

Gene expression is tightly regulated during cell cycle progression. DeCaprio and colleagues now provide a comprehensive model of the MuvB complex's dynamic control of cell cycle gene expression from quiescence through mitosis. They find that MuvB sequentially recruits B-Myb and then FoxM1 to gene promoters during G2/M. The study provides a definitive description of MuvB's central role as a part of three distinct transcription complexes in the coordinated expression of a large number of genes throughout the cell cycle.
Keywords: DREAM complex, Myb–MuvB complex, B-Myb, FoxM1, ChIP sequencing, cell cycle
Abstract
Cell cycle progression is dependent on two major waves of gene expression. Early cell cycle gene expression occurs during G1/S to generate factors required for DNA replication, while late cell cycle gene expression begins during G2 to prepare for mitosis. Here we demonstrate that the MuvB complex—comprised of LIN9, LIN37, LIN52, LIN54, and RBBP4—serves an essential role in three distinct transcription complexes to regulate cell cycle gene expression. The MuvB complex, together with the Rb-like protein p130, E2F4, and DP1, forms the DREAM complex during quiescence and represses expression of both early and late genes. Upon cell cycle entry, the MuvB complex dissociates from p130/DREAM, binds to B-Myb, and reassociates with the promoters of late genes during S phase. MuvB and B-Myb are required for the subsequent recruitment of FoxM1 to late gene promoters during G2. The MuvB complex remains bound to FoxM1 during peak late cell cycle gene expression, while B-Myb binding is lost when it undergoes phosphorylation-dependent, proteasome-mediated degradation during late S phase. Our results reveal a novel role for the MuvB complex in recruiting B-Myb and FoxM1 to promote late cell cycle gene expression and in regulating cell cycle gene expression from quiescence through mitosis.
Transcriptional profiling of a variety of eukaryotes has revealed the periodic expression of many genes required for progression through the cell cycle (Cho et al. 1998; Spellman et al. 1998; Whitfield et al. 2002; van der Meijden et al. 2002). Typically, the products of genes required for DNA replication and mitosis are expressed just prior to and during the phase when they are needed, setting the stage for the orderly progression of events during the cell division cycle. In yeast, such regulated expression is accomplished by transcription factors that are expressed in consecutive, interdependent waves (Breeden 2003). In mammalian cells, several transcription factors, including the E2F family (for reviews, see Dimova and Dyson 2005; Blais and Dynlacht 2007), B-Myb (MYBL2) (Zhu et al. 2004), FoxM1 (Laoukili et al. 2005; Wang et al. 2005), and NF-Y (Caretti et al. 2003), have been reported to regulate the transcription of cell cycle genes. However, how these proteins coordinate the periodic expression of their target genes at distinct phases of the cell cycle remains largely unknown.
Recent studies identified the mammalian DREAM complex comprised of DP1 (TFDP1), the Rb-related protein p130 (RBL2), E2F4, and the MuvB core complex (containing LIN9, LIN37, LIN52, LIN54, and RBBP4) as a global repressor of cell cycle genes during G0 or quiescence. ChIP–chip (chromatin immunoprecipitation on microarray) for the DREAM subunits p130, E2F4, LIN9, and LIN54 revealed specific and cooperative binding of the DREAM complex to the promoters of >800 cell cycle-regulated genes (Litovchick et al. 2007). Upon cell cycle entry from quiescence, the intact MuvB core complex was released from the p130–E2F4–DP1-containing DREAM complex and independently bound to B-Myb (Litovchick et al. 2007; Schmit et al. 2007; Pilkinton et al. 2007). Knockdown of DREAM subunits led to the loss of repression of cell cycle genes during quiescence (Litovchick et al. 2007), while knockdown of the MuvB core complex or B-Myb in proliferating cells reduced mRNA levels of genes with peak expression during G2 and M phase, including Cyclin B1 (CCNB1) and Polo-like kinase (PLK1) (Osterloh et al. 2007; Kittler et al. 2007; Knight et al. 2009). This suggested that the MuvB core complex serves at least two independent roles by associating with distinct sets of transcription factors in quiescent and cycling cells.
While the DREAM complex appears to be a global repressor of cell cycle genes in quiescence, the extent of cell cycle gene regulation by the mammalian MuvB complex in association with B-Myb and its universe of targets has not been determined so far. Moreover, many genes required for mitotic progression, including CCNB1 and PLK1, are known to be regulated by other transcription factors, such as FoxM1. Hence, determining whether B-Myb and FoxM1 cooperate or act independently in regulating these genes is important for understanding the program of G2/M gene expression required for mitosis.
Here, we investigated the dynamics of the MuvB complex during the cell cycle. By ChIP sequencing of B-Myb and LIN9 and cell cycle expression profiling, we determined that many late cell cycle genes were direct targets of the B-Myb–MuvB complex. Furthermore, we identified a critical role for the MuvB complex and B-Myb in recruitment of FoxM1 to promoters of late cell cycle genes, thereby unifying two pathways regulating the program of mitotic gene expression and revealing a central role for the MuvB complex in regulating cell cycle gene expression from quiescence into mitosis.
Results
B-Myb binds to the MuvB core complex during S phase
The MuvB core complex associates with p130 and B-Myb during the cell cycle. To determine when in the cell cycle the MuvB core binds to p130 and B-Myb, we performed immunoprecipitation (IP) followed by Western blot with lysates from cells synchronized throughout the cell cycle. Primary IMR-90 fibroblasts were serum-starved to enrich for G0 and then fed with serum and aphidicolin to block cells in G1/S. Cells released from aphidicolin block for 5 and 10.5 h were enriched for S phase and G2/M, respectively (Supplemental Fig. S1A). Western blots of cell lysates revealed that B-Myb was expressed during the aphidicolin block (B) and during S phase (R5), but was absent in G0 and reduced in G2/M (R10.5) (Fig. 1A). B-Myb was coprecipitated by LIN9 during the aphidicolin block and in S phase but not during G0 and was markedly reduced in G2/M, consistent with its expression pattern during the cell cycle. In contrast, LIN9 coprecipitated p130 during G0 but not during S phase or G2/M (Fig. 1A), consistent with the observation that the DREAM complex was only detectable during the G0 quiescent state (Litovchick et al. 2007).
Figure 1.

The switch from DREAM to B-Myb–MuvB is accompanied by dynamic changes in the promoter occupancies of these complexes on DREAM target promoters. (A) Lysates from IMR-90 cells in serum-starved quiescence (G0), or released from G0 by serum readdition and blocked in early S phase with aphidicolin (24 h treatment, B) and released from the aphidicolin block for 5 and 10.5 h (R5 and R10.5) were immunoprecipitated with antibodies to LIN9 followed by Western blot. (*) Nonspecific band. (B) Lysates prepared from HeLa cells blocked in early S phase by double thymidine block (0) and then released from the block for the indicated times (2, 5, 6, 7, and 8 h) were immunoprecipitated with antibodies to B-Myb and LIN9 followed by Western blot. (C) Lysates from T98G cells in serum-starved quiescence (G0) and 10 or 18 h after serum addition (S10 and S18) were immunoprecipitated with antibodies to LIN9 and B-Myb followed by Western blot. (*) Nonspecific band. (D–F) Chromatin from cells as in C was immunoprecipitated with antibodies to E2F4, LIN9, and B-Myb. Differential site occupancy on DREAM target promoters indicated in the X-axis across G0, G1/S, and S was normalized to G0 values. Average values and SD (standard deviation) from three biological replicates are shown.
We explored the cell cycle-dependent association of B-Myb and the MuvB core in other cell types using different methods of synchronization. HeLa cells were synchronized at the G1/S boundary by double thymidine block and then released into the cell cycle (Supplemental Fig. S1B). Similar to what was observed in IMR-90 cells, B-Myb protein levels were highest in HeLa cells during S phase and decreased during G2 and M (Fig. 1B, input). Conversely, LIN9 was expressed throughout the S, G2, and M phases. IP for B-Myb and LIN9 revealed that their binding to each other was restricted to S phase (Fig. 1B). LIN54 also coprecipitated B-Myb together with LIN9 and LIN37, indicating that B-Myb bound to the MuvB core complex (Supplemental Fig. S1C). Notably, no p130 was coprecipitated by LIN9 or LIN54 in these experiments, consistent with the absence of a quiescent state in HeLa cells. In contrast, T98G cells exit into a G0 quiescent state when serum-starved and can re-enter the cell cycle upon serum addition (Supplemental Fig. S1D). An IP for LIN9 readily coprecipitated p130 from quiescent T98G cells but had reduced binding to p130 in extracts from serum-stimulated S-phase cells (Fig. 1C). B-Myb was poorly expressed in G0 but was markedly increased in S phase, with strong LIN9 coprecipitation apparent in the S-phase extracts but not in the G0 or G1/S extracts (Fig. 1C). These results indicate that B-Myb binds to the MuvB core complex during the S phase of the cell cycle in multiple cell types. In addition, they validate the use of HeLa cells for studying the B-Myb–MuvB complex in isolation from the p130/DREAM complex, and of T98G or IMR90 cells for studying the transition from p130/DREAM to B-Myb–MuvB.
B-Myb–MuvB binds to a subset of DREAM targeted promoters during S phase
During G0, the DREAM complex binds specifically to the promoters of >800 cell cycle genes (Litovchick et al. 2007). We examined the promoters of four genes—E2F1, CDC6, CCNB1, and PLK1—repressed by the DREAM complex during G0 (Litovchick et al. 2007) but whose expression patterns upon cell cycle entry are distinct. Expression of E2F1 and CDC6 increases early in the cell cycle and peaks during G1/S, while CCNB1 and PLK1 expression is late during the cell cycle and peaks during G2/M (Whitfield et al. 2002). We wanted to determine whether the switch of the MuvB core complex binding from p130–E2F4 in G0 to B-Myb in S phase affected binding to these promoters. We performed ChIP for E2F4 and LIN9, two components of the DREAM complex, with chromatin isolated from quiescent and serum-stimulated T98G cells. As expected, the occupancy of E2F4 on all four DREAM target promoters was readily detected in G0 cells, but diminished to background levels as cells progressed into G1 and S phase (Fig. 1D). Similarly, LIN9 bound to all four promoters in G0 cells, and its binding also decreased to near background levels during G1/S (Fig. 1E). However, LIN9 binding reappeared on the CCNB1 and PLK1 promoters during S phase (S18), with occupancy levels comparable with those observed in G0. In contrast, LIN9 did not bind to the E2F1 and CDC6 promoters during S phase (Fig. 1E). Using antibodies specific for LIN54, another component of the MuvB core complex, chromatin-binding profiles similar to LIN9 were obtained (Supplemental Fig. S2). Given that LIN9 could coprecipitate B-Myb in extracts from S-phase cells (Fig. 1C), we determined whether B-Myb could also bind to these DREAM target promoters. B-Myb was enriched on the CCNB1 and PLK1 promoters during S phase (S18) but not on the E2F1 and CDC6 promoters (Fig. 1F). Together, these results indicate a dynamic association of the MuvB core complex and B-Myb on a subset of DREAM targeted promoters.
Genome-wide identification of B-Myb–MuvB targets
To identify the universe of genes targeted by B-Myb and MuvB, we performed ChIP-seq (ChIP followed by deep sequencing) for B-Myb and LIN9. The specificity of the antibodies for the targets was verified by IP-Westerns and mass spectrometry (Litovchick et al. 2007). Two biologically independent replicate experiments for B-Myb, LIN9, and input DNA control were performed using chromatin prepared from proliferating HeLa cells. The total number of reads obtained from these ChIP-seq experiments ranged from 18 million to 28 million, and 60%–90% of the reads could be uniquely aligned with the human genome (Supplemental Table S1). Visualization using the University of California at Santa Cruz (UCSC) Genome Browser revealed that specific regions were enriched for reads in both LIN9 and B-Myb samples compared with input chromatin. Strikingly, many gene loci, including the promoter regions of CCNB1 and PLK1, showed enrichment of both LIN9 and B-Myb ChIP-seq reads (Supplemental Fig. S3A,B). In contrast, E2F1 and CDC6 promoter regions revealed no significant enrichment of LIN9 or B-Myb ChIP-seq reads (Supplemental Fig. S3C,D). The absence of LIN9 binding to the E2F1 and CDC6 promoters is consistent with the lack of interaction between LIN9 and p130 in proliferating HeLa cells.
To assess the extent of reproducibility between the two biological replicates, ChIP-seq reads from each experiment were used to call peaks individually using CisGenome (Ji et al. 2008). We found that the top 40% of peaks identified from the first ChIP-seq experiment for LIN9 and B-Myb overlap by 57% and 59%, respectively, with the complete set of results from the biological replicate for LIN9 and B-Myb ChIP-seq, suggesting reasonable concordance between the two replicates. ChIP-seq reads from the replicates for LIN9 or B-Myb were then pooled to call peaks using CisGenome (Ji et al. 2008). All peaks with a false discovery rate (FDR) < 0.1 and having at least 10 unique reads were considered as regions that were significantly bound by B-Myb or LIN9. Using these criteria, 2777 LIN9-bound and 2056 B-Myb-bound regions were identified (Supplemental Table S2). Notably, 61% of all B-Myb-bound regions were also bound by LIN9, and 45% of all LIN9-bound regions were also bound by B-Myb. A total of 1247 regions were bound by both B-Myb and LIN9 (Fig. 2A; Supplemental Table S3). In addition to having a significant number of shared targets, a rank correlation plot of LIN9-bound versus B-Myb-bound regions revealed a strong correlation in their binding to these shared set of targets, consistent with the model that B-Myb cooperated with the MuvB core complex for specific DNA binding (Fig. 2B).
Figure 2.

Genome-wide in vivo binding sites of LIN9 and B-Myb overlap significantly and are highly enriched in promoters of mitotic genes. (A) Venn diagram showing the overlap between significant peaks (FDR < 0.1) for LIN9 and B-Myb binding, with size and overlap drawn to scale. Numbers indicate the number of binding sites (or peaks) for each factor. LIN9 is shown in light blue, B-Myb is in green, and the overlap is in yellow. The color scheme of LIN9 in light blue and B-Myb in green is preserved throughout the rest of the figures. (B) Rank correlation plot of sites bound by B-Myb and LIN9. Sites bound by both factors are shown as black circles, sites bound by LIN9 only are shown in blue, and sites bound by B-Myb only are shown in green. The rs value represents the Spearman's rank correlation coefficient of the overlapping sites. (C) Location of regions bound by B-Myb and LIN9 relative to the TSS. 0 indicates TSS, negative values indicate regions upstream of the TSS, and positive values indicate regions downstream from the TSS. (D) Genes targeted by both B-Myb and LIN9 or uniquely targeted by any one factor were functionally annotated using DAVID gene ontologies, biological pathway (BP), and cellular component (CC). The top two functional clusters obtained using each of the three gene sets are indicated along with the P-values.
B-Myb–MuvB targets are enriched for mitotic genes
To determine whether B-Myb–MuvB bound to gene promoters or other regulatory regions, genomic locations bound by B-Myb and LIN9 were mapped with respect to known transcription start sites (TSSs). This location analysis revealed that densities of B-Myb and LIN9 ChIP-seq peaks were highest near the TSS (Fig. 2C). We annotated 1376 LIN9 and 1215 B-Myb target genes from the corresponding ChIP-seq peaks (Supplemental Tables S2, S3) and determined the extent of overlap between the target genes of the two factors. This analysis revealed that 828 genes were common targets of both B-Myb and LIN9 (Fig. 2D). Analysis of this set of common target genes using the DAVID (Database for Annotation, Visualization, and Integrated Discovery) gene ontology program showed significant enrichment for genes with functional roles in mitosis (Fig. 2D; Supplemental Table S4). In contrast, the analysis of genes that were uniquely targeted by B-Myb or LIN9 yielded fewer functional clusters with much higher P-values (lower significance) compared with those obtained with the common gene set (Fig. 2D; Supplemental Table S4).
We validated the binding of B-Myb and LIN9 to promoters of several newly identified target genes using ChIP-qPCR. B-Myb and LIN9 ChIPs were enriched for all of the target promoters tested but not for a control Actin promoter in both HeLa and T98G cells (Supplemental Figs. S4A,B). To determine whether expression of these genes was also dependent on the presence of both B-Myb and the MuvB core complex, we performed siRNA-mediated depletion of B-Myb, LIN9, and LIN54 in HeLa cells. RT-qPCR revealed that knockdown of LIN9, LIN54, or B-Myb resulted in decreased expression levels of PLK1, CENPF, CCNB1, ASPM, and GTSE1, as well as several additional target genes (Supplemental Fig. S4C). In contrast, expression levels of CDC6 and E2F1 were unaffected by these knockdowns, consistent with the observation that promoters of these genes were not bound by the B-Myb–MuvB complex (Supplemental Fig. S3C,D).
B-Myb–MuvB target genes are predominantly expressed late in the cell cycle
Many genes required for mitosis are expressed exclusively during G2/M (Whitfield et al. 2002). To determine whether B-Myb–MuvB targets have peak expression at specific points during the cell cycle, we generated expression profiles from three independent replica experiments of HeLa cells synchronized by thymidine block and release and compared these profiles with ChIP-seq data obtained for B-Myb and LIN9. A box plot of the average expression of B-Myb and LIN9 target genes identified by ChIP-seq indicated that these genes had higher expression at 4, 6, and 8 h, corresponding to late S, G2, and M phases, respectively, compared with 0, 2, and 12 h, corresponding to G1/S, early S, and G1 phases (Fig. 3A). Gene set enrichment analysis (GSEA) comparing the expression of B-Myb and LIN9 target genes at the 6-h versus 0-h time points revealed strong enrichment for genes with peak expression at 6 h (G2/M phase) compared with 0 h (early S phase) (Fig. 3B). These results demonstrate that the genes whose promoters are bound by B-Myb and the MuvB core complex have peak expression during the G2/M phase of the cell cycle.
Figure 3.

B-Myb–MuvB binds to a subset of DREAM targets expressed late in the cell cycle. (A) Box plot depicting the average expression of 278 B-Myb and LIN9 target genes, defined by FDR < 0.001 and fold change > 5, in HeLa cells after double thymidine block and release. Expression values of the 278 target genes derived from microarray analysis of the 0-, 2-, 4-, 6-, 8-, and 12-h time points were averaged. The difference in the means of the three replicates for each time point is displayed as a box plot. (B) GSEA was used to test whether the 278 B-Myb and LIN9 target genes (same as in A) were randomly distributed or tended to occur toward the extremes of the list of genes differentially expressed between 6 and 0 h after double thymidine block and release. The enrichment score of 0.6 reflects the degree to which these B-Myb and LIN9 target genes are overrepresented at the extreme top of the entire ranked list of genes differentially expressed between the 6- and 0-h time points. (C) Heat maps depicting the expression of a cluster of genes with an expression pattern similar to PLK1 (late cell cycle cluster) or CDC6 (early cell cycle cluster) at different times after double thymidine block and release. Genes in the clusters that were targets of DREAM (Litovchick et al. 2007) or B-Myb–MuvB (this study) are indexed by black lines on either side of the heat map. P-values of overlaps (Fisher's exact test) are also indicated. (D) Box plot depicting the average expression of 92 genes that are common targets of DREAM and B-Myb–MuvB. The analysis description is the same as in A. (E) GSEA plot comparing the expression of 92 target genes common to DREAM and B-Myb–MuvB at the 6- and 0-h time points. The analysis description is the same as in B. Arrows in A, C, and D indicate when mitosis was observed.
B-Myb–MuvB targets include a subset of DREAM targets expressed late in the cell cycle
Next, we sought to determine whether the B-Myb–MuvB target genes were a subset of the DREAM target genes. Using CDC6 and PLK1 expression patterns as standards for early and late cell cycle genes, respectively (Supplemental Fig. S5A), clusters of genes with cell cycle expression similar (with a correlation of >0.9) to these standards were generated. Genes in these clusters were overlapped with DREAM and B-Myb–MuvB targets identified by ChIP–chip (Litovchick et al. 2007) and ChIP-seq, respectively. This analysis revealed that B-Myb–MuvB targets were significantly enriched in the late cell cycle cluster compared with the early cell cycle cluster (P-value 9.06 × 10−12 vs. 0.006) (Fig. 3C). In contrast, DREAM targets were equally and significantly enriched in both clusters (P-value 7.17 × 10−16 vs. 2.2 × 10−16) (Fig. 3C). Similar results were obtained when E2F1 or RFC4 and CCNB1 or Cyclin A2 expression patterns were used as standards to derive the early and late cell cycle clusters, respectively (Supplemental Fig. S5A,B). To explore these differences, we compared the overall cell cycle-dependent expression of 92 genes that were common targets of DREAM and B-Myb–MuvB. Box plots showing the average expression of this gene set in different cell cycle phases and GSEA comparing their expression at the 6-h versus 0-h time points indicated that these genes had higher expression during G2 and M phases compared with early S and G1 phases of the cell cycle (Fig. 3D,E). These results indicate that the B-Myb–MuvB targeted genes represent a functional subset of the DREAM targeted genes expressed during the late cell cycle in G2/M.
Reciprocal requirement of B-Myb and MuvB for binding to promoters
Given the high degree of overlap in B-Myb and LIN9 target genes, we asked whether B-Myb and the MuvB core required each other for specific binding to promoters using specific siRNAs targeting LIN9, LIN54, and B-Myb in HeLa cells. The knockdown for each factor was highly effective, as determined by RT-qPCR, and did not affect the mRNA levels of the other members of the complex (Fig. 4A). Notably, knockdown of LIN54 or B-Myb did not affect the protein levels of each other or of LIN9. In contrast, LIN9 depletion led to decreased protein levels of LIN9 as well as LIN54 and B-Myb (Fig. 4B). Given these observations, we depleted LIN54 and performed an IP for LIN9 and found coprecipitation with LIN37, LIN52, and B-Myb, indicating that LIN9 and the other MuvB components could retain binding to B-Myb in the absence of LIN54 (Fig. 4C). Although LIN9 was capable of coprecipitating B-Myb in LIN54-depleted cells, LIN9 and B-Myb binding to the PLK1 promoter was markedly reduced (Fig. 4D). Furthermore, although LIN9 and LIN54 protein levels were unaffected by B-Myb depletion, their association with the PLK1 promoter was also significantly reduced in the absence of B-Myb (Fig. 4D). We observed similar effects with binding to the CCNB1 promoter in LIN54-depleted cells (Supplemental Fig. S6A). Therefore, B-Myb and the MuvB core complex require each other for specific binding to target promoters.
Figure 4.

B-Myb and LIN54 are both required for the B-Myb–MuvB complex to bind to the PLK1 promoter. (A) RNA isolated from untransfected (U), control (siCon), and LIN9, LIN54, or B-Myb siRNA transfected HeLa cells was analyzed by RT-qPCR with specific primers for genes indicated in the X-axis. The fold change was calculated relative to GAPDH mRNA levels and plotted with respect to the value for the untransfected sample, which was taken as 1. (B) HeLa cells were transfected with the indicated siRNAs to B-Myb and MuvB or control siRNA (Con) or left untransfected (U). The expression of B-Myb, LIN54, LIN9, and Vinculin (loading control) in whole-cell lysates was detected by Western blot. (C) Lysates from untransfected (U), control (Con), or LIN54 siRNA transfected HeLa cells were immunoprecipitated with LIN9 antibody. Protein levels of the MuvB proteins and B-Myb were assayed by Western blots. (D) Chromatin from HeLa cells transfected with control siRNA (siCon), B-Myb siRNA (siB-Myb), or LIN54 siRNA (siLIN54) was immunoprecipitated with antibodies to LIN9, LIN54, B-Myb, and Histone H3. ChIP enrichment of the PLK1 promoter was calculated relative to siCon. ChIP for H3 was used as a control to compare ChIP efficiencies between the two sets of chromatin. Average values and SD from three experimental data points are shown.
B-Myb phosphorylation correlates with the start but not the peak of target gene expression in vivo
To determine whether B-Myb and MuvB chromatin binding was linked to the cell cycle-dependent expression of target genes, we prepared chromatin and RNA from HeLa cells synchronized by double thymidine block and release. ChIP for LIN54 revealed that its binding to the PLK1 promoter was enriched throughout S phase and G2 (Fig. 5A). In contrast, ChIP for B-Myb revealed that its binding to the PLK1 promoter was enriched during S phase and then rapidly diminished during G2 to reach its lowest levels during mitosis (Fig. 5A). Notably, B-Myb bound to the PLK1 promoter during S phase when PLK1 mRNA levels were low (0-, 2-, and 4-h time points in Fig. 5A), followed by the loss of its binding during peak PLK1 expression in G2/M (7- and 8-h time points in Fig. 5A). Similar temporal binding profiles were also obtained for B-Myb on the CCNB1 promoter (Supplemental Fig. S6B). These results revealed that occupancies of B-Myb and LIN54 on the PLK1 promoter reached maximal levels during S phase, and while LIN54 binding persisted in G2, B-Myb binding was lost. This suggested that once recruited to promoters in a B-Myb-dependent manner, LIN54 continued to bind to these promoters even after B-Myb binding was lost.
Figure 5.

B-Myb phosphorylation at the end of S phase correlates with the start of target gene expression. (A) Chromatin and RNA from HeLa cells synchronized by double thymidine block and released for the indicated times were collected. Chromatin was used for ChIP with B-Myb and LIN54 antibodies to determine the enrichment of the PLK1 promoter at different time points. RNA was used to determine the level of expression of PLK1 relative to GAPDH. Average values and SD from three experimental data points are shown. (B) Lysates prepared from HeLa cells treated with DMSO or Velcade (1 μM) 2 h after release from double thymidine block and allowed to progress for 6 h (i.e., 8 h after release from the block) were Western-blotted with the indicated antibodies. (*) Nonspecific band. (C) Lysates from HeLa cells at several intervals after double thymidine block and release were Western-blotted or immunoprecipitated with a phospho-specific antibody to B-Myb that recognized pT487. Western blots of the pT487 IP were used to detect the presence of coprecipitated B-Myb and MuvB. (D) Lysates from HeLa cells at several intervals after double thymidine block and release were immunoprecipitated with LIN54 antibody and Western-blotted to detect pT487 B-Myb and the rest of the MuvB core proteins. (E) Chromatin from HeLa cells after double thymidine block and release was used for ChIP with total B-Myb and phospho-specific pT487 B-Myb antibodies to determine the enrichment of the PLK1 promoter. RNA collected at the same intervals was assayed to determine the level of expression of PLK1 relative to GAPDH. Average values and SD from three experimental data points are shown. Arrows indicate when mitosis was observed.
Decreased binding of B-Myb to the PLK1 and CCNB1 promoters during G2 and M phases correlated with reduced B-Myb protein levels (Fig. 5B; Supplemental Fig. S7A). The loss of B-Myb expression and binding to CCNB1 and PLK1 promoters during G2 and M phases could be rescued by treatment of cells with proteasome inhibitors (Fig. 5B; Supplemental Fig. S7A,B), indicating that B-Myb was degraded in a proteasome-dependent manner during G2.
Previous studies have reported that B-Myb is phosphorylated during S phase (Robinson et al. 1996) and that its phosphorylation by Cyclin A can promote both B-Myb-dependent transactivation in reporter assays and B-Myb degradation (Lane et al. 1997; Sala et al. 1997; Johnson et al. 2002). To explore this, lysates prepared from synchronized HeLa cells were treated with λ phosphatase. Western blots for B-Myb revealed the presence of slower-migrating forms of B-Myb at 4 and 5 h after release from thymidine block that were sensitive to phosphatase treatment (Supplemental Fig. S7C). Western blot with an antibody specific for B-Myb phosphorylated on residue Thr 487 (pT487) detected a strong signal for phosphorylated B-Myb at 4 and 5 h after release from thymidine block (Fig. 5C, top panel). To determine whether B-Myb phosphorylation affected binding to the MuvB core complex, we performed IP for pT487 B-Myb followed by Western blot. The MuvB proteins were robustly coprecipitated by pT487 B-Myb at 4 and 5 h and to a somewhat lesser extent at 6 h after release from thymidine block (Fig. 5C). Similarly, an IP for LIN54 coprecipitated the phosphorylated form of B-Myb at 4 and 5 h after release from thymidine block (Fig. 5D). Although association with phosphorylated B-Myb was transient, LIN54 remained bound to other components of the MuvB core complex throughout this experiment (Fig. 5D). In addition, ChIP with the pT487 B-Myb antibody indicated that phosphorylated B-Myb was associated with the PLK1 promoter peaking at 4 h after release from the thymidine block and preceding the peak expression of PLK1 mRNA (Fig. 5E). These results demonstrate that phosphorylation of B-Myb during S phase does not disrupt coprecipitation with the MuvB core complex or specific binding to target promoters and immediately precedes the degradation of B-Myb, the loss of B-Myb binding to promoters, and the peak expression of PLK1 and CCNB1 mRNA during G2/M.
Motif analysis of B-Myb and LIN9 ChIP-seq regions identifies FoxM1 as a transcriptional co-regulator of late cell cycle genes
Because B-Myb binding to promoters occurred prior to the corresponding peak of target gene expression, we suspected that transcription factors in addition to B-Myb might contribute to activating late cell cycle gene expression. We further reasoned that if this was the case, promoters of target genes may contain consensus binding sites for these transcription factors coenriched with B-Myb- and MuvB-binding sites. Therefore, we sought to identify motifs enriched in B-Myb- and LIN9-bound genomic regions. All significantly bound target regions with FDR < 0.01 were used for de novo motif discovery. Consensus sequences for Myb, CHR (cell cycle gene homology region), FoxM1, NF-Y, and AP-1 were among the top 10 de novo motifs identified (Fig. 6A). Remarkably, each of these five motifs was identified independently as significantly enriched in both the B-Myb and LIN9 ChIP-seq data sets (Supplemental Table S5). De novo identification of the Myb and CHR consensus sequences, the latter recently shown to be bound by LIN54 (Schmit et al. 2009), provided additional validation that B-Myb and LIN9 bound specifically and cooperatively to these promoters.
Figure 6.

FoxM1 is required for late cell cycle gene expression and binds to MuvB during G2 and mitosis. (A) De novo motif discovery identifies consensus sequences for Myb, CHR, FoxM1, NF-Y, and AP-1 as significantly enriched in genomic regions targeted by B-Myb and LIN9. Web logos representing motifs that were derived from B-Myb- or LIN9-bound genomic regions are shown. (B) Chromatin from asynchronous HeLa cells was used for ChIP with FoxM1 antibody followed by qPCR to amplify promoter regions of a panel of cell cycle genes indicated in the X-axis. Enrichments were calculated as percentage of the total input chromatin. Average values and SD from three experimental data points are shown. (C) RNA was isolated from siRNA transfected HeLa cells blocked in thymidine and released for different times (0, 2, 4, 6 and 8 h) and was analyzed by RT-qPCR with primers specific for CCNB1. The fold change was calculated relative to GAPDH mRNA levels. Average values and SD from three experimental data points are shown. (D) Lysates from untransfected (U) or FoxM1 siRNA (siFoxM1) transfected HeLa cells were immunoprecipitated with antibodies to FoxM1 and rabbit IgG (IgG). The presence of LIN9 and LIN37 was assayed by Western blot. The bottom panel shows protein levels of FoxM1, LIN37 and LIN9 in the lysates that were used for IP. (Right panel) Lysates from asynchronously growing HeLa cells were immunoprecipitated with two different antibodies to FoxM1 (Ab-1 and Ab-2) and rabbit IgG (IgG). The presence of FoxM1, LIN9, LIN37, and LIN52 was assayed by Western blot. (*) Antibody chains. (E) T7-FoxM1 or Myc-FoxM1 transfected cells (+) or untransfected cells (−) were left untreated (Asynchronous) or, 48 h later, were treated with nocodazole at a concentration of 1μg/mL for 18 h. Lysates from these cells were used for IP with T7 tag or Myc tag antibody-conjugated agarose beads. Protein levels of T7-FoxM1, Myc-FoxM1, LIN9, and LIN37 were assayed by Western blots. (*) IgG heavy chain. (F,G) Lysates from HeLa cells at several intervals after double thymidine block and release were immunoprecipitated with the LIN9 antibody (F) and two different FoxM1 antibodies (G) and were Western-blotted to detect FoxM1, B-Myb, and the MuvB proteins LIN9 or LIN37. (*) IgG light chain. Arrows in the panels indicate when mitosis was observed.
NF-Y and FoxM1 are both known to be regulators of cell cycle gene expression (Caretti et al. 2003; Laoukili et al. 2005; Wang et al. 2005). To determine whether these factors cooperated specifically with B-Myb–MuvB, we analyzed their target genes. We analyzed a previously reported ChIP–chip experiment for NF-Y target genes (Reed et al. 2008) and found significant overlap with DREAM target genes (P-value of overlap = 2.20 × 10−16). Notably, 355 genes shared by DREAM and NF-Y revealed marked enrichment for cell cycle genes by DAVID gene ontology (Supplemental Fig. S8A,B). Within this group were genes involved in mitosis as well as DNA replication, indicating that both early and late cell cycle genes were targeted by NF-Y (Supplemental Fig. S8B). To confirm this observation, we performed ChIP with an antibody for NF-YB and observed specific binding to both early (CDC6 and E2F1) and late (CCNB1 and RACGAP1) cell cycle gene promoters (Supplemental Fig. S8C), indicating that NF-Y-dependent transactivation was unlikely to be specific for the late cell cycle genes. In contrast, ChIP for FoxM1 revealed specific enrichment for promoters of several late cell cycle genes (PLK1, AURKA, RACGAP1, and CCNB1), but not promoters of early cell cycle genes (CDC6 or E2F1) (Fig. 6B). This is consistent with prior reports that FoxM1 could bind specifically to the promoters of at least 10 genes linked to mitosis (Laoukili et al. 2005; Wang et al. 2005).
Given the ability of FoxM1 to bind specifically to the same promoters bound by B-Myb–MuvB, we compared how their knockdown affected target gene expression. Knockdowns for B-Myb and FoxM1 were highly effective (Supplemental Fig. S9). We also observed that FoxM1 mRNA levels were modestly reduced by knockdown of B-Myb, while B-Myb levels were slightly increased upon FoxM1 knockdown (Supplemental Fig. S9). We transfected siRNAs targeting B-Myb, LIN9, LIN54, or FoxM1 or nontargeting control siRNA and performed double thymidine block and release followed by RT-qPCR for CCNB1 mRNA. Knockdown of B-Myb, LIN9, LIN54, or FoxM1 resulted in decreased expression levels of CCNB1 compared with nontargeting control siRNA (Fig. 6C). Knockdown of FoxM1 and B-Myb together had similar effects compared with the individual knockdowns of FoxM1, LIN9, or LIN54 and was only slightly more efficient in lowering CCNB1 mRNA levels than the knockdown of B-Myb alone (Fig. 6C). The absence of an additive effect upon knockdown of both B-Myb and FoxM1 suggested the possibility that these factors fall into a common pathway regulating late cell cycle gene expression.
MuvB proteins bind to FoxM1 during G2 and M phases of the cell cycle
Results from motif analysis of B-Myb and LIN9 ChIP-seq (Fig. 6A) were consistent with previous bioinformatics analysis that has indicated enrichment of the CHR, NF-Y, and B-Myb triplet module in promoters of the late cell cycle genes (Linhart et al. 2005). However, the FoxM1 consensus has not been previously linked with any of these motifs. Analysis of high-confidence FoxM1 and CHR motifs in B-Myb–MuvB targeted promoters revealed an unanticipated co-occurrence of these two motifs (Supplemental Fig. S10). The close proximity of FoxM1 and CHR sites prompted us to test whether FoxM1 and the MuvB complex associated with each other. An IP for FoxM1 coprecipitated LIN37 and LIN9 in extracts from asynchronously growing HeLa cells (Fig. 6D). Depletion of FoxM1 by siRNA reduced FoxM1 protein levels without affecting the levels of LIN37 and LIN9 (Fig. 6D, input panel) and effectively abolished the coprecipitation between FoxM1 and LIN37 (Fig. 6D), demonstrating specificity of the interaction. In addition, both LIN9 and LIN37 were coprecipitated by two different FoxM1 antibodies, and LIN52, another component of the MuvB core complex, was coprecipitated by one of the FoxM1 antibodies (Fig. 6D, right panel). Furthermore, exogenously expressed T7-FoxM1 coprecipitated LIN9 and LIN37 from asynchronous HeLa cells as well those treated with nocodazole to enrich for mitosis (Fig. 6E). Similar results were obtained using Myc-tagged FoxM1 (Fig. 6E, right panel).
We then examined whether the FoxM1–MuvB association occurred in particular phases or throughout the cell cycle. An IP for LIN9 coprecipitated FoxM1 predominantly at 6 h (G2) and 8 h (mitosis) after release from double thymidine block (Fig. 6F). Little, if any, FoxM1 was coprecipitated by LIN9 during the block in early S phase (0 h) or during S phase (2 and 4 h after release). This contrasted with the coprecipitation of B-Myb that was detected during S phase but reduced in G2 and mitosis (Fig. 6F). LIN37 was coprecipitated by LIN9 in all of the cell cycle phases (Fig. 6F), consistent with our previous observation that interaction between members of the MuvB core complex remained intact throughout the cell cycle (Fig. 5D). Importantly, two different antibodies to FoxM1 coprecipitated LIN37 robustly at the 4-, 6-, and 8-h time points (Fig. 6G). These results demonstrate a specific interaction between the MuvB complex and FoxM1 during late S phase, G2, and mitosis.
B-Myb and MuvB recruit FoxM1 to activate late cell cycle gene expression
FoxM1 undergoes progressive phosphorylation during the cell cycle, which in turn is required for its transcriptional activity (Fu et al. 2008; Laoukili et al. 2008; Chen et al. 2009). Consistent with this, we observed mobility shifts in FoxM1 indicative of phosphorylation at 4, 6, and 8 h after release from double thymidine block (Figs. 6G, 7A). At these times, B-Myb also underwent extensive phosphorylation followed by proteasome-mediated degradation (Fig. 5B,C). We tested whether FoxM1 phosphorylation was affected by B-Myb or LIN9 depletion and, vice versa, whether B-Myb degradation was affected by FoxM1 depletion. The appearance of FoxM1 phosphorylation after double thymidine block and release was slightly delayed in B-Myb siRNA knockdown cells, but reached levels comparable with nontargeting control siRNA transfected cells at 6 and 8 h after release from the thymidine block (Fig. 7A). In addition, FoxM1 phosphorylation appeared to be slightly attenuated in LIN9 siRNA transfected cells (Supplemental Fig. S11). B-Myb degradation at the 6- and 8-h time points was unaffected by FoxM1 (Fig. 7A) or LIN9 knockdown (Supplemental Fig. S11). Notably, increases in CCNB1 protein levels were delayed and reduced during progression from S to G2/M in B-Myb, LIN9, and FoxM1 siRNA transfected cells compared with control (Fig. 7A; Supplemental Fig. S11), mirroring the effect observed at the mRNA level (Fig. 6C).
Figure 7.

MuvB and B-Myb recruit FoxM1 to cooperatively activate late cell cycle gene expression. (A) Whole-cell lysates were isolated from siRNA transfected HeLa cells blocked in thymidine and released for different times (0, 2, 4, 6, and 8 h) and were Western-blotted with the indicated antibodies. Vinculin was used as a loading control. (B) Chromatin from HeLa cells after double thymidine block and release was used for ChIP with B-Myb, LIN9, and FoxM1 antibodies. ChIP DNA was used to amplify a region from the promoter of PLK1 (top) and CCNB1 (bottom). Average values and SD from three experimental data points are shown. (C) Chromatin from asynchronously growing HeLa cells was used to immunoprecipitate LIN9 or pT487 B-Myb. Immunoprecipitated chromatin was used for reChIP with either no antibody (control) or antibodies to B-Myb, pT487 B-Myb, LIN54, and FoxM1. Average values and SD from three biological replicates are shown. The enrichment values are expressed as a percentage of the first ChIP (LIN9 or pT487 B-Myb). (D) Chromatin from HeLa cells transfected with control siRNA (siCon), B-Myb siRNA (siB-Myb), LIN9 siRNA (siLIN9), or FoxM1 siRNA (siFoxM1) was immunoprecipitated with antibodies to B-Myb and FoxM1. ChIP enrichments of the PLK1 and CCNB1 promoters were calculated relative to siCon. Average values and SD from three experimental data points are shown. (E) Schematic of our proposed model illustrating an essential role for the MuvB complex in regulating cell cycle gene expression from quiescence into mitosis.
We then determined whether the binding of FoxM1 to promoters was cell cycle-dependent. ChIP with antibodies to FoxM1 revealed specific binding to the PLK1 promoter at 4, 6, and 8 h after release from double thymidine block, peaking slightly after B-Myb binding (Fig. 7B). Importantly, FoxM1 ChIP showed persistent binding to the PLK1 promoter at the 6- and 8-h time points, when B-Myb occupancy was decreased (Fig. 7B). LIN9 binding to the PLK1 promoter peaked concurrently with B-Myb binding, but in contrast to the dramatic loss of B-Myb binding in G2, LIN9 binding to the PLK1 promoter persisted during G2 (Fig. 7B). Similar results were obtained on the CCNB1 promoter (Fig. 7B, bottom panel). These results are consistent with our previous observation that LIN54 remained on the PLK1 promoter after B-Myb binding was lost (Fig. 5A). Together, these data suggest a MuvB complex-dependent handover of promoter occupancy from B-Myb during S phase to FoxM1 during G2/M, while the MuvB complex binding to the promoter remained largely unchanged. However, it was unclear whether B-Myb and FoxM1 bound to promoters in an overlapping manner or whether they bound mutually exclusively to their target promoters.
In order to distinguish between these two possibilities, we performed ChIP-reChIP using antibodies to LIN9, LIN54, B-Myb, and FoxM1. As expected, LIN54 was able to chromatin-immunoprecipitate to the CCNB1, PLK1, and AURKA promoters using chromatin obtained after the first ChIP with LIN9 or pT487 B-Myb (Fig. 7C). B-Myb or phosphorylated B-Myb was also able to chromatin-immunoprecipitate to these promoters using chromatin from the first ChIP with LIN9 (Fig. 7C), consistent with the model that the MuvB complex and B-Myb co-occupy target promoters. Importantly, FoxM1 was robustly enriched on the CCNB1, PLK1, and AURKA promoters in chromatin obtained after the first ChIP with LIN9 as well pT487 B-Myb (Fig. 7C), indicating that the MuvB complex and FoxM1 co-occupy target promoters and that FoxM1 and B-Myb binding can co-occur on promoters.
Given these observations, we tested whether the MuvB complex and B-Myb were required for the recruitment of FoxM1 to these promoters. Using chromatin from HeLa cells transfected with siRNAs specific to B-Myb, LIN9, and FoxM1, we performed ChIP for B-Myb and FoxM1. Consistent with earlier results (Fig. 4B,D), B-Myb binding to the PLK1 and CCNB1 promoters was significantly reduced in B-Myb and LIN9 siRNA transfected cells. However, B-Myb binding was unaffected by the depletion of FoxM1 (Fig. 7D). In sharp contrast, FoxM1 binding to the PLK1 and CCNB1 promoters was significantly reduced upon depletion of either B-Myb or LIN9, indicating that the MuvB complex and B-Myb were required for recruiting FoxM1 to these promoters (Fig. 7D).
These results are summarized in a model (Fig. 7E) that illustrates the timing and requirement of the B-Myb–MuvB complex and FoxM1 binding to promoters and their activation by phosphorylation for expression of the late cell cycle genes.
Discussion
In this study, we demonstrate that the MuvB core complex and B-Myb are directly involved in recruiting FoxM1 to promoters of the late cell cycle genes, providing a new rationale for their requirement in mitotic progression. These results unify two separate fields that have demonstrated important contributions from B-Myb and FoxM1 to mitosis. Furthermore, they suggest a far more ubiquitous role for the MuvB core complex in regulating cell cycle gene expression than previously suspected. The MuvB core, at a minimum, serves as a platform to recruit three distinct transcription factors to overlapping sets of cell cycle gene promoters during the various cell cycle phases. The MuvB core complex with p130–E2F4–DP1 binds during quiescence to the promoters of genes expressed early and late in the cell cycle, and the complex with B-Myb binds during S phase and the complex with FoxM1 binds during G2 and mitosis to late cell cycle gene promoters. Our results provide evidence for sequential recruitment—first of B-Myb and later of FoxM1—in a MuvB-dependent manner to promoters of genes expressed during the G2/M phase of the cell cycle. In addition, this study reveals that the CHR sequence, which is bound by LIN54 and the MuvB core complex, cooperates with the specific DNA-binding elements for B-Myb and FoxM1 to coordinate late cell cycle gene expression.
Using global approaches, we demonstrate that the transcriptional program governed by B-Myb, MuvB, and FoxM1 controls the expression of a large set of genes with peak expression during G2 and mitosis. Remarkably, more than a quarter of the genes with cell cycle expression similar to PLK1 or CCNB1 were identified as direct targets of B-Myb and LIN9 by ChIP-seq. Consistent with previous reports, knockdown of MuvB, B-Myb, or FoxM1 resulted in decreased expression levels of the targeted mitotic genes. For example, 32 out of 78 genes significantly down-regulated in LIN54-depleted cells (Kittler et al. 2007) and 14 out of 31 genes significantly down-regulated in LIN9-depleted cells (Osterloh et al. 2007) were direct targets of B-Myb and LIN9 in ChIP-seq experiments. These data indicate that the mitotic defects observed in B-Myb-depleted (Tarasov et al. 2008), MuvB-depleted (Kittler et al. 2007), or FoxM1-depleted (Laoukili et al. 2005) cells stem, at least in part, from the loss of their cooperative binding to the promoters of genes whose expression is required for mitosis.
In addition to mitosis, the MuvB core complex, as part of the DREAM complex, is required for exit from the cell cycle and entry into quiescence and senescence. This activity is dependent on the DYRK1A kinase-mediated phosphorylation of Ser 28 (S28) in LIN52. We showed that p130 binds specifically to the MuvB complex through an interaction mediated by phosphorylated residue S28-LIN52 (Litovchick et al. 2011). Alanine point substitution of this residue, S28A-LIN52, disrupted MuvB binding to p130 but did not affect binding to B-Myb. Although the C terminus of Drosophila B-Myb is necessary and sufficient for binding to MuvB (Andrejka et al. 2011), the specific requirements for MuvB binding to B-Myb or FoxM1 are not known. Notably, we demonstrated LIN54 is not required for B-Myb binding to LIN37, LIN9, and LIN52.
During quiescence, the MuvB core, as part of the DREAM complex, binds to the promoters of most, if not all, cell cycle-dependent genes (Litovchick et al. 2007). In contrast, the MuvB core complex, together with B-Myb or FoxM1, binds exclusively to the promoters of a subset of DREAM target genes that are expressed late in the cell cycle. Promoters of genes expressed during G1/S for the most part were not bound by B-Myb–MuvB or FoxM1–MuvB, indicating that distinct transcriptional mechanisms governed the expression of early and late cell cycle genes in proliferating cells. Previous studies have shown that expression of the early cell cycle genes was dependent on the activating E2F transcription factors E2F1, E2F2, and E2F3a (Wu et al. 2001). We found no evidence that the activating E2F transcription factors contributed to the expression of the late cell cycle genes. For example, de novo motif analysis of B-Myb and LIN9 ChIP-seq did not reveal enrichment for the E2F consensus sequence. Instead, our study demonstrates that expression of the late cell cycle genes is dependent on the integrative actions of at least three factors: B-Myb, FoxM1, and the MuvB complex.
Motif analysis of B-Myb and LIN9 ChIP-seq targeted genomic regions revealed consensus sites for multiple transcription factors that could serve as potential coregulators of the late cell cycle genes. Enrichment for the FoxM1 consensus site prompted us to investigate whether there was any dependency on B-Myb and MuvB, leading to the discovery of an interaction between FoxM1 and MuvB during G2 and mitosis on the B-Myb–MuvB targeted late cell cycle gene promoters. Interestingly, FoxM1 and C-Myb, a Myb family protein exclusively expressed in hematopoietic cells, were recently described as synergistic master regulators of proliferation in the germinal center (Lefebvre et al. 2010), supporting the idea of a pervasive link between FoxM1 and the Myb family of proteins. Identification of the CHR, NF-Y, and Myb motifs by ChIP-seq was consistent with the ChIP validation of LIN54, NF-Y, and B-Myb on promoters of cell cycle genes and with previous bioinformatics analysis that revealed enrichment of a CHR, NF-Y, and B-Myb triplet module in promoters of the late cell cycle genes, but not in promoters of the early cell cycle genes (Linhart et al. 2005). NF-Y appears to be a more general transcription factor, since it could bind to the promoters of both early and late cell cycle genes. NF-Y also has other roles that extend beyond the cell cycle; for example, it cooperates with SREBP1 to regulate genes important for lipid metabolism and insulin signaling (Reed et al. 2008). Enrichment of the AP-1 consensus in B-Myb–MuvB targeted regions was more curious. c-Jun and c-Fos, components of the AP-1 transcription factor, bind to several thousand sites in the human genome, and AP-1 sites are present within 100 base pairs (bp) of the vast majority of RNA polymerase II (Pol II)- and Pol III-regulated promoters (Raha et al. 2010). This suggests that AP-1 sites are ubiquitous at promoters and hence are enriched in B-Myb–MuvB targeted genomic regions. However, several recent studies have shown that JNK (c-Jun N-terminal kinase) activity and the phosphorylation of c-Jun, which increases AP-1 target gene transcription, are elevated during G2/M (Oktay et al. 2008; Gutierrez et al. 2010), raising the possibility that there could be a G2/M-specific component to AP-1 function that may potentially cooperate with B-Myb–MuvB.
The MuvB complex and its cognate CHR sequence may contribute to sequence-specific binding of B-Myb and FoxM1 to the late cell cycle promoters. The lack of B-Myb binding to promoters in the absence of LIN54 and vice versa is suggestive of cooperativity that is likely dependent on both B-Myb and CHR consensus sites on promoters. In the case of FoxM1, its winged-helix domain has been reported to have a surprisingly low affinity for the consensus element TAAACA (Littler et al. 2010). It is plausible that MuvB binding to its consensus sequence and to FoxM1 increases the specific affinity of FoxM1 for the late cell cycle promoters, thereby serving an essential role in recruitment of FoxM1 to the late cell cycle promoters.
It is clear from several lines of evidence, including the genome-wide ChIP data, that B-Myb and MuvB are critical for G2/M gene expression. Both the work reported here and that in previous literature have consistently observed decreased mRNA and protein levels of mitotic genes in B-Myb- or MuvB-depleted cells. The observation that mitotic gene expression is decreased after RNAi-mediated knockdown of B-Myb or MuvB has been interpreted to indicate that B-Myb serves as a transcriptional activator for late gene expression (Osterloh et al. 2007; Knight et al. 2009). An alternate explanation is suggested by the observation that B-Myb and MuvB are required for the recruitment of FoxM1. In this scenario, FoxM1 may serve as the specific transcriptional activator, while the B-Myb–MuvB complex serves to facilitate recruitment of FoxM1 to the late cell cycle gene promoters. Furthermore, the absence of late cell cycle gene expression during S phase suggests that the B-Myb–MuvB complex could serve as a specific repressor of the late cell cycle genes in addition to its role in recruiting FoxM1. Along these lines, the MuvB core complex may serve as a repressor when it is part of the DREAM complex during quiescence. Knockdown or loss of activity of MuvB components reduces the ability of cells to repress cell cycle genes upon entry into quiescence (Litovchick et al. 2007, 2011). Although there is no direct evidence that B-Myb and MuvB serve as repressors of late cell cycle gene expression during S phase, it is clear that expression appears to be delayed until B-Myb undergoes phosphorylation-dependent proteasome-mediated degradation, while FoxM1, together with MuvB, is present on promoters when these genes become fully activated. Further experiments are required to detail the mechanisms of action of the MuvB complex when bound to these three distinct transcription factors.
Interestingly, the mammalian B-Myb, MuvB, and FoxM1 pathway shares several similarities with the budding yeast Yox1, Mcm1, and Fkh2 pathway that controls the expression of mitotic genes. In this system, the Mcm1 protein appears to serve as a platform that can switch between states of repression and activation (for a preview, see Leatherwood and Futcher 2010). The Yox1–Mcm1 complex acts as a repressor, while the Fkh2–Mcm1 complex functions as an activator on the same promoter. Furthermore, transcriptional activation by Fkh2–Mcm1 is dependent on the loss of Yox1 (Pramila et al. 2002), drawing an interesting parallel with B-Myb degradation. However, there appear to be interesting differences as well. In contrast to our results, which indicate that FoxM1 and phospho-B-Myb can bind simultaneously to promoters, biochemical studies in yeast have revealed that Yox1 and Fkh2 may in fact directly compete for binding to the Mcm1 platform, suggesting that their association with Mcm1 could be mutually exclusive (Darieva et al. 2010).
Both Caenorhabditis elegans and Drosophila contain complexes similar to the mammalian DREAM complex, albeit with some interesting differences. The worm DRM (DP/Rb/MuvB) complex does not contain Myb, as there appears to be no Myb ortholog in C. elegans (Harrison et al. 2006). The fly dREAM (RBF, dE2F2, and dMyb-interacting proteins)/MMB (Myb–MuvB) complex contains dMyb, which is present in the same complex along with RBF1, RBF2, dE2F2, dDP, and the Myb-interacting proteins (Korenjak et al. 2004; Lewis et al. 2004). The dREAM/MMB complex simultaneously contains both fly orthologs of Rb and Myb, while in mammalian cells, p130–E2F4–DP1 and B-Myb bind mutually exclusively to the MuvB complex in a cell cycle-dependent manner (Litovchick et al. 2007; Pilkinton et al. 2007; Schmit et al. 2007). Interestingly, ChIP–chip studies of the fly complex revealed that although dE2F2 and dMyb were part of the same complex, they functioned as mutually exclusive targeting factors (Georlette et al. 2007). Given the central role of FoxM1 in the MuvB complex control of the cell cycle, it will be important to determine whether there are FoxM1 orthologs in flies and worms that are required for late cell cycle gene expression and whether their activity is dependent on the fly or worm MuvB complex.
In summary, our study demonstrates the integration of MuvB, B-Myb, and FoxM1 in regulating late cell cycle gene expression and mitosis. The analogies to the pathway in budding yeast that governs expression of the late mitotic genes are striking and suggest that this regulatory module likely has been preserved through evolution for the precise temporal regulation of genes critical for mitosis.
Materials and methods
Cell lines and antibodies
T98G, HeLa, and IMR-90 cell lines were obtained from American Type Culture Collection. Antibodies specific to LIN9, LIN37, LIN52, and LIN54 (Bethyl Laboratories) have been described previously (Litovchick et al. 2007). The following commercial antibodies were used for IP and Western blot analysis: anti- Rb2/p130 (BD Biosciences), anti-p130 (sc-317, Santa Cruz Biotechnology), anti-B-Myb (sc-724, Santa Cruz Biotechnology), anti-CCNB1 (sc-752, Santa Cruz Biotechnology), anti-B-Myb phospho-pT487 (Epitomics), anti-FoxM1 (sc-502, Santa Cruz Biotechnology), anti-FoxM1 (A301-532A, Bethyl Laboratories), T7 tag antibody agarose (69026, EMD Chemicals), c-myc antibody agarose immobilized (S190-104, Bethyl Laboratories), T7 tag monoclonal antibody (69522-3, EMD Chemicals), Myc tag (9B11), mouse mAb (2276, Cell Signaling Technology), and anti-Vinculin (V9131, Sigma-Aldrich). For control IPs, normal anti-rabbit serum (111-001-001, Jackson ImmunoResearch) was used. The following commercial antibodies were used for ChIPs: anti-E2F4 (sc-1082, Santa Cruz Biotechnology), anti-B-Myb (sc-725, Santa Cruz Biotechnology), anti-B-Myb (A301-656A, Bethyl Laboratories), anti-B-Myb phospho-pT487 (Epitomics), anti-FoxM1 (sc-502, Santa Cruz Biotechnology), anti-NF-YB (PAb001, GeneSpin), and anti-Histone H3 (ab1791, Abcam).
Plasmids and transfections
T7-FoxM1 and Myc-FoxM1 were kind gifts from Dr. Pradip Raychaudhuri (University of Illinois at Chicago) and Dr. Zheng Fu (Virginia Commonwealth University), respectively. Plasmid transfection was performed using Lipofectamine LTX and Plus Reagent (Invitrogen) according to the manufacturer's protocol. Transfection of SMARTpool siRNA (Dharmacon) was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Cell cycle synchronization and flow cytometry
Synchronization to obtain cells enriched in different cell cycle phases was performed for IMR-90 (Abdurashidova et al. 1998), HeLa (Whitfield et al. 2000), and T98G (Litovchick et al. 2004) as described previously. Cell cycle distribution was assessed by flow cytometry using FACSCanto (BD Biosciences) after staining with propidium iodide (Sigma-Aldrich).
qPCR
qPCRs were performed using SYBR Green (Agilent). Fold enrichments in ChIP-qPCR experiments were calculated as percentage of input. When a comparison across different conditions was required, relative occupancies were calculated.
Calculation of fold enrichment as percentage of input was as follows:
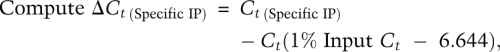 |
where 6.644 = log2 of 100 (because 1% input represents a 100-fold diluted sample).
% Input = 100 × 2(−ΔCt).
Calculation of relative occupancy was as follows:
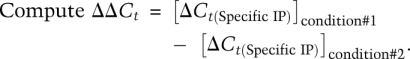 |
Relative (or differential) site occupancy across these conditions is 2(−ΔΔCt).
For RT-qPCR, RNA was isolated using Trizol (Invitrogen) and purified with RNeasy columns (Qiagen). RNA was reverse-transcribed using SuperScript III RT (Invitrogen), and fold change was calculated relative to GAPDH using the 2–ΔΔCt method. Sequences of all of the primers used in this study are available on request.
ChIP, ChIP-reChIP, and ChIP-seq
ChIP was performed as described (Rayman et al. 2002). ChIP-reChIP was performed using the Re-ChIP-IT kit (Active Motif). For ChIP-seq, 30 ng of DNA from ChIP experiments and similar amounts of input DNA were prepared for sequencing with the Illumina ChIP-Seq Sample Prep kit. ChIP-seq DNA libraries were quantified using Nanodrop (Thermo-Scientific), and a sharp peak at 200–400 nucleotides (nt) was confirmed on the BioAnalyzer (Agilent) before sequencing. Diluted libraries were used for cluster generation and single-end sequencing using the Genome Analyzer II (Illumina) at the Center for Cancer Computational Biology (Dana-Farber Cancer Institute) following the manufacturer's protocol. These ChIP-seq data can be accessed from the Gene Expression Omnibus (GEO) repository GSE27031.
Analysis of ChIP-seq data
Base calling and mapping of the 36-bp reads to the human genome (Build 37, hg19) was done using Bowtie (Langmead et al. 2009) using very strict alignment criteria: only perfect matches (−n 0) and only unique alignments (−m 1). Although sequence duplication levels were low (<10%) in the ChIP-seq runs, deduplicated reads were generated and used in subsequent analysis. The Bowtie output bam and index files were loaded into the UCSC Genome Browser (Kent et al. 2002) for sequence view. Enriched genomic regions from ChIP-seq experiments were determined using CisGenome version 1.2 (Ji et al. 2008) by two-sample analysis using the input signal as negative control. This analysis determined regions where ChIP signals are enriched relative to the input using a conditional binomial model that involves a 100-bp window and 100-bp step passing a 0.1 FDR cut-off. Peaks common to LIN9 and B-Myb were defined as having at least a one-base overlap between peaks obtained for the two factors. Fold change values were used to plot the correlation graph and calculate the Spearman's rank correlation coefficient rs. For location analysis, Chip-seq peaks were mapped with respect to individual gene TSSs extracted from the UCSC hg19 RefSeq annotation database, and the peaks located within ±10 kb of the TSSs were plotted. Target genes of B-Myb and LIN9 were defined as those having ChIP-seq peaks within 10 kb upstream of the TSSs and up to 10 kb downstream from the TESs (transcription end sites). Gene ontology analysis with specific gene sets was performed using DAVID with default parameters (Dennis et al. 2003; Huang et al. 2009).The Gibbs sampler, a de novo motif discovery tool integrated with CisGenome (Ji et al. 2008), was used to predict motifs that were significantly enriched in the ChIP-seq peak regions. Initially, targets passing FDR < 0.1, 0.01, or 0.001 were used independently for deriving motifs. Many motifs were shared between the different categories of targets. Motifs from targets with FDR < 0.01 had the highest overlap between B-Myb and LIN9 ChIP-seq data sets and are reported here. For analyzing co-occurrence of FoxM1 and CHR motifs, the MochiView suite was used. A third-order Markov model (MM) was created using LIN9 and B-Myb targeted promoters, and each motif was scanned along this set of promoters using the MM as background. Only hits with a −log (P-value) ≥ 2, equivalent to an α value of 0.01 (99% confidence), were accepted. The 5′ ends of all hits for a single motif (referred as the anchor) were anchored to position 0, and the distribution of the other motif was plotted relative to that position.
Microarray analysis
Total RNA was extracted with Trizol reagent (Invitrogen) followed by DNase I digestion and cleanup using RNAeasy columns (Qiagen). RNA was hybridized to Human Gene 1.0 ST arrays following procedures recommended by the manufacturer (Affymetrix). The R Bioconductor Affy package was used to analyze the microarray files. After loading the raw CEL files in R, RMA (robust multiarray averaging) was used to subtract background, normalize intensities, and summarize gene expression levels (Irizarry et al. 2003). These microarray data can be accessed from the GEO repository GSE27031. Values from the normalized probe sets were used for performing GSEA (Subramanian et al. 2005) downloaded from http://www.broadinstitute.org/gsea. Genes common to DREAM and B-Myb–MuvB were obtained by overlapping the DREAM target gene list containing 818 genes (Litovchick et al. 2007) and the B-Myb–MuvB target gene list containing 828 genes (this study). The R time-course package was used to calculate correlation of the expression pattern of individual genes with the expression pattern of the standards (Tai and Speed 2006). Genes used as standards for deriving the early and late cell cycle clusters were chosen based on their expression profiles, reported previously (Whitfield et al. 2002).
Acknowledgments
We thank Eric McIntush (Bethyl Laboratories) for antibodies. We thank the Microarray Core and the Center for Cancer Computational Biology (CCCB) at Dana-Farber Cancer Institute for excellent assistance with expression profiling and ChIP-seq, respectively. We thank all members of the DeCaprio laboratory, N. Dyson (Massachusetts General Hospital), and W. Tansey (Vanderbilt University) for discussions. We thank N. Dyson and W. Wei (Beth Israel Deaconess Medical Center) for critically reading the manuscript. S.D. thanks S. Bentink and B. Haibe-Kains (Dana-Farber Cancer Institute) for discussions. We thank M. Correll (CCCB) for critically reviewing the bioinformatics analysis presented here. We thank Dustin Holloway (CCCB) for the bioinformatics analysis on FoxM1 and CHR motifs. This work was supported in part by Public Health Service grants P01CA050661, RO1CA93804, and R01CA63113 to J.A.D., and a research grant from the Susan F. Smith Center for Women's Cancers, Dana-Farber Cancer Institute to S.S. S.S. designed and performed all of the experiments, analyzed data, and wrote the paper; S.D. performed bioinformatics analysis and helped with generating Figures 2 and 3; and J.A.D. designed experiments, gave conceptual advice, and wrote the paper.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.181933.111.
References
- Abdurashidova G, Riva S, Biamonti G, Giacca M, Falaschi A 1998. Cell cycle modulation of protein-DNA interactions at a human replication origin. EMBO J 17: 2961–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejka L, Wen H, Ashton J, Grant M, Iori K, Wang A, Manak JR, Lipsick JS 2011. Animal-specific C-terminal domain links myeloblastosis oncoprotein (Myb) to an ancient repressor complex. Proc Natl Acad Sci 108: 17438–17443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais A, Dynlacht BD 2007. E2F-associated chromatin modifiers and cell cycle control. Curr Opin Cell Biol 19: 658–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breeden LL 2003. Periodic transcription: a cycle within a cycle. Curr Biol 13: R31–R38 doi: 10.1016/S0960-9822(02)01386-6 [DOI] [PubMed] [Google Scholar]
- Caretti G, Salsi V, Vecchi C, Imbriano C, Mantovani R 2003. Dynamic recruitment of NF-Y and histone acetyltransferases on cell-cycle promoters. J Biol Chem 278: 30435–30440 [DOI] [PubMed] [Google Scholar]
- Chen YJ, Dominguez-Brauer C, Wang Z, Asara JM, Costa RH, Tyner AL, Lau LF, Raychaudhuri P 2009. A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J Biol Chem 284: 30695–30707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, et al. 1998. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell 2: 65–73 [DOI] [PubMed] [Google Scholar]
- Darieva Z, Clancy A, Bulmer R, Williams E, Pic-Taylor A, Morgan BA, Sharrocks AD 2010. A competitive transcription factor binding mechanism determines the timing of late cell cycle-dependent gene expression. Mol Cell 38: 29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: 3 doi: 10.1186/gb-2003-4-5-p3 [PubMed] [Google Scholar]
- Dimova DK, Dyson NJ 2005. The E2F transcriptional network: Old acquaintances with new faces. Oncogene 24: 2810–2826 [DOI] [PubMed] [Google Scholar]
- Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, Tindall DJ, Chen J 2008. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol 10: 1076–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georlette D, Ahn S, MacAlpine DM, Cheung E, Lewis PW, Beall EL, Bell SP, Speed T, Manak JR, Botchan MR 2007. Genomic profiling and expression studies reveal both positive and negative activities for the Drosophila Myb MuvB/dREAM complex in proliferating cells. Genes Dev 21: 2880–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez GJ, Tsuji T, Chen M, Jiang W, Ronai ZA 2010. Interplay between Cdh1 and JNK activity during the cell cycle. Nat Cell Biol 12: 686–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MM, Ceol CJ, Lu X, Horvitz HR 2006. Some C. elegans class B synthetic multivulva proteins encode a conserved LIN-35 Rb-containing complex distinct from a NuRD-like complex. Proc Natl Acad Sci 103: 16782–16787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP 2003. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: e15 doi: 10.1093/nar/gng015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH 2008. An integrated software system for analyzing ChIP–chip and ChIP-seq data. Nat Biotechnol 26: 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LR, Johnson TK, Desler M, Luster TA, Nowling T, Lewis RE, Rizzino A 2002. Effects of B-Myb on gene transcription: Phosphorylation-dependent activity and acetylation by p300. J Biol Chem 277: 4088–4097 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler R, Pelletier L, Heninger AK, Slabicki M, Theis M, Miroslaw L, Poser I, Lawo S, Grabner H, Kozak K, et al. 2007. Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat Cell Biol 9: 1401–1412 [DOI] [PubMed] [Google Scholar]
- Knight AS, Notaridou M, Watson RJ 2009. A Lin-9 complex is recruited by B-Myb to activate transcription of G2/M genes in undifferentiated embryonal carcinoma cells. Oncogene 28: 1737–1747 [DOI] [PubMed] [Google Scholar]
- Korenjak M, Taylor-Harding B, Binne UK, Satterlee JS, Stevaux O, Aasland R, White-Cooper H, Dyson N, Brehm A 2004. Native E2F/RBF complexes contain Myb-interacting proteins and repress transcription of developmentally controlled E2F target genes. Cell 119: 181–193 [DOI] [PubMed] [Google Scholar]
- Lane S, Farlie P, Watson R 1997. B-Myb function can be markedly enhanced by cyclin A-dependent kinase and protein truncation. Oncogene 14: 2445–2453 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH 2005. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 7: 126–136 [DOI] [PubMed] [Google Scholar]
- Laoukili J, Alvarez M, Meijer LA, Stahl M, Mohammed S, Kleij L, Heck AJ, Medema RH 2008. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol Cell Biol 28: 3076–3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherwood J, Futcher B 2010. King of the castle: Competition between repressors and activators on the Mcm1 platform. Mol Cell 38: 1–2 [DOI] [PubMed] [Google Scholar]
- Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, Wang K, Sumazin P, Kustagi M, Bisikirska BC, et al. 2010. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol 6: 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Beall EL, Fleischer TC, Georlette D, Link AJ, Botchan MR 2004. Identification of a Drosophila Myb-E2F2/RBF transcriptional repressor complex. Genes Dev 18: 2929–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhart C, Elkon R, Shiloh Y, Shamir R 2005. Deciphering transcriptional regulatory elements that encode specific cell cycle phasing by comparative genomics analysis. Cell Cycle 4: 1788–1797 [DOI] [PubMed] [Google Scholar]
- Litovchick L, Chestukhin A, DeCaprio JA 2004. Glycogen synthase kinase 3 phosphorylates RBL2/p130 during quiescence. Mol Cell Biol 24: 8970–8980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, Chen R, Washburn MP, Liu XS, DeCaprio JA 2007. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell 26: 539–551 [DOI] [PubMed] [Google Scholar]
- Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA 2011. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev 25: 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler DR, Alvarez-Fernandez M, Stein A, Hibbert RG, Heidebrecht T, Aloy P, Medema RH, Perrakis A 2010. Structure of the FoxM1 DNA-recognition domain bound to a promoter sequence. Nucleic Acids Res 38: 4527–4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oktay K, Buyuk E, Oktem O, Oktay M, Giancotti FG 2008. The c-Jun N-terminal kinase JNK functions upstream of Aurora B to promote entry into mitosis. Cell Cycle 7: 533–541 [DOI] [PubMed] [Google Scholar]
- Osterloh L, von Eyss B, Schmit F, Rein L, Hubner D, Samans B, Hauser S, Gaubatz S 2007. The human synMuv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. EMBO J 26: 144–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilkinton M, Sandoval R, Colamonici OR 2007. Mammalian Mip/LIN-9 interacts with either the p107, p130/E2F4 repressor complex or B-Myb in a cell cycle-phase-dependent context distinct from the Drosophila dREAM complex. Oncogene 26: 7535–7543 [DOI] [PubMed] [Google Scholar]
- Pramila T, Miles S, GuhaThakurta D, Jemiolo D, Breeden LL 2002. Conserved homeodomain proteins interact with MADS box protein Mcm1 to restrict ECB-dependent transcription to the M/G1 phase of the cell cycle. Genes Dev 16: 3034–3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha D, Wang Z, Moqtaderi Z, Wu L, Zhong G, Gerstein M, Struhl K, Snyder M 2010. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc Natl Acad Sci 107: 3639–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpole S, Watson RJ, te Riele H, Dynlacht BD 2002. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev 16: 933–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M 2008. Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet 4: e1000133 doi: 10.1371/journal.pgen.1000133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson C, Light Y, Groves R, Mann D, Marias R, Watson R 1996. Cell-cycle regulation of B-Myb protein expression: Specific phosphorylation during the S phase of the cell cycle. Oncogene 12: 1855–1864 [PubMed] [Google Scholar]
- Sala A, Kundu M, Casella I, Engelhard A, Calabretta B, Grasso L, Paggi MG, Giordano A, Watson RJ, Khalili K, et al. 1997. Activation of human B-MYB by cyclins. Proc Natl Acad Sci 94: 532–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmit F, Korenjak M, Mannefeld M, Schmitt K, Franke C, von Eyss B, Gagrica S, Hanel F, Brehm A, Gaubatz S 2007. LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle 6: 1903–1913 [DOI] [PubMed] [Google Scholar]
- Schmit F, Cremer S, Gaubatz S 2009. LIN54 is an essential core subunit of the DREAM/LINC complex that binds to the cdc2 promoter in a sequence-specific manner. FEBS J 276: 5703–5716 [DOI] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 9: 3273–3297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YC, Speed TP 2006. A multivariate empirical Bayes statistic for replicated microarray time course data. Ann Stat 34: 2387–2412 [Google Scholar]
- Tarasov KV, Tarasova YS, Tam WL, Riordon DR, Elliott ST, Kania G, Li J, Yamanaka S, Crider DG, Testa G, et al. 2008. B-MYB is essential for normal cell cycle progression and chromosomal stability of embryonic stem cells. PLoS ONE 3: e2478 doi: 10.1371/journal.pone.0002478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meijden CM, Lapointe DS, Luong MX, Peric-Hupkes D, Cho B, Stein JL, van Wijnen AJ, Stein GS 2002. Gene profiling of cell cycle progression through S-phase reveals sequential expression of genes required for DNA replication and nucleosome assembly. Cancer Res 62: 3233–3243 [PubMed] [Google Scholar]
- Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH 2005. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2–Cks1) ubiquitin ligase. Mol Cell Biol 25: 10875–10894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield ML, Zheng LX, Baldwin A, Ohta T, Hurt MM, Marzluff WF 2000. Stem-loop binding protein, the protein that binds the 3′ end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms. Mol Cell Biol 20: 4188–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. 2002. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell 13: 1977–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, et al. 2001. The E2F1-3 transcription factors are essential for cellular proliferation. Nature 414: 457–462 [DOI] [PubMed] [Google Scholar]
- Zhu W, Giangrande PH, Nevins JR 2004. E2Fs link the control of G1/S and G2/M transcription. EMBO J 23: 4615–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]