1073/pnas.180290497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego E.M., Wang Z.G., Peruzzi D., He L.Z., Cordon-Cardo C., Pandolfi P.P. 2001. Role of promyelocytic leukemia (PML) protein in tumor suppression. J. Exp. Med. 193:521–529 10.1084/jem.193.4.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley J.D., Golomb H.M., Dougherty C. 1977. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet. 309:549–550 10.1016/S0140-6736(77)91415-5 [DOI] [PubMed] [Google Scholar]
- Sawyers C.L. 2008. The cancer biomarker problem. Nature. 452:548–552 10.1038/nature06913 [DOI] [PubMed] [Google Scholar]
- Scaglioni P.P., Yung T.M., Cai L.F., Erdjument-Bromage H., Kaufman A.J., Singh B., Teruya-Feldstein J., Tempst P., Pandolfi P.P. 2006. A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell. 126:269–283 10.1016/j.cell.2006.05.041 [DOI] [PubMed] [Google Scholar]
- Shen T.H., Lin H.K., Scaglioni P.P., Yung T.M., Pandolfi P.P. 2006. The mechanisms of PML-nuclear body formation. Mol. Cell. 24:331–339 10.1016/j.molcel.2006.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z.X., Shi Z.Z., Fang J., Gu B.W., Li J.M., Zhu Y.M., Shi J.Y., Zheng P.Z., Yan H., Liu Y.F., et al. 2004. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 101:5328–5335 10.1073/pnas.0400053101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiels C., Islam S.A., Vatcheva R., Sasieni P., Sternberg M.J., Freemont P.S., Sheer D. 2001. PML bodies associate specifically with the MHC gene cluster in interphase nuclei. J. Cell Sci. 114:3705–3716 [DOI] [PubMed] [Google Scholar]
- Shtutman M., Zhurinsky J., Oren M., Levina E., Ben-Ze’ev A. 2002. PML is a target gene of beta-catenin and plakoglobin, and coactivates beta-catenin-mediated transcription. Cancer Res. 62:5947–5954 [PubMed] [Google Scholar]
- Sohal J., Phan V.T., Chan P.V., Davis E.M., Patel B., Kelly L.M., Abrams T.J., O’Farrell A.M., Gilliland D.G., Le Beau M.M., Kogan S.C. 2003. A model of APL with FLT3 mutation is responsive to retinoic acid and a receptor tyrosine kinase inhibitor, SU11657. Blood. 101:3188–3197 10.1182/blood-2002-06-1800 [DOI] [PubMed] [Google Scholar]
- Stadler M., Chelbi-Alix M.K., Koken M.H.M., Venturini L., Lee C., Saïb A., Quignon F., Pelicano L., Guillemin M.-C., Schindler C., de Thé H. 1995. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene. 11:2565–2573 [PubMed] [Google Scholar]
- Stehmeier P., Muller S. 2009. Phospho-regulated SUMO interaction modules connect the SUMO system to CK2 signaling. Mol. Cell. 33:400–409 10.1016/j.molcel.2009.01.013 [DOI] [PubMed] [Google Scholar]
- Sternsdorf T., Phan V.T., Maunakea M.L., Ocampo C.B., Sohal J., Silletto A., Galimi F., Le Beau M.M., Evans R.M., Kogan S.C. 2006. Forced retinoic acid receptor alpha homodimers prime mice for APL-like leukemia. Cancer Cell. 9:81–94 10.1016/j.ccr.2005.12.030 [DOI] [PubMed] [Google Scholar]
- Stuurman N., de Graaf A., Floore A., Josso A., Humbel B., de Jong L., van Driel R. 1992a. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J. Cell Sci. 101:773–784 [DOI] [PubMed] [Google Scholar]
- Stuurman N., Floore A., Colen A., de Jong L., van Driel R. 1992b. Stabilization of the nuclear matrix by disulfide bridges: identification of matrix polypeptides that form disulfides. Exp. Cell Res. 200:285–294 10.1016/0014-4827(92)90174-7 [DOI] [PubMed] [Google Scholar]
- Sung K.S., Lee Y.A., Kim E.T., Lee S.R., Ahn J.H., Choi C.Y. 2011. Role of the SUMO-interacting motif in HIPK2 targeting to the PML nuclear bodies and regulation of p53. Exp. Cell Res. 317:1060–1070 10.1016/j.yexcr.2010.12.016 [DOI] [PubMed] [Google Scholar]
- Szostecki C., Guldner H.H., Netter H.J., Will H. 1990. Isolation and characterization of cDNA encoding a human nuclear antigen predominantly recognized by autoantibodies from patients with primary biliary cirrhosis. J. Immunol. 145:4338–4347 [PubMed] [Google Scholar]
- Tallman M.S., Altman J.K. 2009. How I treat acute promyelocytic leukemia. Blood. 114:5126–5135 10.1182/blood-2009-07-216457 [DOI] [PubMed] [Google Scholar]
- Tallman M.S., Kwaan H.C. 1992. Reassessing the hemostatic disorder associated with acute promyelocytic leukemia. Blood. 79:543–553 [PubMed] [Google Scholar]
- Tatham M.H., Geoffroy M.C., Shen L., Plechanovova A., Hattersley N., Jaffray E.G., Palvimo J.J., Hay R.T. 2008. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 10:538–546 10.1038/ncb1716 [DOI] [PubMed] [Google Scholar]
- Tsukamoto T., Hashiguchi N., Janicki S.M., Tumbar T., Belmont A.S., Spector D.L. 2000. Visualization of gene activity in living cells. Nat. Cell Biol. 2:871–878 10.1038/35046510 [DOI] [PubMed] [Google Scholar]
- Vernier M., Bourdeau V., Gaumont-Leclerc M.F., Moiseeva O., Bégin V., Saad F., Mes-Masson A.M., Ferbeyre G. 2011. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 25:41–50 10.1101/gad.1975111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Chen X.Y., Wang B.S., Rong Z.X., Qi H., Chen H.Z. 2011. The efficacy and safety of arsenic trioxide with or without all-trans retinoic acid for the treatment of acute promyelocytic leukemia: a meta-analysis. Leuk. Res. 35:1170–1177 10.1016/j.leukres.2011.06.002 [DOI] [PubMed] [Google Scholar]
- Wang Z.Y., Chen Z. 2008. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 111:2505–2515 10.1182/blood-2007-07-102798 [DOI] [PubMed] [Google Scholar]
- Warrell R.P., Jr., de Thé H., Wang Z.Y., Degos L. 1993. Acute promyelocytic leukemia. N. Engl. J. Med. 329:177–189 10.1056/NEJM199307153290307 [DOI] [PubMed] [Google Scholar]
- Wartman L.D., Larson D.E., Xiang Z., Ding L., Chen K., Lin L., Cahan P., Klco J.M., Welch J.S., Li C., et al. 2011. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J. Clin. Invest. 121:1445–1455 10.1172/JCI45284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidtkamp-Peters S., Lenser T., Negorev D., Gerstner N., Hofmann T.G., Schwanitz G., Hoischen C., Maul G., Dittrich P., Hemmerich P. 2008. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 121:2731–2743 10.1242/jcs.031922 [DOI] [PubMed] [Google Scholar]
- Weis K., Rambaud S., Lavau C., Jansen J., Carvalho T., Carmo-Fonseca M., Lamond A., Dejean A. 1994. Retinoic acid regulates aberrant nuclear localization of PML-RAR alpha in acute promyelocytic leukemia cells. Cell. 76:345–356 10.1016/0092-8674(94)90341-7 [DOI] [PubMed] [Google Scholar]
- Welch JS, Yuan W., Ley T.J. 2008. Expression of PML-RAR alpha by the murine PML locus leads to myeloid self-renewal, clonal expansion and morphologic promyelocytic leukemia. Blood. 112:344–345 [Google Scholar]
- Yang S., Kuo C., Bisi J.E., Kim M.K. 2002. PML-dependent apoptosis after DNA damage is regulated by the checkpoint kinase hCds1/Chk2. Nat. Cell Biol. 4:865–870 10.1038/ncb869 [DOI] [PubMed] [Google Scholar]
- Ye X., Zerlanko B., Zhang R., Somaiah N., Lipinski M., Salomoni P., Adams P.D. 2007. Definition of pRB- and p53-dependent and -independent steps in HIRA/ASF1a-mediated formation of senescence-associated heterochromatin foci. Mol. Cell. Biol. 27:2452–2465 10.1128/MCB.01592-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Poustovoitov M.V., Ye X., Santos H.A., Chen W., Daganzo S.M., Erzberger J.P., Serebriiskii I.G., Canutescu A.A., Dunbrack R.L., et al. 2005. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell. 8:19–30 10.1016/j.devcel.2004.10.019 [DOI] [PubMed] [Google Scholar]
- Zhang X.W., Yan X.J., Zhou Z.R., Yang F.F., Wu Z.Y., Sun H.B., Liang W.X., Song A.X., Lallemand-Breitenbach V., Jeanne M., et al. 2010. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 328:240–243 10.1126/science.1183424 [DOI] [PubMed] [Google Scholar]
- Zhao Z., Zuber J., Diaz-Flores E., Lintault L., Kogan S.C., Shannon K., Lowe S.W. 2010. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 24:1389–1402 10.1101/gad.1940710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H., Ying H., Yan H., Kimmelman A.C., Hiller D.J., Chen A.J., Perry S.R., Tonon G., Chu G.C., Ding Z., et al. 2008. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 455:1129–1133 10.1038/nature07443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Koken M.H.M., Quignon F., Chelbi-Alix M.K., Degos L., Wang Z.Y., Chen Z., de Thé H. 1997. Arsenic-induced PML targeting onto nuclear bodies: implications for the treatment of acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 94:3978–3983 10.1073/pnas.94.8.3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Gianni M., Kopf E., Honoré N., Chelbi-Alix M., Koken M., Quignon F., Rochette-Egly C., de Thé H. 1999. Retinoic acid induces proteasome-dependent degradation of retinoic acid receptor alpha (RARalpha) and oncogenic RARalpha fusion proteins. Proc. Natl. Acad. Sci. USA. 96:14807–14812 10.1073/pnas.96.26.14807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Lallemand-Breitenbach V., de Thé H. 2001. Pathways of retinoic acid- or arsenic trioxide-induced PML/RARalpha catabolism, role of oncogene degradation in disease remission. Oncogene. 20:7257–7265 10.1038/sj.onc.1204852 [DOI] [PubMed] [Google Scholar]
- Zhu J., Chen Z., Lallemand-Breitenbach V., de Thé H. 2002. How acute promyelocytic leukaemia revived arsenic. Nat. Rev. Cancer. 2:705–713 10.1038/nrc887 [DOI] [PubMed] [Google Scholar]
- Zhu J., Zhou J., Peres L., Riaucoux F., Honoré N., Kogan S., de Thé H. 2005. A sumoylation site in PML/RARA is essential for leukemic transformation. Cancer Cell. 7:143–153 10.1016/j.ccr.2005.01.005 [DOI] [PubMed] [Google Scholar]