

Abstract
The survival of cardiomyocytes must be ensured as the myocardium adjusts to a myriad of competing physiologic and pathophysiologic demands. A significant loss of these contractile cells, together with their replacement by stiff fibrillar collagen in the form of fibrous tissue accounts for a transition from a usually efficient muscular pump into one that is failing. Cellular and subcellular mechanisms involved in the pathogenic origins of cardiomyocyte cell death have long been of interest. This includes programmed molecular pathways to either necrosis or apoptosis which are initiated from ischemic or nonischemic origins. Herein we focus on the central role played by a mitochondriocentric signal-transducer-effector pathway to nonischemic cardiomyocyte necrosis which is common to acute and chronic stressor states. We begin by building upon the hypothesis advanced by Albrecht Fleckenstein and coworkers some 40 years ago based on the importance of calcitropic hormone- mediated intracellular Ca2+ overloading which predominantly involves subsarcolemmal mitochondria and is the signal to pathway activation. Other pathway components, which came to be recognized in subsequent years, include the induction of oxidative stress and opening of the mitochondrial inner membrane permeability transition pore. The ensuing loss of cardiomyocytes and consequent replacement fibrosis, or scarring, represents a disease of adaptation and a classic example of when homeostasis begets dyshomeostasis.
Keywords: Mitochondria, Cardiomyocytes, Necrosis, Calcium overloading, Oxidative stress, mPTP opening
Introduction
The heart has classically been viewed as a postmitotic organ: its parenchyma, composed of terminally differentiated highly specialized cardiomyocytes, is unable to replicate. In essence, the myocardium has a fixed number of cardiomyocytes. This perspective, however, has more recently been challenged. For example, Anversa and coworkers [30] and Houser et al. [3] have given evidence of cell cycle activation and documented nuclear and cellular division of a subpopulation of cardiomyocytes. They would argue such endogenous progenitor cells exist and could be mobilized to regenerate myocardium following injury [30,3]. Dawn et al. [66] have focused on the mobilization of progenitor cells derived from bone marrow and their homing to the site of myocardial injury, where they would transdifferentiate and replace lost cardiomyocytes. One remains hopeful that in the future such pluripotent cells, irrespective of their origin, could be strategically mobilized to rescue and rebuild injured myocardium.
These provocative possibilities notwithstanding, the survival of cardiomyocytes must be ensured as this muscular pump adapts to diverse physiologic and pathophysiologic stimuli. These include: the hemodynamic burden imposed by short-term pressure or volume loading, which respectively accompanies the physiologic stress of isometric and isotonic exercise; the provocation incurred during elevations in circulating catecholamines associated with acute bodily injury; and the cumulative and persistent hemodynamic overloading and neurohormonal activation which are integral to chronic stressor states, such as congestive heart failure. Its pumping efficiency is compromised when the cumulative loss of cardiomyocytes crosses the biologic threshold and their replacement by stiff fibrillar collagen in the form of fibrous tissue poses an impediment to both the stretching and contraction of the myocardium while the hypertrophy of remaining muscle cells reaches its physiologic limits. Such pathologic remodeling accounts for the clinical transition to heart failure and holds true irrespective of the etiologic origins of cell loss or whether necrotic or apoptotic cell death pathways, or both, are involved. A critical loss of cardiomyocytes and the appearance of fibrosis are fundamental structural abnormalities accounting for the appearance of a failing muscular pump while an ongoing loss of cardiomyocytes contributes to the progressive nature of heart failure (HF) [9,18].
The cellular and subcellular mechanisms involved in the pathogenic origins of cardiomyocyte cell death have long been of interest, be they related to reduced coronary blood flow with myocardial infarction, ischemia/reperfusion injury, or nonischemic necrosis. In a series of studies, many appearing in Pflügers Archives 40 years ago [83,20,44], Fleckenstein and coworkers drew attention to the importance of excessive intracellular Ca2+ accumulation (EICA) and the role of Ca2+ overloaded mitochondria in particular, together with ATP depletion, in leading to nonischemic cardiomyocyte necrosis. Herein, we build upon their hypothesis and the central role played by a mitochondriocentric signal-transducer-effector pathway in accounting for necrotic cell loss and which is common to both acute and chronic stressor states. In so doing, we draw attention to mitochondria-targeted interventions and their potential as cardioprotective strategies in salvaging myocardium. For the reader interested in cardiomyocyte apoptosis several reviews are recommended [47,28,77].
The Death of Cardiomyocytes: Necrosis and Apoptosis
The death of cardiomyocytes involves complex programmed molecular pathways: one is termed necrosis [48]; the other apoptosis [47,28,77]. The potential contribution of another yet-to-be-fully-elucidated pathway, termed autophagy with lysosomal-mediated autocannibalism, is in evolution and its biologic significance in the failing heart remains to be fully established [46,58,12,4].
Necrosis
Necrotic cell death is viewed as “dirty” death because the hyperpermeable cell on its terminal trajectory spills intracellular contents (e.g., troponins) which serve as “danger” signals to the innate immune system [54]. The ensuing invasion of inflammatory cells to the site of necrosis, together with recruited myofibroblasts, results in tissue repair and a subsequent reparative fibrosis. This myocardial scarring is a morphologic footprint of previous necrosis while elevated serum troponin levels represent molecular markers of recent cardiomyocyte necrosis.
Foci of fibrosis are found scattered throughout the myocardium of the right and left heart in the explanted failing human heart, where fibrosis is considered the major component to the adverse structural remodeling of myocardium [6]. It follows pari passu that necrosis is a major form of cardiomyocyte loss in the failing heart. The diffuse patchy distribution to myocardial scarring, often most prevalent in the endomyocardium, suggests that cardiomyocyte necrosis is widespread and likely to be an ongoing process. Each episode of necrosis and tissue repair is coupled to myofibroblast-derived collagen fibrillogenesis and the accumulation of stiff type I collagen [89]. This fibrillar collagen, which predominates in scar tissue, has the tensile strength equivalent to steel. In time, the progressive fibrosis of myocardium contributes to diastolic and systolic ventricular dysfunction [90,9,18].
Apoptosis
Apoptosis, on the other hand, is considered a “sterile” form of cell death with cardiomyocytes rapidly engulfed by macrophages never to spill their contents, nor to provoke the immune system or to invoke the invasion of inflammatory cells to the site of injury. Hence, tissue repair eventuating in fibrous tissue does not take place after apoptotic cell death. Accordingly, the apoptotic loss of cardiomyocytes leaves neither a morphologic trail nor a biochemical footprint. Some would question the pathophysiologic role of apoptosis in the loss of cardiomyocytes and the progression to HF [69,81,31,80].
Mitochondriocentric Pathway to Nonischemic Cardiomyocyte Necrosis
Historical Perspective
Research conducted by Albrecht Fleckenstein and coworkers at the Physiologisches Institute of the University of Freiburg some 40 years ago, led to the novel hypothesis that catecholamine-mediated EICA in cardiomyocytes and mitochondrial Ca2+ overloading in particular, combined with ATP depletion, lead to necrosis of these cells. Because the calcitropic hormones, parathyroid hormone (PTH) and vitamin D, potentiate Ca2+ overloading and ATP loss, they sensitize cardiomyocytes to catecholamine-induced necrosis [22]. This contrasts to Ca2+ channel blockers, cations (e.g., Zn2+) which compete with Ca2+ entry, or β1 adrenergic receptor antagonists, each of which were cardioprotective in response to catecholamine treatment of rats using isoproterenol (Isop) [45,36,75,16,76]. The multidisciplinary team of talented investigators from the Institute contributed importantly to this “new principle in cardiac pathophysiology” [21]. They included H. Antonio, H. J. Doering; J. Janke, H. Kammermeier, R. Kaufmann, H. Krause, and H. Tritthart. The Ca2+ overloading-to-cardiomyocyte-necrosis hypothesis was extended by these investigators and by Bhattacharya et al. to the hereditary cardiomyopathy of the Syrian Hamster, where the cardioprotective properties of Ca2+ channel blockade were likewise demonstrated [51,19,8].
The Fleckenstein hypothesis of nonischemic cardiomyocyte necrosis has recently been broadened into a central mitochondriocentric signal-transducer-effector (MSTE) pathway and found to be common to both acute and chronic stressor states [74,41]. As Fleckenstein envisaged, the signal for this pathway includes catecholamine- or PTH-mediated intracellular Ca2+ overloading (see Figure 1). This initial event involves the subsarcolemmal mitochondria spatially closest to Ca2+ channels (vis-à-vis the interfibrillar population closer to contractile proteins) [23,41,63,17,11]. The pathway transducer is embodied by induction of oxidative stress within Ca2+ overloaded organelles and where their rate of reactive oxygen species generation overwhelms their rate of detoxification by endogenous metalloenzyme-based antioxidant defenses whose activity depends on intracellular Cu2+, Zn2+ and Se2+. A more terminal event and pathway effector is a cyclophilin D-dependent opening of the mitochondrial inner membrane permeability transition pore (mPTP) [34]. The consequent osmotic-based entry of solutes and ongoing swelling accounts for the degeneration of these organelles, loss of ATP synthesis, and ensuing cardiomyocyte necrosis.
Fig. 1.
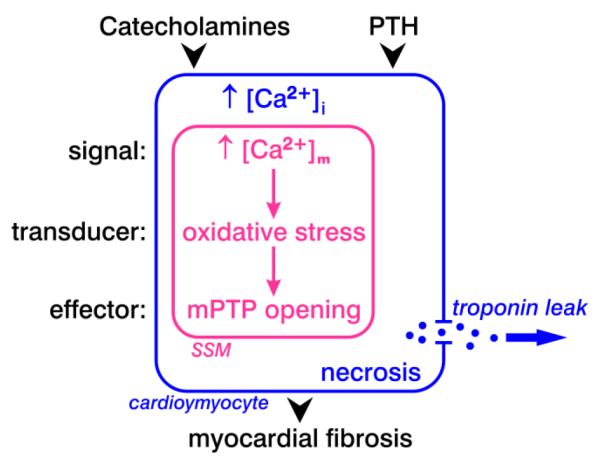
A mitochondriocentric signal-transducer-effector pathway to nonischemic cardiomyocyte necrosis. Catecholamines and the calcitropic hormone, parathyroid hormone (PTH), each raises Ca2+ entry leading to intracellular Ca2+ overloading with the ensuing increment in cytosolic [Ca2+]i and mitochondrial [Ca2+]m, specifically the subsarcolemmal (SSM) population of these organelles. In response to mitochondrial Ca2+ overloading and ATP depletion, there follows the induction of oxidative stress by these organelles and subsequent opening of their inner membrane permeability transition pore (mPTP) with solute entry, osmotic swelling and structural degeneration of these organelles. This programmed sequence of events leads to cardiomyocyte necrosis with a leakage of intracellular contents, including troponins. Tissue repair follows the loss of necrotic cardiomyocytes. This healing eventuates in a reparative fibrosis that preserves the structural integrity of the myocardium. Microscopic scarring represents a morphologic trail, with elevated serum troponins, the biochemical footprints of cardiomyocyte necrosis
How cardiomyocytes determine the activation of apoptotic or necrotic cell death pathways is not fully understood and is under investigation [34,56,60]. The functional roles of these specific pathway components was selectively validated (vide infra) in acute and chronic stressor states using pharmacologic agents that interrupted the cascade at specific downstream targets in rats receiving either Isop or 4 wks aldosterone/salt treatment (ALDOST), respectively [74,73,38,39]. We hypothesize that a lesser level of pathway activation, without ATP depletion, may lead to apoptosis whereas a more severe activation and ATP depletion would result in irreparable damage with cardiomyocyte necrosis and reparative fibrosis in the heart as observed in our animal studies.
Acute Stressor States
Following a single subcutaneous dose of Isop, ionized hypocalcemia is evident within 2 h and attributable to the translocation of Ca2+ to diverse tissues that includes the heart, skeletal muscle and peripheral blood mononuclear cells [74]. In cardiomyocytes this EICA includes increased mitochondrial [Ca2+]m coupled with increased H2O2 production and increased plasma and cardiac tissue 8-isoprostane, a biomarker of lipid peroxidation, and enhanced propensity to mPTP opening, as evidenced by the response to experimental CaCl2 stimulation. Intracellular Ca2+ overloading and consequent necrosis with myocardial scarring that accompanies Isop-mediated hyperadrenergic state favors the endomyocardium of the left ventricular apex [59,88,64,7,74]. We believe this propensity to catecholamine-induced injury at the apex is in keeping with its greater β1 adrenergic receptor density [74,7,14] and which is driven by the apical to basal activation of the myocardium needed for the sequential peristaltic-like emptying of the ventricle into the aorta [65,70,10]. These pathophysiologic events were abrogated by pretreatment with either carvedilol, a β-adrenergic receptor antagonist, or quercetin, a flavonoid, each of which have mitochondria-targeted antioxidant properties [74,67,42,72,84]. In transgenic mice with enhanced sarcolemmal L-type Ca2+ channel activity, a progressive necrosis of cardiomyocytes is seen together with consequent appearance of reparative fibrosis and pump dysfunction which are accentuated by provocation with Isop [5]. In other rodent models, the loss of cyclophilin D blocked Ca2+ overloading-based cardiomyocyte necrosis while overexpression of Bcl-2, an antiapoptotic factor, proved ineffective [57]. Collectively, these findings support the central role of the MSTE pathway in nonischemic cardiomyocyte necrosis during acute hyperadrenergic states.
Chronic Stressor States
Chronic inappropriate (relative to dietary Na+) aldosteronism appears with congestive heart failure (CHF), low-renin hypertension, or the adrenal glands’ autonomous production of aldosterone. ALDOST has been used in rats to induce a chronic stressor state to interrogate the specific MSTE pathway components. However, we have been unable to reproduce the expected cardiac remodeling in mice receiving ALDOST. Hence, we have not pursued genetic manipulations to further elucidate the pathway components in mice.
Chronic ALDOST in rats is associated with marked increments in fecal and urinary Ca2+ excretion, and which are already evident at wk 1. For the fixed Ca2+ intake attendant with laboratory chow provided to these rats, these excretory losses quickly lead to the appearance of plasma ionized hypocalcemia at wk 1 generating a major stimulus to the parathyroid glands’ increased secretion of PTH. The accompanying secondary hyperparathyroidism (SHPT) is further evidenced by bone resorption and a marked fall in bone mineral density and strength seen at wks 4–6 ALDOST [14,15]. PTH-mediated intracellular Ca2+ overloading includes both cardiomyocyte cytosolic free [Ca2+]i and subsarcolemmal mitochondrial [Ca2+]m, and is evident at wk 1 and persistent over 4 wks, together with increased production of H2O2 by these mitochondria.
This prooxidant state is counterbalanced by an increment in antioxidant defenses which also appears at wk 1. This includes the increased activities of Cu/Zn-superoxide dismutase and glutathione peroxidase and total antioxidant capacity, which consists of nonenzymatic low molecular weight antioxidants [38]. However, the persistent Ca2+ overloading of cardiomyocytes present over 4 wks leads to a dysequilibrium between pro- and antioxidants and altered redox state. In the absence of additional increments in endogenous defenses, a marked rise in reactive oxidant species generation by these organelles follows coupled with the subsequent opening of their inner membrane mPTP leading to cardiomyocyte necrosis and consequent tissue repair. Cardiac pathology with microscopic scarring is first evident at 4 wks ALDOST [79,41].
The crucial importance of PTH-mediated Ca2+ overloading-induced oxidative stress in leading to the proinflammatory cardiac failure phenotype of this model is evidenced by the upregulation of the redox-sensitive nuclear transcription factor (NF)-κB and the proinflammatory gene cascade it regulates, including ICAM-1, MCP1 and TNF-α, and which are abrogated by cotreatment with an antioxidant N-acetylcysteine [79]. The importance of NFκB signaling in the heart and its role as a signaling integrator has led some to propose targeting this mediator of cytokine responses in preventing pathologic remodeling with HF [37,26].
The crucial importance of SHPT in mediating this pathophysiologic scenario was demonstrated by a number of interventions. They included: cotreatment with spironolactone, an aldosterone receptor antagonist, which attenuated the marked excretory losses of Ca2+ in feces and urine, and thereby prevented ionized hypocalcemia with SHPT [15]; cotreatment with a Ca2+ and vitamin D3-supplemented diet to prevent ionized hypocalcemia and SHPT [25]; surgical parathyroidectomy performed prior to initiating ALDOST [87]; medical parathyroidectomy using cotreatment with a calcimimetic to raise the threshold of the Ca2+-sensing receptor of the parathyroids and prevent SHPT [71]; and cotreatment with a Ca2+ channel blocker [1]. In subsequent studies the MSTE pathway components were addressed using mitochondria-targeted interventions (see Figure 2). They included quercetin and cyclosporine A, an mPTP opening inhibitor. We found the marked increments in [Ca2+]m and mitochondrial H2O2 production, together with increased 8-isoprostane and propensity to their mPTP opening and myocardial scarring seen at wk 4 ALDOST, were each attenuated by cotreatment with either of these agents. Mitochondrial electron transport as a source of reactive oxygen species has also been targeted for cardioprotection [85] while in the vasculature, NADPH oxidase as a source of oxygen radical generation has been targeted [61,49].
Fig. 2.
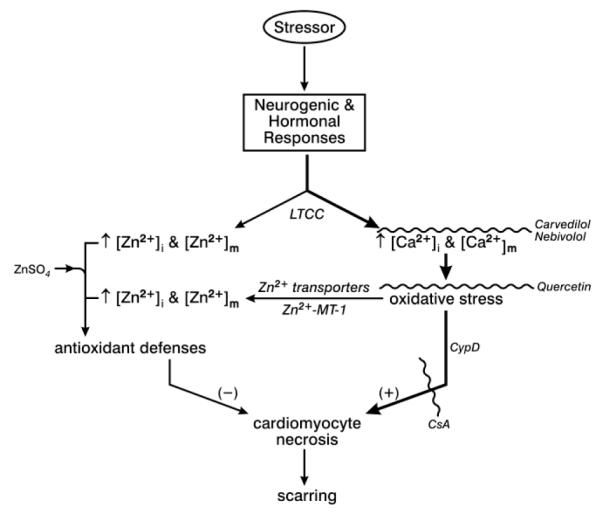
Acute and chronic stressor states are accompanied by neurogenic and hormonal responses which lead to intracellular Ca2+ and Zn2+ entry via L-type Ca2+ channels (LTCC) with Ca2+ acting as prooxidant and Zn2+ as antioxidant. Cytosolic [Ca2+]i and [Zn2+]i, as well as mitochondrial [Ca2+]m and [Zn2+]m are each increased. Intracellular Ca2+ overloading and the ensuing induction of oxidative stress upregulates the expression of Zn2+ transporters that further raises Zn2+ entry while releasing inactive intracellular Zn2+ which had been bound to its binding protein metallothionein (MT)-1. Collectively, these responses further raise [Zn2+]i and [Zn2+]m and antioxidant defenses. Maintenance of this critical equilibrium between pro- and antioxidants determine the fate of cardiomyocytes with oxidative stress leading to necrosis and myocardial scarring.
Mitochondria-targeted interventions can be used as cardioprotective strategies. For example, the prooxidant pathway to necrosis can be interrupted by pharma- and nutriceuticals: a) reducing mitochondrial Ca2+ uptake with either carvedilol or nebivolol; b) a flavonoid, quercetin, as antioxidant; and c) with cyclosporine A, a mPTP opening inhibitor. Antioxidant defenses, on the other hand, can be upregulated by ZnSO4 supplementation that raises [Zn2+]i and [Zn2+]m. See text. Adapted with permission from [40]
Intrinsically Coupled Ca2+ and Zn2+ Dyshomeostasis: Prooxidant and Antioxidant
An excessive accumulation of cytosolic [Ca2+]i and mitochondrial [Ca2+]m do occur in cardiomyocytes under acute and chronic stressor states, in which each operates as endogenous prooxidant. Mg2+ and Mn2+ are natural antagonists to Ca2+ entry. Subsequently, we identified Zn2+ to be intrinsically coupled to cardiomyocyte Ca2+ entry whereas Zn2+ is distributed within the cytosol and mitochondria functioning as antioxidant [41,13] (see Table 1).
Table 1.
Coupled dyshomeostasis of Ca2+ and Zn2+ in cardiomyocytes: response to chronic aldosteronism [38,13]
| Cytosolic Free | Mitochondrial | |||
|---|---|---|---|---|
| [Ca2+]i (nM) |
[Zn2+]i (nM) |
[Ca2+]m (nM) |
[Zn2+]m (ng/mg mitochondrial protein) |
|
| Control | 29±4 | 0.76±0.12 | 82±11 | 26±3 |
| ALDOST (4 wks) | 80±5* | 1.64±0.08* | 139±9* | 42±3* |
p<0.05 vs. control
Acute Stressor States
Soon after a single subcutaneous dose of Isop, cardiac tissue Ca2+ levels rise. Because of cell necrosis, Zn2+ transporters are no longer operative and therefore tissue Zn2+ levels decline at the site of injury [74,16,76]. In surrounding tissues, however, Zn2+ levels rise with the upregulation in its binding protein, metallothionein (MT)-1 [82]. Pretreatment with supplemental ZnSO4 enhances [Zn2+]i levels and antioxidant potential, and has proven cardioprotective against Isop-induced nonischemic cardiomyocyte necrosis [16,76].
Chronic Stressor States
At 4 wks ALDOST, the expression of Zn2+ transporters and MT-1 is upregulated and correspondingly cytosolic and mitochondrial Zn2+ levels are increased (see Table 1). In an effort to raise antioxidant defenses, cotreatment with a ZnSO4 supplement was used. It raised cardiomyocyte [Zn2+]i to 2.0 nM, which was significantly greater than that seen with ALDOST alone (see Table 1) [24,39]. The attendant increased antioxidant capacity of these cells abrogated the rise in mitochondrial H2O2 production and levels of 8-isoprostane in cardiac tissue and mitochondria. Moreover, the rise in [Zn2+]i attenuated intracellular Ca2+ overloading. Given the attenuation in scarring, expressed as reduced myocardial collagen volume fraction, myocardium was salvaged from necrotic cell death by the ZnSO4 supplement [38].
The response in [Zn2+]i was examined in rats receiving ALDOST and simultaneously cotreated with either carvedilol or nebivolol [13]. These β1 receptor antagonists are unique in that they have an additional property as a β3 receptor agonist, where the latter provokes nitric oxide (NO) formation from endothelial NO synthase which, in turn, releases Zn2+ which had been inactive being bound to MT-1 (see Figure 3). In response to carvedilol, [Zn2+]i rose to 2.5 nM and with nebivolol to 2.8 nM, which were greater than that seen with ZnSO4. The rise in [Zn2+]i activates its sensor, metal-responsive transcription factor (MTF)-1, which upon its translocation to the nucleus upregulates the expression of antioxidant defense genes, including Cu/Zn-SOD, glutathione synthase and MT-1, that collectively raise endogenous antioxidant defenses [38]. Furthermore, the increase in [Zn2+]i gained with either carvedilol or nebivolol served to attenuate Ca2+ entry, and consequently [Ca2+]i and [Ca2+]m levels were each reduced. By enhancing endogenous antioxidant capacity and reducing EICA, the multifactorial antioxidant potential of these newer beta antagonists served to markedly attenuate mitochondrial and cardiac tissue 8-isoprostane levels, and the appearance of myocardial scarring [13].
Fig. 3.
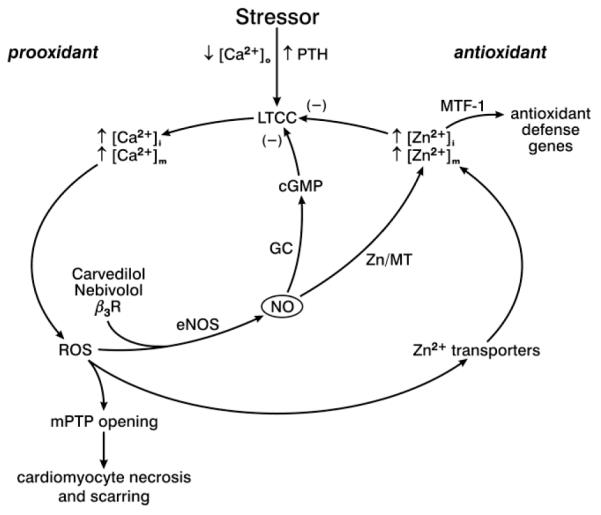
Acute and chronic stressor states are accompanied by fallen levels of ionized [Ca2+]o with elevated plasma PTH, or secondary hyperparathyroidism. Cardiomyocyte prooxidant pathway represented by increments in [Ca2+]i and [Ca2+]m while augmentations in [Zn2+]i and [Zn2+]m serve as an antioxidant pathway. Increments in cytosolic free [Zn2+]i activate its sensor, metal-responsive transcription factor (MTF)-1, which upon its translocation to the nucleus induces the transcription of antioxidant defense genes, including Cu/Zn-superoxide dismutase, glutathione synthase and MT-1. Nebivolol and carvedilol are unique β1 adrenergic receptor blockers in that they are also β3 agonists which upregulates nitric oxide (NO) production from endothelial nitric oxide synthase (eNOS). In turn, NO releases Zn2+ bound to MT-1 which promotes antioxidant defenses while it reduces LTCC-based Ca2+ entry via upregulated cGMP. ROS, reactive oxygen species; GC, guanyl cyclase. Adapted with permission from [13]
Clinical Correlates: Nonischemic Cardiomyocyte Necrosis
Acute Stressor States
A hyperadrenergic response accompanies acute bodily injury, such as acute myocardial infarction (MI), major surgery including coronary artery bypass surgery, thermal burns, head injury or subarachnoid hemorrhage, and musculoskeletal trauma. Sepsis and diabetic ketoacidosis are systemic inflammatory illnesses accompanied by a hyperadrenergic state.
Nonischemic myocardial necrosis secondary to catecholamine-induced cell death can lead to elevations in serum troponins. These elevations, however, are modest in comparison to the segmental loss of myocardium with an acute reduction in coronary blood flow and MI. Elevated troponins have been reported in acute stressor states that include sepsis, gastrointestinal bleeding, pulmonary embolus and subarachnoid hemorrhage [27,35,53,86]. Nonischemic necrosis is a risk factor for multiorgan failure and mortality associated with ventricular dysfunction [2,91].
Chronic Stressor States
Cardiomyocyte necrosis with elevations in serum troponins indicative of cardiomyocyte necrosis but not due to MI or related to renal failure, are found in patients hospitalized with CHF and are associated with increased in-hospital and overall cardiac mortality [33,50,62,92,52,29,78,32,68,55]. A consistent association between troponin elevation and poor outcomes is found in hospitalized and ambulatory patients with HF. A concentration-dependent relationship exists between the magnitude of troponin elevation and adverse outcomes. Consequently, there is the need for serial troponin measurements to assess changing levels of risk. Increased frequency and magnitude of troponin “leak” results in worsened outcomes and transition from compensated to decompensated HF [43].
Summary and Conclusions
Cardiomyocyte survival must be sustained if the efficiency of this muscular pump is to be preserved. Mitochondria play a central role in nonischemic cardiomyocyte necrosis in a pathophysiologic scenario which is common to both acute and chronic stressor states. A mitochondriocentric signal-transducer-effector pathway leads to necrosis. Its major components include: catecholamine or PTH-mediated intracytosolic and intramitochondrial Ca2+ overloading; the induction of oxidative stress by these organelles; and the subsequent opening of their inner mPTP with ensuing osmotic swelling, loss of ATP synthesis, organellar degeneration and a hyperpermeable sarcoplasmic membrane with spillage of cell contents that includes troponins. The necrotic loss of cells leads to a subsequent wound healing response with invading inflammatory cells and myofibroblasts that eventuates in a replacement fibrosis. Necrosis contrasts to apoptosis, which leaves neither a morphologic footprint of fibrosis nor a biochemical trail with elevated troponins. The scattered foci of myocardial scarring found in the explanted failing human heart and the presence of increased serum troponins at the time of hospitalization for episodes of decompensated CHF underscores the importance of cardiomyocyte necrosis and which is ongoing.
Strategic interruption of this pathway by pharmaceuticals and/or nutriceuticals offers a mitochondria-targeted approach toward salvaging cardiomyocytes from necrotic cell death. Experimental evidence gathered to date holds promise for the potential efficacy of such cardioprotective strategies.
Acknowledgements
This work was supported, in part, by NIH grants R01-HL73043 and R01-HL90867 (KTW). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Authors have no conflicts of interest to disclose.
We wish to express our deep gratitude to Richard A. Parkinson, MEd, for editorial assistance and illustrations.
Footnotes
This article is published as part of the Special issue on “Cell-Specific Roles of Mitochondrial Ca2+ Handling”
References
- 1.Ahokas RA, Sun Y, Bhattacharya SK, Gerling IC, Weber KT. Aldosteronism and a proinflammatory vascular phenotype. Role of Mg2+, Ca2+ and H2O2 in peripheral blood mononuclear cells. Circulation. 2005;111:51–57. doi: 10.1161/01.CIR.0000151516.84238.37. [DOI] [PubMed] [Google Scholar]
- 2.Ammann P, Maggiorini M, Bertel O, Haenseler E, Joller-Jemelka HI, Oechslin E, Minder EI, Rickli H, Fehr T. Troponin as a risk factor for mortality in critically ill patients without acute coronary syndromes. J Am Coll Cardiol. 2003;41:2004–2009. doi: 10.1016/s0735-1097(03)00421-2. [DOI] [PubMed] [Google Scholar]
- 3.Angert D, Berretta RM, Kubo H, Zhang H, Chen X, Wang W, Ogorek B, Barbe M, Houser SR. Repair of the injured adult heart involves new myocytes potentially derived from resident cardiac stem cells. Circ Res. 2011;108:1226–1237. doi: 10.1161/CIRCRESAHA.110.239046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arbustini E, Brega A, Narula J. Ultrastructural definition of apoptosis in heart failure. Heart Fail Rev. 2008;13:121–135. doi: 10.1007/s10741-007-9072-8. [DOI] [PubMed] [Google Scholar]
- 5.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 6.Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation. 1994;89:151–163. doi: 10.1161/01.cir.89.1.151. [DOI] [PubMed] [Google Scholar]
- 7.Benjamin IJ, Jalil JE, Tan LB, Cho K, Weber KT, Clark WA. Isoproterenol-induced myocardial fibrosis in relation to myocyte necrosis. Circ Res. 1989;65:657–670. doi: 10.1161/01.res.65.3.657. [DOI] [PubMed] [Google Scholar]
- 8.Bhattacharya SK, Palmieri GM, Bertorini TE, Nutting DF. The effects of diltiazem in dystrophic hamsters. Muscle Nerve. 1982;5:73–78. doi: 10.1002/mus.880050114. [DOI] [PubMed] [Google Scholar]
- 9.Boluyt MO, Bing OH, Lakatta EG. The ageing spontaneously hypertensive rat as a model of the transition from stable compensated hypertrophy to heart failure. Eur Heart J. 1995;16(Suppl N):19–30. doi: 10.1093/eurheartj/16.suppl_n.19. [DOI] [PubMed] [Google Scholar]
- 10.Buchalter MB, Rademakers FE, Weiss JL, Rogers WJ, Weisfeldt ML, Shapiro EP. Rotational deformation of the canine left ventricle measured by magnetic resonance tagging: effects of catecholamines, ischaemia, and pacing. Cardiovasc Res. 1994;28:629–635. doi: 10.1093/cvr/28.5.629. [DOI] [PubMed] [Google Scholar]
- 11.Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. Mitochondrial reactive oxygen species and Ca2+ signaling. Am J Physiol Cell Physiol. 2006;291:C1082–1088. doi: 10.1152/ajpcell.00217.2006. [DOI] [PubMed] [Google Scholar]
- 12.Cao DJ, Gillette TG, Hill JA. Cardiomyocyte autophagy: remodeling, repairing, and reconstructing the heart. Curr Hypertens Rep. 2009;11:406–411. doi: 10.1007/s11906-009-0070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheema Y, Sherrod JN, Shahbaz AU, Zhao W, Zhao T, Ahokas RA, Sun Y, Bhattacharya SK, Gerling IC, Weber KT. Mitochondriocentric pathway to cardiomyocyte necrosis in aldosteronism: cardioprotective responses to carvedilol and nebivolol. J Cardiovasc Pharmacol. 2011;58:80–86. doi: 10.1097/FJC.0b013e31821cd83c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chhokar VS, Sun Y, Bhattacharya SK, Ahokas RA, Myers LK, Xing Z, Smith RA, Gerling IC, Weber KT. Loss of bone minerals and strength in rats with aldosteronism. Am J Physiol Heart Circ Physiol. 2004;287:H2023–H2026. doi: 10.1152/ajpheart.00477.2004. [DOI] [PubMed] [Google Scholar]
- 15.Chhokar VS, Sun Y, Bhattacharya SK, Ahokas RA, Myers LK, Xing Z, Smith RA, Gerling IC, Weber KT. Hyperparathyroidism and the calcium paradox of aldosteronism. Circulation. 2005;111:871–878. doi: 10.1161/01.CIR.0000155621.10213.06. [DOI] [PubMed] [Google Scholar]
- 16.Chvapil M, Owen JA. Effect of zinc on acute and chronic isoproterenol induced heart injury. J Mol Cell Cardiol. 1977;9:151–159. doi: 10.1016/0022-2828(77)90046-3. [DOI] [PubMed] [Google Scholar]
- 17.Coatesworth W, Bolsover S. Spatially organised mitochondrial calcium uptake through a novel pathway in chick neurones. Cell Calcium. 2006;39:217–225. doi: 10.1016/j.ceca.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 18.Conrad CH, Brooks WW, Hayes JA, Sen S, Robinson KG, Bing OHL. Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation. 1995;91:161–170. doi: 10.1161/01.cir.91.1.161. [DOI] [PubMed] [Google Scholar]
- 19.Crawford AJ, Bhattacharya SK. Excessive intracellular zinc accumulation in cardiac and skeletal muscles of dystrophic hamsters. Exp Neurol. 1987;95:265–276. doi: 10.1016/0014-4886(87)90137-3. [DOI] [PubMed] [Google Scholar]
- 20.Doring HJ, Leder O, Jaedicke W, Reindell A, Fleckenstein A. [Limitation of the disappearance of energy-rich phosphate compounds in the hyperactive atrial and ventricular myocardium by divalent Ca-antagonistic inhibitors of electromechanical coupling (iprovdratril, D 600, prenylamine)] Pflügers Arch. 1969;312:R7–R8. [PubMed] [Google Scholar]
- 21.Fleckenstein A, Janke J, Döring HJ, Leder O. Myocardial fiber necrosis due to intracellular Ca overload-a new principle in cardiac pathophysiology. Recent Adv Stud Cardiac Struct Metab. 1974;4:563–580. [PubMed] [Google Scholar]
- 22.Fleckenstein A, Kanke J, Döring HJ, Leder O. Key role of Ca in the production of noncoronarogenic myocardial necroses. Recent Adv Stud Cardiac Struct Metab. 1975;6:21–32. [PubMed] [Google Scholar]
- 23.Gallitelli MF, Schultz M, Isenberg G, Rudolf F. Twitch-potentiation increases calcium in peripheral more than in central mitochondria of guinea-pig ventricular myocytes. J Physiol. 1999;518(Pt 2):433–447. doi: 10.1111/j.1469-7793.1999.0433p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gandhi MS, Deshmukh PA, Kamalov G, Zhao T, Zhao W, Whaley JT, Tichy JR, Bhattacharya SK, Ahokas RA, Sun Y, Gerling IC, Weber KT. Causes and consequences of zinc dyshomeostasis in rats with chronic aldosteronism. J Cardiovasc Pharmacol. 2008;52:245–252. doi: 10.1097/FJC.0b013e3181833eb8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodwin KD, Ahokas RA, Bhattacharya SK, Sun Y, Gerling IC, Weber KT. Preventing oxidative stress in rats with aldosteronism by calcitriol and dietary calcium and magnesium supplements. Am J Med Sci. 2006;332:73–78. doi: 10.1097/00000441-200608000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res. 2011;108:1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 27.Gunnewiek JM, Van Der Hoeven JG. Cardiac troponin elevations among critically ill patients. Curr Opin Crit Care. 2004;10:342–346. doi: 10.1097/01.ccx.0000135514.20538.44. [DOI] [PubMed] [Google Scholar]
- 28.Gustafsson AB, Gottlieb RA. Mechanisms of apoptosis in the heart. J Clin Immunol. 2003;23:447–459. doi: 10.1023/b:joci.0000010421.56035.60. [DOI] [PubMed] [Google Scholar]
- 29.Horwich TB, Patel J, MacLellan WR, Fonarow GC. Cardiac troponin I is associated with impaired hemodynamics, progressive left ventricular dysfunction, and increased mortality rates in advanced heart failure. Circulation. 2003;108:833–838. doi: 10.1161/01.CIR.0000084543.79097.34. [DOI] [PubMed] [Google Scholar]
- 30.Hosoda T, Rota M, Kajstura J, Leri A, Anversa P. Role of stem cells in cardiovascular biology. J Thromb Haemost. 2011;9(Suppl 1):151–161. doi: 10.1111/j.1538-7836.2011.04363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hughes SE. Detection of apoptosis using in situ markers for DNA strand breaks in the failing human heart. Fact or epiphenomenon? J Pathol. 2003;201:181–186. doi: 10.1002/path.1447. [DOI] [PubMed] [Google Scholar]
- 32.Ilva T, Lassus J, Siirilä-Waris K, Melin J, Peuhkurinen K, Pulkki K, Nieminen MS, Mustonen H, Porela P, Harjola VP. Clinical significance of cardiac troponins I and T in acute heart failure. Eur J Heart Fail. 2008;10:772–779. doi: 10.1016/j.ejheart.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 33.Ishii J, Nomura M, Nakamura Y, Naruse H, Mori Y, Ishikawa T, Ando T, Kurokawa H, Kondo T, Nagamura Y, Ezaki K, Hishida H. Risk stratification using a combination of cardiac troponin T and brain natriuretic peptide in patients hospitalized for worsening chronic heart failure. Am J Cardiol. 2002;89:691–695. doi: 10.1016/s0002-9149(01)02341-4. [DOI] [PubMed] [Google Scholar]
- 34.Javadov S, Karmazyn M. Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell Physiol Biochem. 2007;20:1–22. doi: 10.1159/000103747. [DOI] [PubMed] [Google Scholar]
- 35.Jeremias A, Gibson CM. Narrative review: alternative causes for elevated cardiac troponin levels when acute coronary syndromes are excluded. Ann Intern Med. 2005;142:786–791. doi: 10.7326/0003-4819-142-9-200505030-00015. [DOI] [PubMed] [Google Scholar]
- 36.Jin YT, Hasebe N, Matsusaka T, Natori S, Ohta T, Tsuji S, Kikuchi K. Magnesium attenuates isoproterenol-induced acute cardiac dysfunction and beta-adrenergic desensitization. Am J Physiol Heart Circ Physiol. 2007;292:H1593–1599. doi: 10.1152/ajpheart.00985.2006. [DOI] [PubMed] [Google Scholar]
- 37.Jones WK, Brown M, Ren X, He S, McGuinness M. NF-κB as an integrator of diverse signaling pathways: the heart of myocardial signaling? Cardiovasc Toxicol. 2003;3:229–254. doi: 10.1385/ct:3:3:229. [DOI] [PubMed] [Google Scholar]
- 38.Kamalov G, Ahokas RA, Zhao W, Johnson PL, Shahbaz AU, Bhattacharya SK, Sun Y, Gerling IC, Weber KT. Temporal responses to intrinsically coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria during aldosteronism. Am J Physiol Heart Circ Physiol. 2010;298:H385–H394. doi: 10.1152/ajpheart.00593.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamalov G, Ahokas RA, Zhao W, Zhao T, Shahbaz AU, Johnson PL, Bhattacharya SK, Sun Y, Gerling IC, Weber KT. Uncoupling the coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria seen in aldosteronism. J Cardiovasc Pharmacol. 2010;55:248–254. doi: 10.1097/FJC.0b013e3181cf0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamalov G, Bhattacharya SK, Weber KT. Congestive heart failure: where homeostasis begets dyshomeostasis. J Cardiovasc Pharmacol. 2010;56:320–328. doi: 10.1097/FJC.0b013e3181ed064f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamalov G, Deshmukh PA, Baburyan NY, Gandhi MS, Johnson PL, Ahokas RA, Bhattacharya SK, Sun Y, Gerling IC, Weber KT. Coupled calcium and zinc dyshomeostasis and oxidative stress in cardiac myocytes and mitochondria of rats with chronic aldosteronism. J Cardiovasc Pharmacol. 2009;53:414–423. doi: 10.1097/FJC.0b013e3181a15e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kametani R, Miura T, Harada N, Shibuya M, Wang R, Tan H, Fukagawa Y, Kawamura S, Matsuzaki M. Carvedilol inhibits mitochondrial oxygen consumption and superoxide production during calcium overload in isolated heart mitochondria. Circ J. 2006;70:321–326. doi: 10.1253/circj.70.321. [DOI] [PubMed] [Google Scholar]
- 43.Kociol RD, Pang PS, Gheorghiade M, Fonarow GC, O’Connor CM, Felker GM. Troponin elevation in heart failure prevalence, mechanisms, and clinical implications. J Am Coll Cardiol. 2010;56:1071–1078. doi: 10.1016/j.jacc.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 44.Kohlhardt M, Bauer B, Krause H, Fleckenstein A. Differentiation of the transmembrane Na and Ca channels in mammalian cardiac fibres by the use of specific inhibitors. Pflügers Arch. 1972;335:309–322. doi: 10.1007/BF00586221. [DOI] [PubMed] [Google Scholar]
- 45.Kohlhardt M, Bauer B, Krause H, Fleckenstein A. Selective inhibition of the transmembrane Ca conductivity of mammalian myocardial fibres by Ni, Co and Mn ions. Pflügers Arch. 1973;338:115–123. doi: 10.1007/BF00592747. [DOI] [PubMed] [Google Scholar]
- 46.Kostin S, Pool L, Elsässer A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klövekorn WP, Schaper J. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–724. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 47.Kumar D, Lou H, Singal PK. Oxidative stress and apoptosis in heart dysfunction. Herz. 2002;27:662–668. doi: 10.1007/s00059-002-2430-3. [DOI] [PubMed] [Google Scholar]
- 48.Kung G, Konstantinidis K, Kitsis RN. Programmed necrosis, not apoptosis, in the heart. Circ Res. 2011;108:1017–1036. doi: 10.1161/CIRCRESAHA.110.225730. [DOI] [PubMed] [Google Scholar]
- 49.Kuroda J, Sadoshima J. NADPH oxidase and cardiac failure. J Cardiovasc Transl Res. 2010;3:314–320. doi: 10.1007/s12265-010-9184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuwabara Y, Sato Y, Miyamoto T, Taniguchi R, Matsuoka T, Isoda K, Yamane K, Nishi K, Fujiwara H, Takatsu Y. Persistently increased serum concentrations of cardiac troponin in patients with acutely decompensated heart failure are predictive of adverse outcomes. Circ J. 2007;71:1047–1051. doi: 10.1253/circj.71.1047. [DOI] [PubMed] [Google Scholar]
- 51.Lossnitzer K, Janke J, Hein B, Stauch M, Fleckenstein A. Disturbed myocardial calcium metabolism: a possible pathogenetic factor in the hereditary cardiomyopathy of the Syrian hamster. Recent Adv Stud Cardiac Struct Metab. 1975;6:207–217. [PubMed] [Google Scholar]
- 52.Löwbeer C, Gustafsson SA, Seeberger A, Bouvier F, Hulting J. Serum cardiac troponin T in patients hospitalized with heart failure is associated with left ventricular hypertrophy and systolic dysfunction. Scand J Clin Lab Invest. 2004;64:667–676. doi: 10.1080/00365510410003002. [DOI] [PubMed] [Google Scholar]
- 53.Maeder M, Fehr T, Rickli H, Ammann P. Sepsis-associated myocardial dysfunction: diagnostic and prognostic impact of cardiac troponins and natriuretic peptides. Chest. 2006;129:1349–1366. doi: 10.1378/chest.129.5.1349. [DOI] [PubMed] [Google Scholar]
- 54.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 55.Miller WL, Hartman KA, Burritt MF, Grill DE, Jaffe AS. Profiles of serial changes in cardiac troponin T concentrations and outcome in ambulatory patients with chronic heart failure. J Am Coll Cardiol. 2009;54:1715–1721. doi: 10.1016/j.jacc.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 56.Mughal W, Kirshenbaum LA. Cell death signalling mechanisms in heart failure. Exp Clin Cardiol. 2011;16:102–108. [PMC free article] [PubMed] [Google Scholar]
- 57.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishida K, Yamaguchi O, Otsu K. Crosstalk between autophagy and apoptosis in heart disease. Circ Res. 2008;103:343–351. doi: 10.1161/CIRCRESAHA.108.175448. [DOI] [PubMed] [Google Scholar]
- 59.Nykamp D, Titak JA. Takotsubo cardiomyopathy, or broken-heart syndrome. Ann Pharmacother. 2010;44:590–593. doi: 10.1345/aph.1M568. [DOI] [PubMed] [Google Scholar]
- 60.Ong SB, Gustafsson AB. New roles for mitochondria in cell death in the reperfused myocardium. Cardiovasc Res. 2011 doi: 10.1093/cvr/cvr312. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31(Suppl 2):S170–S180. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 62.Peacock WF, 4th, De Marco T, Fonarow GC, Diercks D, Wynne J, Apple FS, Wu AH. Cardiac troponin and outcome in acute heart failure. N Engl J Med. 2008;358:2117–2126. doi: 10.1056/NEJMoa0706824. [DOI] [PubMed] [Google Scholar]
- 63.Rizzuto R, Pinton P, Brini M, Chiesa A, Filippin L, Pozzan T. Mitochondria as biosensors of calcium microdomains. Cell Calcium. 1999;26:193–199. doi: 10.1054/ceca.1999.0076. [DOI] [PubMed] [Google Scholar]
- 64.Rona G. Catecholamine cardiotoxicity. J Mol Cell Cardiol. 1985;17:291–306. doi: 10.1016/s0022-2828(85)80130-9. [DOI] [PubMed] [Google Scholar]
- 65.Rushmer RF, Thal N. The mechanics of ventricular contraction; a cinefluorographic study. Circulation. 1951;4:219–228. doi: 10.1161/01.cir.4.2.219. [DOI] [PubMed] [Google Scholar]
- 66.Sanganalmath SK, Abdel-Latif A, Bolli R, Xuan YT, Dawn B. Hematopoietic cytokines for cardiac repair: mobilization of bone marrow cells and beyond. Basic Res Cardiol. 2011;106:709–733. doi: 10.1007/s00395-011-0183-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Santos DL, Moreno AJ, Leino RL, Froberg MK, Wallace KB. Carvedilol protects against doxorubicin-induced mitochondrial cardiomyopathy. Toxicol Appl Pharmacol. 2002;185:218–227. doi: 10.1006/taap.2002.9532. [DOI] [PubMed] [Google Scholar]
- 68.Sato Y, Nishi K, Taniguchi R, Miyamoto T, Fukuhara R, Yamane K, Saijyo S, Tanada Y, Yamamoto E, Goto T, Takahashi N, Fujiwara H, Takatsu Y. In patients with heart failure and non-ischemic heart disease, cardiac troponin T is a reliable predictor of long-term echocardiographic changes and adverse cardiac events. J Cardiol. 2009;54:221–230. doi: 10.1016/j.jjcc.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 69.Schaper J, Lorenz-Meyer S, Suzuki K. The role of apoptosis in dilated cardiomyopathy. Herz. 1999;24:219–224. doi: 10.1007/BF03044964. [DOI] [PubMed] [Google Scholar]
- 70.Sedmera D, Reckova M, Bigelow MR, Dealmeida A, Stanley CP, Mikawa T, Gourdie RG, Thompson RP. Developmental transitions in electrical activation patterns in chick embryonic heart. Anat Rec A Discov Mol Cell Evol Biol. 2004;280:1001–1009. doi: 10.1002/ar.a.20107. [DOI] [PubMed] [Google Scholar]
- 71.Selektor Y, Ahokas RA, Bhattacharya SK, Sun Y, Gerling IC, Weber KT. Cinacalcet and the prevention of secondary hyperparathyroidism in rats with aldosteronism. Am J Med Sci. 2008;335:105–110. doi: 10.1097/MAJ.0b013e318134f013. [DOI] [PubMed] [Google Scholar]
- 72.Sgobbo P, Pacelli C, Grattagliano I, Villani G, Cocco T. Carvedilol inhibits mitochondrial complex I and induces resistance to H2O2-mediated oxidative insult in H9C2 myocardial cells. Biochim Biophys Acta. 2007;1767:222–232. doi: 10.1016/j.bbabio.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 73.Shahbaz AU, Kamalov G, Zhao W, Zhao T, Johnson PL, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, Weber KT. Mitochondria-targeted cardioprotection in aldosteronism. J Cardiovasc Pharmacol. 2011;57:37–43. doi: 10.1097/FJC.0b013e3181fe1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shahbaz AU, Zhao T, Zhao W, Johnson PL, Ahokas RA, Bhattacharya SK, Sun Y, Gerling IC, Weber KT. Calcium and zinc dyshomeostasis during isoproterenol-induced acute stressor state. Am J Physiol Heart Circ Physiol. 2011;300:H636–H644. doi: 10.1152/ajpheart.00900.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shechter M. Magnesium and cardiovascular system. Magnes Res. 2010;23:60–72. doi: 10.1684/mrh.2010.0202. [DOI] [PubMed] [Google Scholar]
- 76.Singal PK, Dhillon KS, Beamish RE, Dhalla NS. Protective effect of zinc against catecholamine-induced myocardial changes electrocardiographic and ultrastructural studies. Lab Invest. 1981;44:426–433. [PubMed] [Google Scholar]
- 77.Singh SS, Kang PM. Mechanisms and inhibitors of apoptosis in cardiovascular diseases. Curr Pharm Des. 2011 doi: 10.2174/138161211796390994. In press. [DOI] [PubMed] [Google Scholar]
- 78.Sukova J, Ostadal P, Widimsky P. Profile of patients with acute heart failure and elevated troponin I levels. Exp Clin Cardiol. 2007;12:153–156. [PMC free article] [PubMed] [Google Scholar]
- 79.Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart. Role of oxidative stress. Am J Pathol. 2002;161:1773–1781. doi: 10.1016/S0002-9440(10)64454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takemura G, Fujiwara H. Morphological aspects of apoptosis in heart diseases. J Cell Mol Med. 2006;10:56–75. doi: 10.1111/j.1582-4934.2006.tb00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tamura T, Said S, Lu W, Harris J, Neufeld D, Burbach JA, Gerdes AM. Is apoptosis present in progression to chronic hypertensive heart failure? J Card Fail. 2000;6:37–42. doi: 10.1016/s1071-9164(00)00010-5. [DOI] [PubMed] [Google Scholar]
- 82.Thomas M, Vidal A, Bhattacharya SK, Ahokas RA, Sun Y, Gerling IC, Weber KT. Zinc dyshomeostasis in rats with aldosteronism. Response to spironolactone. Am J Physiol Heart Circ Physiol. 2007;293:H2361–H2366. doi: 10.1152/ajpheart.00200.2007. [DOI] [PubMed] [Google Scholar]
- 83.Tritthart H, Fleckenstein A, Kaufmann R. [Specific increase in relaxation velocity of the isolated ventricular myocardium of rhesus monkeys, guinea pigs and frogs produced by sympathetic transmitters, and the neutralization of this effect by beta-receptor blockade] Pflügers Arch. 1968;303:350–365. doi: 10.1007/BF00596391. [DOI] [PubMed] [Google Scholar]
- 84.Trumbeckaite S, Bernatoniene J, Majiene D, Jakstas V, Savickas A, Toleikis A. The effect of flavonoids on rat heart mitochondrial function. Biomed Pharmacother. 2006;60:245–248. doi: 10.1016/j.biopha.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 85.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81:449–456. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 86.Vasile VC, Babuin L, Rio Perez JA, Alegria JR, Song LM, Chai HS, Afessa B, Jaffe AS. Long-term prognostic significance of elevated cardiac troponin levels in critically ill patients with acute gastrointestinal bleeding. Crit Care Med. 2009;37:140–147. doi: 10.1097/CCM.0b013e318192faa3. [DOI] [PubMed] [Google Scholar]
- 87.Vidal A, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, Weber KT. Calcium paradox of aldosteronism and the role of the parathyroid glands. Am J Physiol Heart Circ Physiol. 2006;290:H286–H294. doi: 10.1152/ajpheart.00535.2005. [DOI] [PubMed] [Google Scholar]
- 88.Weber HW, Van Der Walt JJ. Cardiomyopathy in crowded rabbits. Recent Adv Stud Cardiac Struct Metab. 1975;6:471–477. [PubMed] [Google Scholar]
- 89.Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–1652. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- 90.Weber KT, Brilla CG, Janicki JS. Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc Res. 1993;27:341–348. doi: 10.1093/cvr/27.3.341. [DOI] [PubMed] [Google Scholar]
- 91.Wu TT, Yuan A, Chen CY, Chen WJ, Luh KT, Kuo SH, Lin FY, Yang PC. Cardiac troponin I levels are a risk factor for mortality and multiple organ failure in noncardiac critically ill patients and have an additive effect to the APACHE II score in outcome prediction. Shock. 2004;22:95–101. doi: 10.1097/01.shk.0000132484.97424.32. [DOI] [PubMed] [Google Scholar]
- 92.Zairis MN, Tsiaousis GZ, Georgilas AT, Makrygiannis SS, Adamopoulou EN, Handanis SM, Batika PC, Prekates AA, Velissaris D, Kouris NT, Mytas DZ, Babalis DK, Karidis KS, Foussas SG. Multimarker strategy for the prediction of 31 days cardiac death in patients with acutely decompensated chronic heart failure. Int J Cardiol. 2009;141:284–290. doi: 10.1016/j.ijcard.2008.12.017. [DOI] [PubMed] [Google Scholar]