

Abstract
We conducted a systematic analysis to determine the reason for the apparent disparity of success of immunotherapy between clinical and experimental cancers. To do this, we performed a search of PubMed using the keywords “immunotherapy” AND “cancer” for the years of 1980 and 2010. The midspread of experimental tumors used in all the relevant literature published in 2010 were between 0.5–121 mm3 in volume or had grown for four to eight days. Few studies reported large tumors that could be considered representative of clinical tumors, in terms of size and duration of growth. The predominant effect of cancer immunotherapies was slowed or delayed outgrowth. Regression of tumors larger than 200 mm3 was observed only after passive antibody or adoptive T cell therapy. The effectiveness of other types of immunotherapy was generally scattered. By comparison, very few publications retrieved by the 1980 search could meet our selection criteria; all of these used tumors smaller than 100 mm3, and none reported regression. In the entire year of 2010, only 13 used tumors larger than 400 mm3, and nine of these reported tumor regression. Together, these results indicate that most recent studies, using many diverse approaches, still treat small tumors only to report slowed or delayed growth. Nevertheless, a few recent studies indicate effective therapy against large tumors when using passive antibody or adoptive T cell therapy. For the future, we aspire to witness the increased use of experimental studies treating tumors that model clinical cancers in terms of size and duration of growth.
Keywords: cancer immunotherapy, inflammation, large tumors, mouse model, systematic review
Introduction
Clinical success of cancer immunotherapy has been frustratingly evasive. Successful experimental therapies have rarely led to effective clinical therapies, and reported experimental successes are largely limited to therapies against small tumors. Rejection of large, established disease is rare. Summarizing three decades of studies on immunity to cancer, William Woglom concluded in 1929 that immunotherapy is futile against an established tumor, and “nothing may accordingly be hoped for at present in respect to a successful therapy from this direction.”1 Eighty-two years later, the outlook is not quite so grim, as future successes can be expected to evolve from clinically relevant experimental models.
The difficulty of combating an established disease is partly founded in fundamental Darwinian principles. Statistically, the cellular heterogeneity within a tumor becomes relevant when the tumor accumulates one billion (109) cancer cells, in man and in mouse, equivalent to a tumor with a diameter of 1 cm.2,3 At this point, the accumulation of cancer cell variants results in miniscule probability that all cancer cells are susceptible to a single chemotherapeutic agent.2,3 This statement is based on models of chemotherapy, but there is nothing to suggest that immunotherapies cannot be modeled in the same way; for example, antigen-loss variants can be considered analogous to drug-resistant mutants. The relevant variables that could be considered are innumerable, but the nature of the problem is fundamentally the same in both cases.
Apart from matters of size and cell number, the physiology of tumors is also a critical feature in cancer therapy. Indeed, the growth and establishment of a transplanted tumor is distinct from the development of an autochthonous cancer. Histological examination reveals that the early growth of a transplanted tumor following inoculation is marked by substantial inflammation, necrosis, and only a thin rim of viable cancer cells.4 But the formation of an established billion cell mass of one centimeter diameter renders a tumor histologically indistinguishable from those presented clinically.4 At this time, the tumor presents as a formidable adversary against any sort of therapy.
While Woglom's declaration was perhaps true in 1929, eight decades of experimentation has yielded a more promising outlook. Present attitudes toward immunotherapy are best described as heterogenous: the field has been characterized as everything from premature to blossoming. On one hand, the approval of immunotherapies such as IL-2, rituximab, ipilumimab, and sipuleucel-T (Provenge) is testament to a level of acceptance of immunotherapy for oncological treatment. Nevertheless, definitive efficacy and compelling effectiveness of broader applicability for such therapies has yet to be demonstrated. In any case, where immunotherapy is reported to be effective experimentally, the results have not been translated to success in the clinic.
David Weiss argued in 1980 that the failure of clinical immunotherapy is due not to categorical difference between human and animal cancers, but rather that an irrelevant laboratory tumor model was extrapolated for clinical application.5 Similar assertions continue to be made, but have yet to be substantiated by systematic analysis of the literature. Ultimately, good experimental models of cancer are designed to represent as closely as possible clinical cancers with the hope to mimic a clinical setting. Here we analyze systematically the effects of experimental immunotherapies on tumors differing in size and duration of growth and determine the relative efficacy of different immunotherapies. We hope to highlight the strengths and shortcomings of experimental tumor models with regards to their clinical relevance and also shed light on the potential advances that have been made in experimental cancer immunotherapy.
Results
Quantity of evidence
All searches were conducted on PubMed using the keywords “immunotherapy” AND “cancer” for the years 2010 and 1980. Reviews, clinical trials, and case reports were excluded by the search engine. Further selection criteria were followed as described in the Materials and Methods. Two different types of analyses were conducted. In the first type of analysis encompassing the entire year of 1980 or 2010, we only extracted data reporting tumor size and duration of growth at the beginning of therapy for a general survey of experimental models. The second type of analysis used those studies published in April, June, or November of 2010, and in the entire year of 1980, as long as studies provided extractable data on tumor growth and efficacy of the treatment. These three months of 2010 were chosen to represent about one fourth of the publications for the year and ensure that quarterly publications would be represented.
In the first type of analysis, a total of 282 publications were retrieved by the search in 1980. Of this number, ten publications reported usable tumor size related data. A total of 789 publications were retrieved by the search in 2010. Of this number, 195 publications reported experiments using solid tumors and presented usable data on tumor growth. In the second, more detailed analysis, 99, 100, and 90 publications were retrieved for the months of April, June, and November 2010, respectively. Of these, 32, 26, and 16 publications reported extractable growth data for the same respective months.
The size of tumors at the beginning of therapy
We estimated an average tumor size at the beginning of therapy for all experiments reported in 2010 (195 publications). Figure 1 shows the size distribution of tumors employed in experimental immunotherapies reported in 2010. In the bulk of publications, tumors are treated ranging in volume between 0.5–121 mm3. The median reported tumor volume was 45 mm3. The number of publications falls off extremely rapidly with increasing tumor size. The incidence of publications becomes sparse for tumor volumes upwards of 375 mm3.
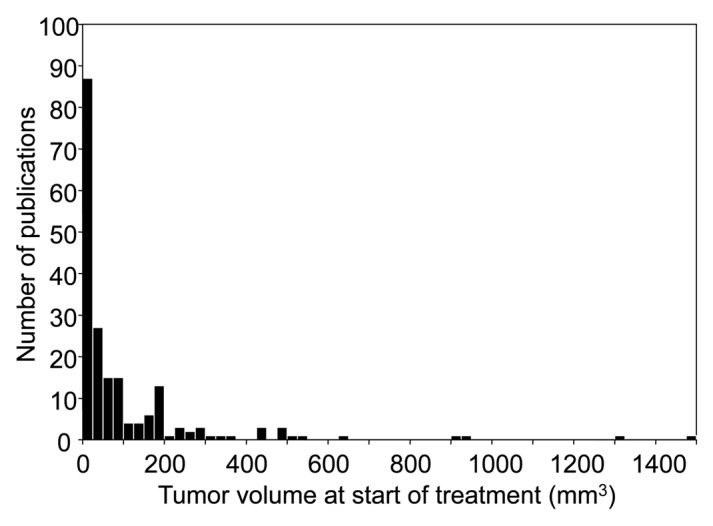
Figure 1. Precipitous falloff in the number of publications in the year 2010 with increasing size of tumors treated by immunotherapy. A search of PubMed using the keywords “immunotherapy” AND “cancer” recovered 195 experimental studies that met our selection criteria. 75% of tumors treated in these studies were smaller than 121 mm3. Note the sparseness of publications presenting tumors larger than 375 mm3. (n = 195; Q1 = 0.5, Q2 = 45, Q3 = 121.4)
The starting time of treatment after tumor cell inoculation
From the 195 publications reporting size data in 2010, we extracted the time given for tumors to establish before treatment was given. Xenotransplanted tumors were excluded only from this particular analysis due to the irregular growth kinetics of xenografts in immunocompromised hosts. After excluding xenotransplants, 158 publications reported the time when treatment was given after tumor cell inoculation. The distribution of times given for tumors to grow before treatment is presented Figure 2. The median time was 5 d. The bulk of publications cluster around tumors that have grown for 4–8 d before treatment. Only 15 publications reported tumors that had grown for 14 d or longer before treatment.
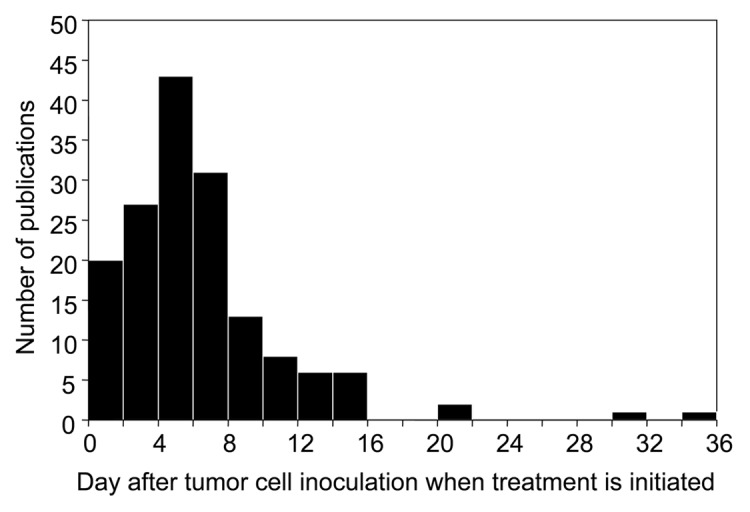
Figure 2. Most experimental tumors are treated less than a week after tumor cell inoculation. The median time reported was five days. All 158 cancer immunotherapy studies listed in PubMed meeting our selection criteria for 2010 using syngeneic murine tumor models are presented. Only nine tumors have grown for 14 d or longer before treatment. (n = 158; Q1 = 4, Q2 = 5, Q3 = 8)
The predominant effect of cancer immunotherapies is slowed or delayed outgrowth
Having characterized the distribution of tumor sizes employed in experimental immunotherapies, we asked what sort of therapeutic effect could be achieved on these tumors of varying sizes. The magnitude of the effect size (E) was expressed as a ratio of linearized growth rates of treated tumors against control tumors (Eqn. 1) (see Materials and Methods). The subset of publications from 2010 reporting therapeutically related experiments was used for detailed analyses. Equation 1 was applied to these experiments reported in 74 papers from April, June and November 2010, and the maximum effect size of multiple reported experiments for each respective paper was selected to represent that paper. In Figure 3, the effect size on tumor growth is plotted against tumor volume at the start of treatment. Despite the prevalence of experiments using small tumors, most succeed only at slowing or delaying tumor growth (0 < E ≤ 1). By contrast, of the four experiments using large tumors reported during these three months, two induce tumor regression (E > 1), and one arrests tumor growth (E = 1). Although the number of publications using large tumors is too few to draw definitive conclusions, we note that the therapies that induce regression of large tumors both use adoptive immune cell transfer. Other types of immunotherapy do not seem to follow a trend, and their effects are generally scattered.

Figure 3. Most experimental immunotherapies published treat small tumors yet succeed only at slowing or delaying tumor growth, but in several recent reports, larger tumors are being treated and a few reports present tumor regression. An effect size (E) of 1 indicates the treatment arrested tumor growth. An E < 1 indicates that the treated tumor still grew progressively, but only slower than the control or in a delayed fashion, i.e., a reduction of the growth rate of the tumor. An E > 1 indicates tumor regression. (Left panel) Detailed analysis was done for all experimental cancer immunotherapy publications listed in PubMed for April, June, and November of 2010. Regression of tumors larger than 200 mm3 is observed only after passive antibody or adoptive T cell therapy. (n = 74). (Right panel) The same analysis was performed for those publications in the entire year of 1980. Very few publications presented analyzable data. No publication uses tumors larger than 200 mm3, and regression is not observed at all. (n = 10).
Twelve out of 74 publications reported tumor regression. Ten of these 12 used tumors smaller than 500 mm3 at the beginning of therapy, and eight used tumors less than 200 mm3. Only two papers reported regression of tumors larger than 500 mm3. We also examined the effectiveness of immunotherapies reported in the entire year of 1980. Only ten publications reported analyzable tumor growth data, all of which employed tumors less than ~100 mm3 in size. None of the publications retrieved from 1980 reported tumor regression (E > 1, Fig. 3, left panel).
Comparing the data from both years, an enormous difference in the quantity of analyzable data is clear; in the three months of 2010 alone, there were more than seven times the number of publications from the entire year of 1980 that presented analyzable data. Both sets of data appear cluster around small tumors less than 200 mm3 in volume at the time of treatment (Fig. 3). However, a critical difference between the two is that the scatter of the 2010 data reaches in the vertical direction toward effect sizes greater than one, indicative of tumor regression. Furthermore, the 2010 data also includes publications using larger tumors, some of which even regress. Neither of these features is present in the 1980 data.
The efficacy of immunotherapy against large tumors
The paucity of experimentation using large tumors cannot yield definitive conclusions for the effectiveness of immunotherapies. We therefore proceeded to examine all experiments using large tumors in 2010. While the standard size for an established tumor is a 1 cm diameter or 500 mm3 as discussed, a 400 mm3 cutoff was selected to accommodate for a reasonable amount of measurement and unit conversion error (see Materials and Methods). As a further justification for this cutoff, we observed a break in continuity between 375–425 mm3 in the distribution of tumor sizes (Fig. 1), so no publication would be included at the expense of another using similarly sized tumors. In 195 publications reporting tumor size data for the year 2010, 13 were found to use tumors with a volume greater than 400 mm3.7-19
Table 1 summarizes the reported experimental immunotherapies against large tumors retrieved in the year 2010 by our search. Of these 13 publications, nine reported tumor regression or eradication8,9,12,13,15-19 and four reported slowed, delayed, or arrested growth.7,10,11,14 The four demonstrating arrested growth, employed passive antibody therapy (Rituximab), adjuvant therapy (IL-1 receptor agonist, TLR-2 agonist), or dendritic cell vaccination. Six of the nine publications demonstrating regression used adoptive cell transfer therapy. Of the other three, one employed passive antibody therapy (Rituximab), one used radioimmunotherapy (90Y-Veltuzumab), and the last transferred sporulating C. novyi intravenously. Three publications reported eradication and of these, animals were monitored for relapse for 0, 15 and 126 d. In the nine publications reporting tumor regression, five used syngeneic tumors and three used human xenotransplanted tumors in immunodeficient hosts.
Table 1. Effects and types of experimental immunotherapies reported in the year 2010 treating tumors > 400 mm3 in size.
| Author | Tumor model | Type of immunotherapy | Size of tumor and duration of growth at start of treatment* | Strongest reported treatment effect |
|---|---|---|---|---|
| Sharkey RM, et al.15 |
xenotransplant (Ramos) |
radioimmunotherapy 90Y-Veltuzumab |
0.5 cm3 (time not reported) |
rejection in 9/10 mice (monitored 18 wks post- rejection, 1 relapse at wk 7) |
| Maletzki K, et al.19 |
syngeneic (Panc02) |
adjuvant bacteriolytic C. novyi |
450 mm3 (32 d) |
0 mm3 (monitored 15 d post-rejection) |
| Garcia-Hernandez Mde L, et al.9 |
syngeneic (B16-OVA) |
adoptive transfer Tc17 T cells |
500 mm3 (7 d) |
rejection, no long-term growth data |
| Paulos CM, et al.13 |
xenotransplant (M108) |
adoptive transfer Th17 cells |
190 mm2 (6–7 weeks) |
regression to 50 mm2 |
| Kerkar S, et al.12 |
syngeneic (B16) |
adoptive transfer IL-12 engineered pmel-1 CD8+ |
80–100 mm2 (14 d) |
regression 10 mm2 |
| Xie Y, et al.17 |
syngeneic (B16) |
adoptive transfer TRP-1 CD4+ |
100 mm2 (11 d) |
regression 16 cm2 |
| Zhao Y, et al.18 |
xenotransplant (M108) |
adoptive transfer RNA CAR T cells |
100 mm2 (66 d) |
regression 50 mm2 |
| Shrimali RK, et al.16 |
syngeneic (B16) |
adoptive transfer, antibody pmel splenocytes, anti-VEGF |
100–200 mm2 (12–18 d) |
regression to 10 mm2 then relapse |
| Buhé V, et al.8 |
xenotransplant (Daudi) |
antibody, adjuvant Rituximab + CpG |
0.94 cm3 (35 d) |
regression 0.51 cm3 |
| Ringshausen I, et al.14 |
xenotransplant (SUDHL4) |
antibody rituximab |
100 mm2 (time not reported) |
no change in volume |
| Kayashima H, et al.11 |
syngeneic (MH134) |
Cytokine and vaccination IL-12 and DC vaccination |
0.5 cm3 (time not reported) |
growth delay |
| Harnack U, et al.10 |
syngeneic (CT26) |
adjuvant IL-1 receptor agonist |
440 mm3 (8 d) |
growth delay |
| Akazawa T, et al.7 | syngeneic (EL4-EG7) |
adjuvant synthetic TLR-2 agonist |
1.4–1.6 cm3 (16 d) |
growth delay |
Sizes are listed as reported. The 13 publications listed had sizes that converted to 400 mm3 or larger.
Discussion
It is frequently claimed that the success of immunotherapy in experimental animal models does not translate to success in the clinic. A survey of the literature reveals that this discrepancy may be attributed to inappropriate experimental design and general indifference toward therapeutic goals of tumor rejection. The vast majority of experimental immunotherapies target very small tumors in mice, and the results are generally not applicable to tumors of clinically detectable size. Experimental models which test therapy when tumors are 0.5–120 mm3 in volume or have only grown for 0–8 d measure the effect of therapy primarily against inflammatory swelling and few viable cancer cells;4 unfortunately this has been the case over decades of experimentation.4,5,20,21 Furthermore, one would expect that successful therapy against these small tumors would be reported often. Yet even when such small tumors are being treated, the best effect is most frequently described as a delayed or slowed growth rate. Size reduction of a tumor is infrequently reported. A weak in vivo “anti-tumor effect” against a small tumor may be helpful in understanding a certain treatment regimen, and may also help the field of immunotherapy to progress; but it is not sufficient cause to celebrate an experimental triumph, which has been all too frequent for a majority of the papers coming from good laboratories and reported in excellent journals and often touted by the lay press.
We cannot deny that moderate effects against small tumors may hold potential relevance. Adjuvant immunotherapy is used effectively after surgical excision against residual disease and micrometastases.20 The significance of immunotherapy against this sort of “secondary” disease should not be neglected, yet the disproportionate amount of data concerned with minimally detectable tumors raises a troubling question: Why isn’t there much greater emphasis by investigators, funding agencies, and journals on using experimental models more relevant to a frequent clinical situation?
It is odd that even of the many publications treating small tumors, few succeed in inducing regression. It seems that there is a misguided attitude that any effect against tumor cells placed in a mouse constitutes a noteworthy clinically interesting result. Perhaps this discrepancy is symptomatic of misinterpreting the purpose and outcome of an assay. Henry Winn, in 1960, described an assay in which suspensions of lymph node cells and tumor cells at varying ratios were injected into mice, and the outgrowth of tumors was taken as a readout.22 Variations of the Winn assay are employed ubiquitously, but the interpretations of results are misappropriated. Winn himself described the assay as “a technique for assaying the activity of immunologically activated lymphoid cells”,22 and not as a readout for therapeutic potential. A handful of publications today reporting Winn-like experiments acknowledge the original purpose of the assay, and instead immediately propose prospective therapeutic potential. Merely performing a cytotoxicity assay in a mouse surrogate is a doppelgänger compared with a rejection assay using a large established tumor.
Despite the prevalence of inappropriate experimental models and weak therapeutic effects, the field has certainly progressed. Our search retrieved about a third as many publications in 1980 compared with 2010. Among these, very few publications reported data sufficiently well to permit comparison and replication. Many of the 1980 studies were more interested in the nature of transplantation immunology, rather than cancer immunotherapy. Of the therapeutically-related publications that were excluded, many had insufficient controls or did not report critical values necessary to generate a measure of effect size (E). The tremendous expansion in accessibly reported data is a testament to quality methodological standards and translational ambitions. It should be noted the standard of reporting experimental therapies remains erratic and compares poorly to the conventions of clinical studies. Nevertheless, a few well-crafted and well-reported studies have emerged in this examination of the literature.
Despite the described failures of experimental cancer immunotherapy, cynicism is truly unwarranted. While all the experimental immunotherapies reported three decades ago proved to be ineffective and implemented minimally relevant tumors, a number of studies from 2010 effectively induced regression of large established disease. Thirteen publications are acknowledged for employing large tumors, and of these 13, nine demonstrated tumor regression, but not eradication to zero volume. Of the four that did not induce regression, one induced a state of stable disease and three accomplished slowed or delayed growth. The latter three treated tumors by the administration of adjuvant, cytokines, or vaccination—all of which constitute active immunotherapy. The former study used Rituximab to arrest tumor growth. Of the nine publications reporting tumor regression, eight employed some form of adoptive or passive immunotherapy and one used intravenous delivery of C. novyi spores. Although this study reported tumor regression within hours, the toxicity of the therapy killed all animals within 24 h. Two studies employed passive antibody therapy, one with a bacterial adjuvant, and one conjugated to yttrium radioisotope. The remaining six studies employed adoptive immune cell transfer.
The success of passive antibody or adoptive T cell immunotherapy compared with active immunotherapy against large tumors speaks for itself. But this is not to say that active immunotherapy should be abandoned—the fact that such therapies have any effect on such large tumors is a success that few other studies have even attempted to report. In combination with passive antibody or adoptive T cell immunotherapy, the therapeutic potential of adjuvants, cytokines, and vaccinations is relatively untapped. Indeed, one study using adoptive cell transfer required the additional support of cytokine expression to achieve tumor regression. Undeniably, the heterogenous nature of cancer will necessarily preclude any single immunotherapy from becoming an archetype for therapeutic efficacy.23
Our study is not intended to forecast either the efficacy of cancer immunotherapy or the relevance of future studies to clinical disease; rather, it is intended to describe the field as it stands today. Our ambition is to bring attention to the current trends in experimental cancer immunotherapy in a manner substantiated by systematic analysis of the literature. For the future, we aspire to witness finer experimental models and superior therapeutic effects, with the hope of finding for patients cancer immunotherapies that are also widely effective against larger and longer established tumors.
Materials and Methods
Search and selection criteria
We conducted all searches of PubMed using the keywords “immunotherapy” AND “cancer.” All studies available via the University of Chicago Library were inspected. Very few studies were unavailable for inspection. Studies were limited to those in mice carrying syngeneic, allogeneic, or xenogeneic tumors. All tumor types were included in each of our analyses, with the exception of the analysis concerning the duration of tumor growth before the start of treatment. Xenotransplants were excluded from this particular analysis due to the irregular growth kinetics of xenograft tumors in immunocompromised mice. One study used autochthonous carcinogen-induced tumors and was excluded because it was impossible to determine how much the carcinogen contributed to inflammation. Studies were also limited to those written in English. Relevance was first determined by inspection of the title and abstract, followed by more detailed reading when necessary. Prophylactic therapies, including vaccinations and other treatments administered before tumor cell inoculation were excluded. Prophylaxis is very different from therapy of established disease, be it cancer or the flu.6 The immunological factors that contribute to this distinction are innumerable (tolerance, suppression, tumor microenvironment, etc.). Prophylactic immunotherapies are capable of producing very impressive successes, but we are concerned here with models which mimic patients in the clinic with preexisting tumors.
Describing tumor size
The effects of therapies were reported either in terms of solid tumor growth or animal survival over time. Survival data were excluded because surrogate endpoints were inconsistent. We expressed changes in size of tumors as an index of the experimental effect on growth relative to control (Eqn. 1). Relevant values were taken from the text as reported or otherwise inspected visually. We describe tumor size as a volume V = 1/2 (a × b × c), where a, b and c are orthogonal diameters.24-26 In some cases, volume was expressed as the product of the square of the major diameter times the minor diameter divided by two. Tumor sizes expressed in two dimensions as area were generally reported as the product of orthogonal diameters. The square root of the area was taken as the geometric mean of diameters, and used as an average diameter for calculating volume as described above. For tumor sizes reported in one dimension, the reported diameter was used accordingly. Tumors described as “palpable” or “detectable” were interpreted as 1 mm in diameter. Tumors characterized by mass were converted to volumes assuming a 1 g/cm3 tumor density; since mass is directly proportional to volume, our description effect size is not biased by this transformation. Although different methods of measurement were employed across the publications, the normalization of the effect of treatment on tumor growth compared with control tumor growth minimizes potential discrepancy.
Describing the effects of immunotherapy (“effect size” E)
We sought to formulate a measure of effect size of treatment on the growth of tumors. This effect size was expressed as a ratio of linearized growth rates of treated tumors against control tumors (Eqn. 1). The tumor growth rate of each group was determined using the tumor size at the time that treatment was given to the experimental group (Vi and ti) and the tumor size at the endpoint for each respective group (Vf and tf).
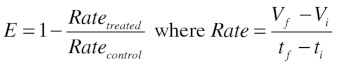 |
(1) |
Untreated mice were used for controls when available. Otherwise, mock treatments or vehicles were used as control (phosphate-buffered saline, saline, normal immunoglobulin, etc.). In principle, we required controls that were designed to have a minimal effect on tumor growth.
A more sophisticated logistic or Gompertzian growth model might better suited to describe tumor growth; however, the reported data is usually insufficient for the application of such models. Our objective was to produce a usable measure of the effect of treatment on the behavior of tumor growth that could be applied across a wide strata of data. Since the effect is expressed relative to the control growth rate, our measure is sufficient to assign an effect size to any controlled experiment.
A qualitative interpretation of our effect size can be derived computationally. All of the following interpretations assume that the control tumor grows out and does not spontaneously regress; this was true for all collected data. An effect size less than zero indicates that the treated tumor grows out faster than the control. An effect size of zero indicates that the treated tumor had the same growth behavior as the control. For an effect size between zero and one, the treated tumor grows out, but either is slowed or delayed compared with the control. An effect size equal to one indicates stable disease. An effect size greater than one indicates regression of the treated tumor.
Acknowledgments
We thank Ainhoa Arina, David Binder, Boris Engels, Christian Idel, and Karin Schreiber for critical review of this manuscript and quality discussion. We are also grateful to Deb Werner for her excellent guidance in the systematic retrieval of publications. This research was supported by the National Institute of Health grants P01-CA97296, R01-CA22677 and R01-CA37516 to H.S. Statistical collaboration (R.T.) was supported in part by grant UL1RR024999 from the National Center for Research Resources. F.T.W. is a recipient of the Biological Sciences Collegiate Division Research Endowments at the University of Chicago.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/18311
References
- 1.Woglom WH. Immunity to transplantable tumours. Cancer Rev. 1929;4:129–214. [Google Scholar]
- 2.Goldie JH, Coldman AJ. A model for tumor response to chemotherapy: an integration of the stem cell and somatic mutation hypotheses. Cancer Invest. 1985;3:553–64. doi: 10.3109/07357908509039817. [DOI] [PubMed] [Google Scholar]
- 3.Spratt JS, Meyer JS, Spratt JA. Rates of growth of human solid neoplasms: Part I. J Surg Oncol. 1995;60:137–46. doi: 10.1002/jso.2930600216. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber K, Rowley DA, Riethmuller G, Schreiber H. Cancer immunotherapy and preclinical studies: why we are not wasting our time with animal experiments. Hematol Oncol Clin North Am. 2006;20:567–84. doi: 10.1016/j.hoc.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Weiss DW. Animal models of cancer immunotherapy: questions of relevance. Cancer Treat Rep. 1980;64:481–5. [PubMed] [Google Scholar]
- 6.Buckwalter MR, Srivastava PK. “It is the antigen(s), stupid” and other lessons from over a decade of vaccitherapy of human cancer. Semin Immunol. 2008;20:296–300. doi: 10.1016/j.smim.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akazawa T, Inoue N, Shime H, Kodama K, Matsumoto M, Seya T. Adjuvant engineering for cancer immunotherapy: Development of a synthetic TLR2 ligand with increased cell adhesion. Cancer Sci. 2010;101:1596–603. doi: 10.1111/j.1349-7006.2010.01583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buhé V, Pers JO, Marianowski R, Berthou C, Youinou P, Loisel S. Development of a Murine model to dissect the CpG-oligonucleotide-enhancement of the killing of human B Cells by rituximab. J Autoimmun. 2010;34:136–44. doi: 10.1016/j.jaut.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Hernandez ML, Hamada H, Reome JB, Misra SK, Tighe MP, Dutton RW. Adoptive transfer of tumor-specific Tc17 effector T cells controls the growth of B16 melanoma in mice. J Immunol. 2010;184:4215–27. doi: 10.4049/jimmunol.0902995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harnack U, Johnen H, Pecher G. IL-1 receptor antagonist anakinra enhances tumour growth inhibition in mice receiving peptide vaccination and beta-(1-3),(1-6)-D-glucan. Anticancer Res. 2010;30:3959–65. [PubMed] [Google Scholar]
- 11.Kayashima H, Toshima T, Okano S, Taketomi A, Harada N, Yamashita Y, et al. Intratumoral neoadjuvant immunotherapy using IL-12 and dendritic cells is an effective strategy to control recurrence of murine hepatocellular carcinoma in immunosuppressed mice. J Immunol. 2010;185:698–708. doi: 10.4049/jimmunol.0900187. [DOI] [PubMed] [Google Scholar]
- 12.Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70:6725–34. doi: 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN, et al. The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells. Sci Transl Med. 2010;2:55ra78. doi: 10.1126/scitranslmed.3000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ringshausen I, Feuerstacke Y, Krainz P, den Hollander J, Hermann K, Buck A, et al. Antifungal therapy with itraconazole impairs the anti-lymphoma effects of rituximab by inhibiting recruitment of CD20 to cell surface lipid rafts. Cancer Res. 2010;70:4292–6. doi: 10.1158/0008-5472.CAN-10-0259. [DOI] [PubMed] [Google Scholar]
- 15.Sharkey RM, Karacay H, Goldenberg DM. Improving the treatment of non-Hodgkin lymphoma with antibody-targeted radionuclides. Cancer. 2010;116:1134–45. doi: 10.1002/cncr.24802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70:6171–80. doi: 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, et al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207:651–67. doi: 10.1084/jem.20091921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70:9053–61. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maletzki C, Gock M, Klier U, Klar E, Linnebacher M. Bacteriolytic therapy of experimental pancreatic carcinoma. World J Gastroenterol. 2010;16:3546–52. doi: 10.3748/wjg.v16.i28.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srivastava PK. Immunotherapy of human cancer: lessons from mice. Nat Immunol. 2000;1:363–6. doi: 10.1038/80795. [DOI] [PubMed] [Google Scholar]
- 21.Finkelstein SE, Heimann DM, Klebanoff CA, Antony PA, Gattinoni L, Hinrichs CS, et al. Bedside to bench and back again: how animal models are guiding the development of new immunotherapies for cancer. J Leukoc Biol. 2004;76:333–7. doi: 10.1189/jlb.0304120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winn HJ. Immune mechanisms in homotransplantation. II. Quantitative assay of the immunologic activity of lymphoid cells stimulated by tumor homografts. J Immunol. 1961;86:228–39. [PubMed] [Google Scholar]
- 23.Skipper HE. Cancer chemotherapy is many things: G.H.A. Clowes Memorial Lecture. Cancer Res. 1971;31:1173–80. [PubMed] [Google Scholar]
- 24.Dethlefsen LA, Prewitt JM, Mendelsohn ML. Analysis of tumor growth curves. J Natl Cancer Inst. 1968;40:389–405. doi: 10.1093/jnci/40.2.389. [DOI] [PubMed] [Google Scholar]
- 25.Budach W, Taghian A, Freeman J, Gioioso D, Suit HD. Impact of stromal sensitivity on radiation response of tumors. J Natl Cancer Inst. 1993;85:988–93. doi: 10.1093/jnci/85.12.988. [DOI] [PubMed] [Google Scholar]
- 26.Rockwell SC, Kallman RF, Fajardo LF. Characteristics of a serially transplanted mouse mammary tumor and its tissue-culture-adapted derivative. J Natl Cancer Inst. 1972;49:735–49. [PubMed] [Google Scholar]