

Abstract
A central goal of biology is understanding and describing the molecular basis of plasticity: the sets of genes that are combinatorially selected by exogenous and endogenous environmental changes, and the relations among the genes. The most viable current approach to this problem consists of determining whether sets of genes are connected by some common theme, e.g. genes from the same pathway are overrepresented among those whose differential expression in response to a perturbation is most pronounced. There are many approaches to this problem, and the results they produce show a fair amount of dispersion, but they all fall within a common framework consisting of a few basic components. We critically review these components, suggest best practices for carrying out each step, and propose a voting method for meeting the challenge of assessing different methods on a large number of experimental data sets in the absence of a gold standard.
Keywords: gene set enrichment analysis, pathway enrichment analysis, expression analysis, GSEA, PWEA, performance evaluation, controlled mutual coverage, CMC
INTRODUCTION
Understanding of complex polygenetic phenotypes—stage of differentiation, disease state, responsiveness to exogenous perturbations and so on—requires a combination of high performance experimental and analytical methods for identifying related sets of genes (e.g. genes in pathways or functional classifications) associated with phenotypic changes. Identification generally means the discovery of gene sets that were not previously known to be related, as well as the determination of which sets among a known collection (e.g. [1]). The former, more difficult problem is also known as the pathway reconstruction or pathway annotation problem and discussed elsewhere [2–5]; here we focus on the latter, including the topological structure of the sets.
Early methods for associating gene sets with phenotype changes first identify individual, potentially relevant genes by making a binary decision based on a quantity that measures the extent of differential expression between the phenotypes (e.g. performing a t-test and requiring P-value <0.01), and then use a Fisher's exact test to determine whether a significant number of these genes belong to a prespecified gene set [6, 7]. An alternative approach begins by ranking all genes according to differential expression, and then determines if a prespecified gene set is significantly overrepresented toward the top or bottom of the ranked list. Such a procedure was first introduced by Subramanian et al. [8] and their particular method was named Gene Set Enrichments Analysis (GSEA). Here we use GSEA broadly to describe all methods for associating gene sets with phenotype changes. We abbreviate prespecified gene sets among a known collection as gene sets or simply sets. We use the terms enrichment and overrepresentation as they are in normal parlance. They do not distinguish method, but frames of reference: the members of a set are said to be overrepresented in a group of genes being examined, and the group is enriched in members of the set.
The GSEA method by Subramanian et al. [8] consists of the following specific steps: (i) rank all genes by the magnitudes of their differential expression and select a window in the ranked list, i.e. a contiguous run of some number of genes starting at any rank, (ii) define an enrichment score based on a weighted Kolmogorov Smirnov (WKS) test that measures the difference between the number of genes in a prespecified gene set that are observed in the window, and the number of occurrences if the genes in the set were uniformly distributed in the list, (iii) simulate a background distribution of the enrichment score by shuffling samples and estimate the statistical significance of the gene set, (iv) repeat steps i–iii for all prespecified gene sets (hypotheses) and for various window sizes, (v) correct for multiple hypotheses testing.
The strategy for performing GSEA has numerous variants, depending on the method for estimating significance (WKS test, mean test, median test, Wilcoxon rank sum test, etc.); background distribution, which is related to the method for estimating significance, but not always dictated by it; choice of the shuffling method when the background distribution is simulated; the method for multiple hypothesis correction; and the choice of weights to account for auxiliary information such as topology of gene sets [8, 9]. Several excellent reviews compared these variants [10–14]. In particular, Ackermann and Strimmer [13] established a general modular framework for performing GSEA. They critically assessed a subset of gene set enrichment procedures using 10 simulated data sets and 2 experimental data sets. In addition, they assessed three so-called global procedures, which do not compute a gene-level enrichment score; rather, they test gene sets as the unit. Ackermann and Strimmer concluded that the choice of gene-level statistics was inconsequential, while the performance of gene set-level statistics was more variable. All of the gene set-level statistics rejected the only simulated negative control data set, and based on the percentage of the other nine simulated data sets being detected, Ackermann and Strimmer concluded that the mean test was more sensitive than the median test and Wilcoxon rank sum test, while GSEA was the least sensitive. Furthermore, they concluded that global procedures yielded worse results than the best of the six gene set-level statistics they tested. Their results on the two experimental data sets were inconsistent with each other and inconsistent with the results based on simulated data. They reported that the results varied greatly depending upon the choice of the null hypothesis, and one global procedure called Hotelling's T2 test was most sensitive for one data set using one type of background distribution, while tests based on conditional FDR and enrichment score, but not the mean test, were more most sensitive in other combinations of experimental data set and null hypothesis. Although the test on experimental data sets is far more biologically relevant than on simulated data sets, the results of the former are harder to interpret due to the lack of a gold standard, i.e. which gene sets are true positives and which are true negatives.
Indeed, the lack of a gold standard has greatly hampered the effort of assessing gene set enrichment methods using experimental data sets. Biological systems are so complex that simulated data sets simply cannot substitute for experimental data sets. Furthermore, it can be argued that the ability of rejecting false positive predictions is even more important than detecting lowly ranked true positives, because it is costly to validate a large number of predictions. Therefore, in this article, we focus on experimental data sets. First we review the core components of gene set enrichment methods in the aspects that are important to experimental data sets. Then we use 132 experimental data sets to critically assess six gene set-level statistics and one global test, which received favorable ratings in previous reviews [10, 13]. To tackle the lack of a gold standard, we introduce the concept of mutual coverage (MC), which reflects the extent to which the gene sets predicted by a particular method are reproduced by other methods. Our results suggest that: (i) the Wilcoxon rank sum test and the WKS test as implemented in GSEA provide the most effective gene set-level statistics for obtaining high MC, (ii) simulated background distributions are more accurate than analytic backgrounds, (iii) the mean and median tests achieve the highest sensitivity but poor MC, and (iv) incorporating topology of gene sets in the analysis increases the sensitivity for all six procedures without reducing MC.
COMPONENTS OF GSEA
In this section, we review the five core components of GSEA methods as illustrated in Figure 1, focusing on the aspects that are particularly important for experimental data sets.
Figure 1:

Key components of performing gene set enrichment analysis.
Data preprocessing
There are two important but frequently overlooked data preprocessing steps. Normalization allows expression values obtained from different experiments to be directly comparable [15, 16]. The expression values of a small, but different set of genes may be missing in different microarray experiments due to technical issues. Imputation of missing data is thus important for maximal data coverage when the results of multiple experiments are compared.
A number of methods are available for normalization [16, 17], yet this critical step is frequently omitted [15]. The most common normalization algorithms—RMA [16] and MAS 5.0 [18]—are designed for expression levels generated with microarrays that follow a lognormal distribution. Thus it is important to log transform the raw intensity values from microarrays. Failure to do so would bias toward high expression values, reducing statistical power because of increase in variance [16]. Log transform is also applied to RNA-seq data. Expression level determined by RNA-seq is usually quantified in Reads Per Kilobase exon Model per million mapped reads (RPKM; density of reads that map to a gene normalized for the length of its mature transcript and for the sequencing depth of the experiment [19]), which after log transform correlates well with normalized intensity measured with microarray, also after log transform [19].
Missing data can be imputed using methods based on K nearest neighbors, singular value decomposition, or least square regression models. Least square regression algorithms were reported to produce lower estimation error than other methods [20, 21]. In this article we use a popular least square regression algorithm, LSimpute_gene [22], to impute missing values in all 132 experimental data sets.
Single gene statistics
The first step in GSEA is to compute a gene-level statistic of differential expression, e.g. a t statistic, a signal to noise ratio (mean to standard deviation ratio), a fold change or a Wilcoxon rank sum statistic. Because phenotype change can affect different genes in opposite directions, i.e. increase the expression levels of some genes while decrease the levels of others, and we want to be able to identify the gene sets that contain both types of genes, it is desirable to eliminate the direction of differential expression by taking the absolute or square of the statistic [13, 23]. However, data transformations that eliminate direction—such as absolute values—lead to asymmetrical distributions, and can nullify some analytical estimates of significance based on analytical background distributions such as the χ2 test [24, 25] (see ‘Estimating significance’ and ‘The validity of analytical background distributions’ sections).
The many-to-many correspondence between genes and probe sets on a microarray creates ambiguity in determining expression levels of genes [26, 27]. A common practice is to calculate the mean or median expression levels of the probe sets that correspond to the same gene; however, doing so usually increases the number of false negatives [28]. An alternative is to perform meta analysis [28], using for example, the method proposed by Fisher [29] or by Stouffer [30, 31]. Rather than merging the expression values directly, these methods merge probe set-level statistics. A similar problem exists in RNA-seq, where some sequencing reads are mapped to multiple genomic locations. Such multi-mappers originate from paralogs, segmentally duplicated regions and low sequence complexity [32]. Ignoring multi-mappers reduces sensitivity and undercounts some genes [19]. Strategies for assigning multi-mappers are discussed in [19, 32, 33].
Gene set-level statistics
The purpose of a gene set-level statistic is to decide whether a gene set is distinct in some statistically significant way. A gene set statistic can be defined in terms of properties of the genes in the set, e.g. the mean, median, variance, etc. of a gene-level statistic (see Table 1 for more details). When a property (and its corresponding statistic) is chosen, the null hypothesis must, of course, also be specified. There are two null hypotheses as defined by Tian et al. [34]. In one case (Q1) the background distribution is obtained by shuffling genes; in the other (Q2), the background distribution is obtained by shuffling phenotypes, i.e. samples (see ‘Estimating significance’ section). The rationale for using Q1 is that a significant gene set should be distinguishable from an equal size set composed of randomly chosen genes. On the other hand, Q2 focuses on a gene set and tests whether its association with the phenotype change is distinguishable from randomly shuffled phenotype changes. Q2 is generally favored because it preserves the relationship of the genes in the set [11, 12, 34] and directly addresses the question of finding gene sets whose expression changes correlates with phenotype changes.
Table 1:
Commonly used gene-set level statistics
| Gene-set statistics | Assumptions for analytical background | Statistics | Description | Analytical background (signed) | Analytical background (absolute) | Note | References |
|---|---|---|---|---|---|---|---|
| χ2-test | Independence normality | χ2 | 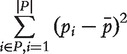 |
χ2 distribution | NA | Variance/Scale test | [25] |
| Mean test | Independence normality | Mean | 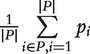 |
Standard normal distribution | Normal distribution | Location test | [25, 34, 52] |
| Median test | Independence same shape | Median | 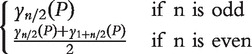 |
Hypergeometic distribution | Hypergeometic distribution | Location test | [13] |
| Wilcoxon rank sum test | Independence same shape | Rank sum | 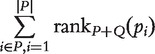 |
Normal distribution | Normal distribution | Location test | [13, 53, 54] |
| WKS test | NA | ES | Maximum deviation (located at position i) between 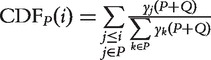 , and , and 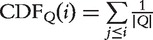
|
NA | NA | Location and shape test | [8, 9, 23] |
P is the given gene-set. pi is the gene statistics of gene i in P. Q is the set of genes not in P. y is the order statistics of P. rankP+Q(x) is the rank of x in the set P + Q. NA: Not applicable.
Gene set-level statistics generally ignore clinical covariates—factors such as age, sex and weight—which can also cause differential expression, confounding the impact of phenotype changes [35]. The effect of covariates can be estimated using, for example, a linear regression model [35]. If a t statistic is used in the gene level, it can be generalized using a linear regression model for covariate correction, after accounting for the increased number of variables to avoid over fitting [35].
Most gene set-level statistics also ignore relationships among genes within the set. For example, if the gene set is a pathway, its topological information is ignored. Including topological information is important for accounting for the effect of genetic buffering [36], which deduces that if a gene fails to propagate its influence to a pathway neighbor, its biological role is buffered. Conversely, a gene that regulates many of its downstream genes may play a pivotal role in the expression changes of the pathway associated with phenotype changes. Methods for including topological information by weighted gene set-level statistics are discussed in [9, 37, 38].
Estimating significance
We use significance in the standard way: the probability that the null hypothesis, evaluated on the background (or null) distribution, is correct. The background distribution can sometimes be written analytically, as in the case of a Gaussian distribution, and it can always be simulated by shuffling experimental data. As noted in the above section, simulated background is dictated by the choice of the null hypothesis (Q1 or Q2), which often leads to different conclusions [34].
Most frequently the gene set-level statistic, e.g. the mean of the t-statistic values of genes in the set, is assumed to follow a normal distribution when expression change has no association with phenotype change [34]. In such case the significance (P-value) of a gene set can be computed analytically [25]. Such an assumption is in question when the expression levels of genes in a set are dependent on one another, which is common for genes in a pathway [34]. In ‘The validity of analytical background distributions’ section we will discuss analytical backgrounds and empirical corrections [25] to make them more useful.
To be concrete in illustrating how significance is estimated using a simulated background distribution, suppose we are interested in estimating the probability that the enrichment score obtained for a particular gene set is a chance occurrence of phenotype changes. The procedure would be to shuffle the phenotype labels, calculate the differential expression of each gene, rank all genes and compute an enrichment score for the same gene set. The entire process is repeated multiple times to obtain a distribution of enrichment scores, and the P-value of the actual enrichment score is simply the fraction of shuffles that produce enrichment scores at least as great as observed. Although simulating the background distribution obviates the need of an analytical background, it can be computational demanding—at least N shuffles need to be performed to achieve a P-value resolution of 1/N [39].
Correction for multiple testing
P-value is the appropriate measure of statistical significance when only one gene set is tested. When a large number of gene sets are tested, there can be many false positives among the gene sets that receive seemingly highly significant P-values; this is called the multiple hypothesis testing problems. The simplest procedure is to choose a P-value which, when multiplied by the number of hypotheses, i.e. the total number of tested gene sets, gives a sufficiently low corrected P-value, e.g. <0.05. This Bonferroni correction [40] is, however, very conservative and sometimes results in an unacceptably large number of false negatives. An alternative is to control the expected fraction of false positives among the predictions, or the false discover rate (FDR), using the method by Benjamini and Hochberg [41]. The original Benjamini–Hochberg procedure assumes a uniform distribution for the P-values [41]. In some cases when there are relatively many ‘non-null’ tests, i.e. when low P-values are prevalent, an FDR variant, positive FDR (pFDR) can be applied [42, 43]. The corrected P-value is called Q-value, defined as the ‘minimum FDR at which a test is called significant’ [42, 44]. The relationship between Q-value and FDR is analogous to that between P-value and type I error [42]. The final significant gene sets are the ones whose Q-values are smaller than an FDR threshold.
THE VALIDITY OF ANALYTICAL BACKGROUND DISTRIBUTIONS
Although appropriate usage of a standard analytical background distribution facilitates rapid computation of P-values with high precision, the critical question is how well the analytical background represents the actual background. One way to test the validity of an analytic background is to examine the distribution of P-values under the null hypothesis, which should be uniform [45, 46]. To address this question, we generated 500 null data sets by shuffling the sample tags of an arbitrarily chosen human data set from the Gene Expression Omnibus (GEO; a public database of high throughput gene expression data), GDS2835 [47]. We constructed the histograms of the null P-value distributions using five gene set-level metrics summarized in Table 1, with analytical and simulated backgrounds.
The results indicate that P-values are uniformly distributed when background distributions are generated by shuffling, irrespective of metric (Figure 2A), whereas the distributions obtained using analytical background distributions are highly nonuniform, and metric dependent (Figure 2B). The null P-value distribution of the χ2 test as shown in Figure 2B deviates greatly from a uniform distribution, because taking the absolute value of the gene-level statistic causes the background to no longer follow the χ2 distribution. Taking the absolute value does not violate the analytical background distributions of the mean, median and Wilcoxon rank sum tests; nonetheless, their null P-value distributions deviate from the uniform distribution. The biased null P-value distribution further violates the assumption of the FDR procedure (‘Correction for multiple testing’ section). Thus we conclude that it is more accurate to use simulated backgrounds than analytical backgrounds.
Figure 2:

P-value distribution of null by (A) simulated background and (B) analytical background. It is clear that analytical backgrounds give biased P-value distributions. WKS (i.e. GSEA) is not shown in (B), because WKS does not follow an analytical background.
COMPARISON OF GENE SET-LEVEL STATISTICS
The problem of comparing different methods for gene set enrichment analysis is made difficult by the lack of a gold standard. One can mine the literature to obtain evidence on whether a gene set is associated with the phenotype change, but this can only be done on a small scale. An alternative is to quantify the number of overlapping predictions (gene sets called significant) by several methods [9, 48]. Because each method can capture a piece of the evidence (from the location of mean, shape of distribution, etc.) of biological perturbation, gene sets predicted by multiple methods should be more reliable than gene sets predicted by only one method. In this article we propose a formal way to use the MC of multiple methods for evaluating gene set-level statistics.
Mutual coverage
We state the concept of MC by multiple methods in precise terms:
Observation 1: given several orthogonal predictors (e.g. gene set-level statistics), a gene set deemed significant by more predictors is less likely to be false than a gene set deemed significant by fewer predictors. The term orthogonal here means that different gene set-level statistics do not use correlated properties to make the prediction. Since ‘mutually supported gene sets’ identified by multiple predictors show statistical significance from multiple distinct properties, they might better reflect the underlying biology of phenotype changes.
Observation 2: if a particular predictor reports a high fraction of ‘mutually supported gene sets’, we assume it has high positive predictive value, especially if the number of predictors is exhaustive. We define MC of a predictor as the ratio of the number of votes from other predictors agreeing with its predictions divided by the maximum number of votes possible (see ‘Methods’ section for details).
Controlled mutual coverage for correlated predictors
The condition that predictors be orthogonal is almost never met. Figure 3 illustrates the Pearson correlation coefficient between the gene set-level statistics in Table 1. For example, mean and median are strongly correlated. If a predictor has many ‘echoes’, its MC can be overestimated. One way to correct for correlations among predictors is to down weight the votes from the echoes. An intuitive approach is to weight each vote according to the probability that two gene set-level statistics agree with each other by chance. In other words, if predictor A has a probability π of voting for all predictions of predictor B by chance, votes from A will be weighted by a function of π; in this article, we simply take it as the inverse of π. Therefore correlated predictors having higher π receive lower weights. To compute π, we generated 500 randomly phenotype-shuffled data sets from the aforementioned experimental data set (GEO GDS2835) and computed the expected frequency that two gene set-level statistics predict the same gene set as significant (see ‘Methods’ section for more details). After weighting and normalization, a controlled MC score (CMC; see ‘Methods’ section) is calculated and the effects of echoes are controlled.
Figure 3:

The Pearson correlation coefficient between all 10 gene-set statistics. The ‘W’- prefix indicates a TIF weighted statistic.
We generated 132 null data sets by randomly shuffling the phenotype tags of an experimental data set (GEO GDS2835), and computed the CMC scores of the 5 original gene set-level statistics and Hotelling's T2 test in one group, and the CMC scores of the 5 topology impact factor (TIF) [9] weighted gene set-level statistics in another group. All 1l statistics show similarly low CMC scores (the first row of Table 2, labeled as ‘Null data set’), indicating that the contributions from correlated predictors have been substantially diminished. We did not compute CMC for a TIF weighted gene set-level statistic with its original statistic in one group, because they are highly correlated (Figure 3).
Table 2:
CMC of null data set compared with filtered data set, using original statistics and TIF weighted statistics at α = 0.01
| Type | χ2-test | Hotelling's T2 test | Mean test | Median test | WKS test | Wilcoxon rank sum test |
|---|---|---|---|---|---|---|
| Null data set (original) | 0.19 | 0.15 | 0.22 | 0.22 | 0.19 | 0.21 |
| Original statistics | 0.21 | 0.23 | 0.24 | 0.28 | 0.40 | 0.44 |
| Original statistics (leave one out) | 0.18; 0.16; 0.16; 0.21; 0.14 | 0.20; 0.17; 0.17; 0.21; 0.17 | 0.21; 0.20; 0.22; 0.18; 0.16 | 0.24; 0.22; 0.25; 0.20; 0.18 | 0.31; 0.33; 0.36; 0.31; 0.29 | 0.38; 0.35; 0.36; 0.33; 0.33 |
| Null data set (weighted) | 0.19 | NA | 0.20 | 0.22 | 0.18 | 0.17 |
| TIF weighted statistics | 0.20 | NA | 0.21 | 0.25 | 0.33 | 0.31 |
| TIF weighted statistics (leave one out) | 0.18; 0.12; 0.12; 0.20 | NA | 0.18; 0.16; 0.20; 0.10 | 0.21; 0.18; 0.22; 0.12 | 0.23; 0.25; 0.29; 0.23 | 0.22; 0.25; 0.19; 0.27 |
WKS and Wilxocon rank sum test show significant higher CMC than in Null data set
Test on 132 experimental data sets
We collected and processed 132 human data sets from GEO, in accordance with the procedure in the ‘Components of GSEA’ section (see ‘Methods’ section for details). The CMC scores of the 6 statistics in Table 1 and 5 TIF weighted statistics are listed in Table 2. The Wilcoxon rank sum test and WKS test show significantly higher CMC in both original (CMC = 0.4 and 0.35, respectively) and TIF weighted forms (CMC = 0.39 and 0.35, respectively) compared with their CMC for the null data sets (0.07). To test whether the CMC score is still biased by dependency, we iteratively removed one statistic at a time and the resulting CMC scores are also listed in Table 2. The results indicate that the Wilcoxon rank sum test and WKS test still perform better than the other statistics, and the order of performance of all predictors does not change (Table 2). We conclude that predictions based on the Wilcoxon rank sum test or WKS test are more likely to be covered by other gene set statistics that use mean, median, χ2 and Hotelling's T2 test, and may imply higher biological inference power. In addition, the TIF weighted WKS test (PWEA [9]) reports 15% more positive prediction and still retains the higher level of CMC (see ‘Supplementary Materials’), suggesting that it is more sensitive than the original WKS test. Note that TIF weighting was not applied to Hotelling's T2 tests since this method performs principal component analysis which can not be applied to the topological information and moreover, one of the purposes of Hotelling's T2 test is to reduce the influence of correlation structure inside a gene set, thus weighting gene based on the correlation of neighbor genes is against its design.
DISCUSSIONS AND CONCLUSIONS
We reviewed approaches to gene set enrichment analysis and attempted to clarify a number of concepts that are important for application to experimental data sets, such as preprocessing of raw data, imputation of missing data, the choice of null hypothesis, and methods for generating null distributions. Our analysis of null P-value distributions indicates that analytical background distributions are less accurate than simulated background distributions. As shown previously [13], the choice of gene set-level statistics is the most important component for gene set enrichment methods; however, and it is difficult to compare the performance of different gene set-level statistics when a gold standard is absent.
In order to address this difficulty we propose a new metric, CMC, essentially a positive predictive value where the true positives are determined by weighted overlaps between different methods. The results of testing 132 experimental data sets suggest that the Wilcoxon rank sum test and WKS test can better cover the predictions of the mean test, the median test, the χ2 test and Hotelling's T2 test, but not vice versa. We postulate that it is because WKS covers not just location shift but also shape changes of the observed distribution of differential expression compared with the background distribution and Wilcoxon rank sum test is robust to the extreme values. Since the WKS test reports more gene-sets than the Wilcoxon rank sum test in our experiments, it is likely to have higher sensitivity (see Supplementary Materials).
To further improve the biological utility of gene set enrichment analysis, we believe that the inclusion of additional biological features such as topology or covariates as discussed in the ‘Gene set-level statistics’ section would be more useful than changing statistics. Utilizing more domain knowledge is likely to reveal more insights in the analysis. The concept of gene set enrichment analysis has been applied to biological features in addition to expression, such as SNPs, copy number variation [49] and protein–protein interaction networks.
METHODS
We collect all human gene expression data sets based on microarrays from GEO, and split each data set according to their phenotypes. All GEO data sets have proper gene name annotations for each probe-set. Subsets within the same GEO entry are scrutinized and only one subset is chosen to avoid redundancy. The data sets with fewer than 10 samples per phenotype were discarded. We imputed missing values of each test set using the LSimpute_gene algorithm [22], which construct weighted multiple regression models based on other genes that best correlated with the genes with missing values. We ensured all expression values were log transformed. Data sets that were normalized by approaches other than RMA or MAS 5.0 were also discarded. In total 132 test sets remained. We then perform t-test and use absolute values of t-statistic as the gene-level statistic of each probe-set. Probe-sets that share the same gene name were combined according to Stouffer's method [31].
We use 201 pathways from KEGG as the collection of gene sets for all analysis. We tested 5 gene set level statistics and one global test, Hotelling T2 test, which were reviewed favorably by Ackermann and Strimmer [13]. A Hotelling T2 statistic for a gene set is calculated as follows:
where 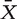 and
and 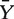 are vectors having k (total number of genes in the gene set) elements, representing the mean expression levels of the genes in the gene set among two phenotypes, with nx and ny samples, respectively, and S−1 is the inverse of the pooled covariance matrix. The problem of Hotelling T2 test in practice is that when k > nx + ny, which is common, the singularity of S makes finding the inverse difficult. Kong et al. [50] solved the issue by using PCA (principal component analysis) to reduce the dimensionality, and we use the same approach to compute the Hotelling T2 statistic.
are vectors having k (total number of genes in the gene set) elements, representing the mean expression levels of the genes in the gene set among two phenotypes, with nx and ny samples, respectively, and S−1 is the inverse of the pooled covariance matrix. The problem of Hotelling T2 test in practice is that when k > nx + ny, which is common, the singularity of S makes finding the inverse difficult. Kong et al. [50] solved the issue by using PCA (principal component analysis) to reduce the dimensionality, and we use the same approach to compute the Hotelling T2 statistic.
For all five gene set-level statistics (Table 1), we applied the method from Hung el al. to weight the gene level statistics by a topological influence factor (TIF). Hotelling's T2 statistic cannot be separated to gene-level and gene set-level, so TIF weighting cannot be performed. The significance levels (P-values) are calculated using simulated backgrounds and corrected for multiple testing using the FDR procedure [51].
To calculate CMC, we define Wk as the prediction profiles of predictor k under the FDR cutoff of 0.05. W is a two dimensional M by N matrix, where M is the total number of data sets (M = 132) and N is the total number of gene sets (N = 201). Wk (i,j) = 1 if predictor k assigns a Q-value below the FDR cutoff for gene set j in data set i, otherwise Wk (i,j) = 0.
The MC of a predictor k is defined as:
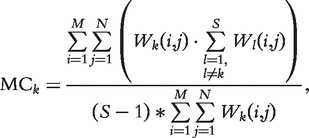 |
where S is the total number of predictors.
The numerator is the total number of votes that predictor k gets from other predictors and the denominator is the maximum votes it can get.
In order to control dependency among different predictors, we first model the probability π that predictor k agrees with predictor l in 500 null data sets, generated by shuffling the sample tags of an arbitrarily chosen human data set from the Gene Expression Omnibus (GEO; a public database of high throughput gene expression data), GDS2835 [47]. Altering the total number of null data sets and the source experimental data set does not change π appreciably. π is defined as:
 |
We use π as the weight of each vote accordingly and define CMC for predictor k as follows:
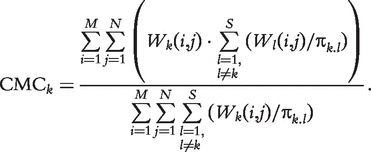 |
CMC is then a weighted ratio that indicates the fraction of predictions supported by other predictors.
SUPPLEMENTARY DATA
Supplementary data are available online at http://bib.oxfordjournals.org/.
Key Points.
Review critical steps in performing a statistically robust gene set enrichment analysis.
Demonstrate the large number of false positives due to inappropriate statistical backgrounds.
Introduce a novel ‘controlled mutual coverage (CMC)’ index to evaluate gene set statistics.
FUNDING
National Institutes of Health (grant RR022971) (partially); National Science Foundation (grant DBI-0850008) (partially).
Supplementary Material
Biographies
Jui-Hung Hung is a post-doc in Zhiping Weng's lab. He develops bioinformatics algorithms and tools.
Tun-Hsiang Yangis a post-doc in Charles DeLisi's lab. He primarily works on statistical genetics.
Zhenjun Hu is a research assistant professor in Charles DeLisi's lab. His research interests are mainly in computational methods for network analyses, integration and visualization.
Zhiping Weng is Director and Professor of Program in Bioinformatics and Integrative Biology at U Mass Medical School. Her lab develops and applies computational methods to study gene regulation, small silencing RNAs and protein docking.
Charles DeLisi is the Metcalf professor of science and engineering at Boston University. His lab focuses on all sorts of topics in systems biology.
References
- 1.Molecular Signatures Database v3.0. Available from: http://www.broadinstitute.org/gsea/msigdb/index.jsp (9 September 2010, date last accessed) [Google Scholar]
- 2.Moriya Y, Itoh M, Okuda S, et al. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–5. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilchrist A, Au CE, Hiding J, et al. Quantitative proteomics analysis of the secretory pathway. Cell. 2006;127(6):1265–81. doi: 10.1016/j.cell.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 4.Koller A, Washburn MP, Lange BM, et al. Proteomic survey of metabolic pathways in rice. Proc Natl Acad Sci USA. 2002;99(18):11969–74. doi: 10.1073/pnas.172183199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ye Y, Doak TG. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput Biol. 2009;5(8):e1000465. doi: 10.1371/journal.pcbi.1000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khatri P, Draghici S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005;21(18):3587–95. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho RJ, Huang M, Campbell MJ, et al. Transcriptional regulation and function during the human cell cycle. Nat Genet. 2001;27(1):48–54. doi: 10.1038/83751. [DOI] [PubMed] [Google Scholar]
- 8.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hung JH, Whitfield TW, Yang TH, et al. Identification of functional modules that correlate with phenotypic difference: the influence of network topology. Genome Biol. 2010;11(2):R23. doi: 10.1186/gb-2010-11-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Efron B, Tibshirani R. On testing the significance of sets of genes. Ann Appl Stat. 2007;1(1):107–29. [Google Scholar]
- 11.Nam D, Kim SY. Gene-set approach for expression pattern analysis. Brief Bioinform. 2008;9(3):189–97. doi: 10.1093/bib/bbn001. [DOI] [PubMed] [Google Scholar]
- 12.Dinu I, Potter JD, Mueller T, et al. Gene-set analysis and reduction. Brief Bioinform. 2009;10(1):24–34. doi: 10.1093/bib/bbn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ackermann M, Strimmer K. A general modular framework for gene set enrichment analysis. BMC Bioinform. 2009;10:47. doi: 10.1186/1471-2105-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu C. Improving the scaling normalization for high-density oligonucleotide GeneChip expression microarrays. BMC Bioinformatics. 2004;5:103. doi: 10.1186/1471-2105-5-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 17.Steinhoff C, Vingron M. Normalization and quantification of differential expression in gene expression microarrays. Brief Bioinform. 2006;7(2):166–77. doi: 10.1093/bib/bbl002. [DOI] [PubMed] [Google Scholar]
- 18.Irizarry RA, Bolstad BM, Collin F, et al. Summaries of affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 20.Celton M, Malpertuy A, Lelandais G, et al. Comparative analysis of missing value imputation methods to improve clustering and interpretation of microarray experiments. BMC Genomics. 2010;11:15. doi: 10.1186/1471-2164-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brock GN, Shaffer JR, Blakesley RE, et al. Which missing value imputation method to use in expression profiles: a comparative study and two selection schemes. BMC Bioinformatics. 2008;9:12. doi: 10.1186/1471-2105-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bo TH, Dysvik B, Jonassen I. LSimpute: accurate estimation of missing values in microarray data with least squares methods. Nucleic Acids Res. 2004;32(3):e34. doi: 10.1093/nar/gnh026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saxena V, Orgill D, Kohane I. Absolute enrichment: gene set enrichment analysis for homeostatic systems. Nucleic Acids Res. 2006;34(22):e151. doi: 10.1093/nar/gkl766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leone FC, Nelson LS, Nottingham RB. The folded normal distribution. Technometrics. 1961;3(4):543–50. [Google Scholar]
- 25.Irizarry RA, Wang C, Zhou Y, et al. Gene set enrichment analysis made simple. Stat Methods Med Res. 2009;18(6):565–75. doi: 10.1177/0962280209351908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leong HS, Yates T, Wilson C, et al. ADAPT: a database of affymetrix probesets and transcripts. Bioinformatics. 2005;21(10):2552–3. doi: 10.1093/bioinformatics/bti359. [DOI] [PubMed] [Google Scholar]
- 27.Harbig J, Sprinkle R, Enkemann SA. A sequence-based identification of the genes detected by probesets on the Affymetrix U133 plus 2.0 array. Nucleic Acids Res. 2005;33(3):e31. doi: 10.1093/nar/gni027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong F, Breitling R. A comparison of meta-analysis methods for detecting differentially expressed genes in microarray experiments. Bioinformatics. 2008;24(3):374–82. doi: 10.1093/bioinformatics/btm620. [DOI] [PubMed] [Google Scholar]
- 29.Fisher R. Statistical Methods for Research Workers. 4th edn. London: Oliver and Boyd; 1932. [Google Scholar]
- 30.Rosenthal R, Hiller JB, Bornstein RF, et al. Meta-analytic methods, the Rorschach, and the MMPI. Psychol Assess. 2001;13(4):449–51. doi: 10.1037//1040-3590.13.4.449. [DOI] [PubMed] [Google Scholar]
- 31.Fundel K, Kuffner R, Aigner T, et al. Normalization and gene p-value estimation: issues in microarray data processing. Bioinform Biol Insights. 2008;2:291–305. doi: 10.4137/bbi.s441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li B, Ruotti V, Stewart RM, et al. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26(4):493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faulkner GJ, Forrest AR, Chalk AM, et al. A rescue strategy for multimapping short sequence tags refines surveys of transcriptional activity by CAGE. Genomics. 2008;91(3):281–8. doi: 10.1016/j.ygeno.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 34.Tian L, Greenberg SA, Kong SW, et al. Discovering statistically significant pathways in expression profiling studies. Proc Natl Acad Sci USA. 2005;102(38):13544–9. doi: 10.1073/pnas.0506577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang Z, Gentleman R. Extensions to gene set enrichment. Bioinformatics. 2007;23(3):306–13. doi: 10.1093/bioinformatics/btl599. [DOI] [PubMed] [Google Scholar]
- 36.Kitami T, Nadeau JH. Biochemical networking contributes more to genetic buffering in human and mouse metabolic pathways than does gene duplication. Nat Genet. 2002;32(1):191–4. doi: 10.1038/ng945. [DOI] [PubMed] [Google Scholar]
- 37.Tarca AL, Draghici S, Khatri P, et al. A novel signaling pathway impact analysis. Bioinformatics. 2009;25(1):75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahnenfuhrer J, Domingues FS, Maydt J, et al. Calculating the statistical significance of changes in pathway activity from gene expression data. Stat Appl Genet Mol Biol. 2004;3:Article 16. doi: 10.2202/1544-6115.1055. [DOI] [PubMed] [Google Scholar]
- 39.Keller A, Backes C, Lenhof HP. Computation of significance scores of unweighted Gene Set Enrichment Analyses. BMC Bioinformatics. 2007;8:290. doi: 10.1186/1471-2105-8-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaffer J. Multiple hypothesis testing: a review. Annu Rev Psychol. 1995;46:561–84. [Google Scholar]
- 41.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 42.Storey JD. The positive false discovery rate: A Bayesian interpretation and the q-value. Ann Stat. 2003;31:2013–35. [Google Scholar]
- 43.Tang Y, Ghosal S, Roy A. Nonparametric bayesian estimation of positive false discovery rates. Biometrics. 2007;63(4):1126–34. doi: 10.1111/j.1541-0420.2007.00819.x. [DOI] [PubMed] [Google Scholar]
- 44.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100(16):9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas Sellke MJB, Berger JO. Calibration of p values for testing precise null hypotheses. Amer Statistician. 2001;55(1):62–71. [Google Scholar]
- 46.Fodor AA, Tickle TL, Richardson C. Towards the uniform distribution of null P values on Affymetrix microarrays. Genome Biol. 2007;8(5):R69. doi: 10.1186/gb-2007-8-5-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hever A, Roth RB, Hevezi P, et al. Human endometriosis is associated with plasma cells and overexpression of B lymphocyte stimulator. Proc Natl Acad Sci USA. 2007;104(30):12451–6. doi: 10.1073/pnas.0703451104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Draghici S, Khatri P, Tarca AL, et al. A systems biology approach for pathway level analysis. Genome Res. 2007;17(10):1537–45. doi: 10.1101/gr.6202607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim TM, Jung YC, Rhyu MG, et al. GEAR: genomic enrichment analysis of regional DNA copy number changes. Bioinformatics. 2008;24(3):420–1. doi: 10.1093/bioinformatics/btm582. [DOI] [PubMed] [Google Scholar]
- 50.Kong SW, Pu WT, Park PJ. A multivariate approach for integrating genome-wide expression data and biological knowledge. Bioinformatics. 2006;22(19):2373–80. doi: 10.1093/bioinformatics/btl401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benjamini Y, Drai D, Elmer G, et al. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1–2):279–84. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 52.Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scheer M, Klawonn F, Munch R, et al. JProGO: a novel tool for the functional interpretation of prokaryotic microarray data using Gene Ontology information. Nucleic Acids Res. 2006;34(Web Server issue):W510–W515. doi: 10.1093/nar/gkl329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barry WT, Nobel AB, Wright FA. Significance analysis of functional categories in gene expression studies: a structured permutation approach. Bioinformatics. 2005;21(9):1943–9. doi: 10.1093/bioinformatics/bti260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.