

Abstract
Background
Cardiovascular complications after traumatic brain injury (TBI) contribute to morbidity and mortality and may provide a target for therapy. We examined blood pressure and left ventricle contractility after TBI, and tested the hypothesis that beta-adrenergic blockade would decrease oxidative stress after TBI.
Material and Methods
Rodents received fluid-percussion injury or sham surgery, confirmed with magnetic resonance imaging (MRI) and histopathology. We followed recovery with sensorimotor coordination testing and blood pressure measurements. We assessed left ventricular ejection fraction using ECG-gated cardiac MRI and measured myocardial reactive oxygen species (ROS) with dihydroethidium. We randomized additional TBI and sham animals to post-operative treatment with propranolol or control, for measurement of ROS.
Results
Blood pressure and cardiac contractility were elevated 48 hours after TBI. Myocardial tissue sections showed increased ROS. Treatment with propranolol diminished ROS levels following TBI.
Conclusions
TBI is associated with increased cardiac contractility and myocardial ROS; decreased myocardial ROS after beta-blockade suggests that sympathetic stimulation is a mechanism of oxidative stress.
Keywords: Traumatic brain injury, reactive oxygen species, adrenergic receptor antagonist
Introduction
Approximately 1.7 million Americans sustain a traumatic brain injury (TBI) each year,1 and cardiovascular complications, such as electrocardiogram (ECG) changes, arrhythmias, ischemia, and heart failure, may delay recovery or contribute to mortality.2 Sympathetic stimulation following head injury causes not only hypertension, but also a marked hyperdynamic cardiac state.3–5 Catecholamines may induce generation of reactive oxygen species (ROS) in vascular and myocardial tissue;6, 7 however, a direct association between myocardial ROS and TBI has not been previously described.
The administration of adrenergic blockade after head trauma in human and animal models has been previously explored but not prospectively validated.8–10 Two retrospective studies suggested that mortality in trauma patients who received beta-adrenergic blockers was significantly decreased, but the mechanism of cardioprotective effects is unknown. 11, 12
We used a rodent survival model of TBI to examine the association between TBI and myocardial oxidative stress. We hypothesized that TBI would increase myocardial ROS, and furthermore, that beta-adrenergic blockade after TBI would mitigate this oxidative stress.
Methods
Animal model
The fluid-percussion injury method was used to produce TBI13. Adult male Sprague Dawley rats (Charles River, Saint Constant, Quebec, Canada; 300–350g) were anesthetized with 2–5% isoflurane. A 3mm diameter craniotomy was drilled and a 2mm internal diameter stainless steel hollow intracranial screw was placed in the skull. Once secured, the intracranial screw was filled with 0.9% normal saline and attached with tubing to the fluid percussion device. TBI was administered using controlled fluid percussion injury. Control animals received anesthesia and scalp incision only. Animals were allowed to recover for at least 36 hours with free access to food and water. Recovery was defined as ability to maintain upright posture, ambulate, and take oral hydration. Animals with hypotension, persistent seizure activity or severe neurological deficits were euthanized and excluded from further analysis. All animals received buprenorphine analgesia while under anesthesia and at 12 hours after surgery. Additional doses of buprenorphine were administered to animals with signs of pain or suffering until the completion of the experiment. All studies were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals (NIH) and approved by the Institutional Animal Care and Use Committee of the University of Vermont.
Propronalol administration
Animals were randomized to receive either propranolol (Sigma-Aldrich, St. Louis, MO) or control. Propranolol was dissolved daily in tap water (460mg/L) and taken ad lib by animals for 10 days after surgery. We assumed a mean intake of 35 mL/ day of water, and administered each animal 50mg/ kg/ day. Previous studies have shown adequate beta-adrenergic receptor blockade in rats with similar oral doses of 4014, 5015, or 8016 mg/kg/day, based on measurements of plasma concentrations of the drug15, effects of the drug on adrenergic receptor binding parameters 14, and effects on heart rate and arterial blood pressure response to isoproterenol infusion 16.
Neurological testing
Sensorimotor coordination of our research animals was assessed using a RotaRod (Med Associates Inc., St. Albans, VT), which is a device that measures the time a rat is able to stay on a rod rotating at a constant acceleration 17. Rather than measuring voluntary exercise, the elevation of the rod above the cage floor serves as motivation to perform the task until failure. The initial velocity of the rod was 4 rpm, and it accelerated every 10 seconds until it reached 40 rpm. The amount of time spent on the rod before falling (maximum: 5 minutes) was recorded for ten trials. Animals were acclimated to the device for five days prior to surgery, and then tested on post-operative day three.
Blood pressure measurement
Blood pressure was measured using both invasive and non-invasive techniques18. For monitoring of blood pressure during surgery, the caudal tail artery was cannulated by either percutaneous approach or a cutdown to place a 24 gauge angiocatheter. Blood pressure measurements using a tail cuff volumetric sensor (CODA NIBP system; Columbus Instruments, Columbus, OH) were obtained after seven days of acclimation18. Rats were warmed during measurements to ensure proper blood flow to the tail. Ten measurements were recorded and then averaged for each time point.
Magnetic resonance imaging
MRI was conducted with an Achieva 3.0 Tesla TX magnet (Philips Medical Systems International B.V., Best, Netherlands); this imaging was performed in vivo with the rats under isoflurane anesthesia. Heart rates were measured, and the level of anesthesia was titrated to achieve heart rates of 150–200 beats per minute for optimal cardiac imaging. A 16 channel Torso-XL receive coil (InVivo Corp., Gainesville, FL) was used in combination with a small single-channel e-Coil (Medtronic Inc., Minneapolis, MN) for image acquisition. Three axially-acquired pulse sequences were used to assess brain injury: T2-weighted (T2W) gradient and spin echo (GRaSE), T2W fluid attenuated inversion recovery (FLAIR), and diffusion-weighted imaging (DWI). The GRaSE acquisition was performed with TR=4266 ms, TE=80 ms, acquired matrix=200×190, field-of-view (FOV)=60 mm ×49 mm, slice thickness=0.8 mm, signal averages (NEX)=3. For the FLAIR sequence, images were acquired with TR=8000 ms, TE=125 ms, acquired matrix=200×171, FOV=60 mm × 49 mm, slice thickness=1 mm, NEX=2. The DWI images were acquired with a diffusion-weighted spin echo EPI (echo planar imaging) sequence using b=1000 s/mm2, TR=1201 ms, TE=81ms, acquired matrix=116×111, FOV=70 mm × 70 mm, slice thickness=1.3 mm, NEX=2. To assess left ventricular function, ECG gated short-axis MR images (8mm thick) of the beating heart were acquired with a balanced steady-state free precession (b-SSFP) acquisition at a matrix size of 144×144 with a FOV of 35–40mm and 2mm slice thickness with 20–30 phases per R-R interval. The TE is 1.3 ms and the TR is 3.0 ms with this sequence. Local B0 and B1 shimming were performed to minimize image artifacts. This provides a temporal resolution of each frame of 40–50 ms. Analysis was performed off-line using a IDL-based software program (Cine Tool). LVEF was estimated by determining the average ratio of biplane area of short-axis images of the ventricle in end-systole and end-diastole at three different LV sections (base, mid-LV, and apex) for each animal.
Immunohistochemistry for detection of glial filbrillary acidic protein (GFAP)
Reactive gliosis was measured in cortical and hippocampal tissue sections after incubation with a fluorescent antibody specific to GFAP, as previously described19, 20. In preparation for histological studies, animals were euthanized by injection with sodium pentobarbital (130 mg/kg), and after resection the desired tissue sample was loaded into a biopsy tray well with Tissue-Tek Optimal Cutting Temperature (Sakura Finetek USA, Inc., Torrance, CA) (OCT) compound. Samples were flash frozen for 30 seconds in a bath of 2-methylbutane cooled by liquid nitrogen, and then immediately transferred to storage at −80°C. Slides were prepared by placing tissue blocks in a cryostat at −20°C and allowed to come to temperature, then removed from biopsy trays and adhered to sectioning chucks with additional OCT. Twenty micron sections were cut in the cryostat, placed on slides, and stored at −20°C. For immunohistochemistry, slides w ere fixed with 3% paraformaldehyde in phosphate buffered saline (PBS), and then blocked with 5% Donkey Serum (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), rinsed with PBS-Tween, and then incubated in rabbit polyclonal IgG AntiGFAP (1:10,000; Dako North America, Inc., Carpinteria, CA) overnight at 4°C. Samples were then incubated in Anti-Rabbit Ig G – Cy3 (1:800 in 5% Donkey Serum; Invitrogen) for 3 hours at room temperature, followed by the application of SlowFade Gold with DAPI (Invitrogen) immediately prior to mounting. Each experiment included negative controls with omission of primary antibody to test for nonspecificity. For each sample sixteen images in 0.5 micron steps were collected using a DeltaVision restoration microscopy system. Deconvolution was completed using SoftWoRx software (Applied Precision Inc., Issaquah, WA) and analysis was done using ImageJ software (National Institute of Health). At least 3 microscope fields per animal were normalized to background and averaged to determine total area of fluorescence per animal
Dihydroethidium staining
Reactive oxygen species were measured in myocardial tissue by confocal microscopy using dihydroethidium (EMD Biosciences, La Jolla, CA) (DHE), as previously described21. Slides were allowed to come to room temperature in a light-protected tray and then incubated for 30 minutes at 37°C with 4μM DHE, and then washed with PBS. Imaging was completed with a Zeiss LSM 510 META Confocal Laser Scanning Imaging System (Carl Zeiss Microimaging, Thornwood, NY) with excitation at 488nm. Negative control slides were incubated for 30 minutes at 37°C with 1000 Units/ mL Super Oxide Dismutase (Sigma) prior to incubation with DHE.
Statistical Analysis
Data are presented as mean +/− standard error of the mean (SEM). Differences between experimental groups in experiments were determined by two-tailed t-test or analysis of variance (ANOVA). In all experiments, P < 0.05 was considered to indicate statistical significance.
Results
To obtain the images shown in Figure 1, we pioneered a novel technology for in vivo, rodent imaging using a 3.0 Tesla magnet. Other investigators have acheived similar resolution using specialized 7.0 or higher Tesla animal magnets, so we now demonstrated a technique that can work on the 3.0 Tesla human magnets found at most medical centers. The key innovation is an inexpensive, single loop disposable receive coil designed for single use prostate imaging that can be easily modified by removal of the balloon to image mice, rats, and very small regions of human anatomy. MRI reveals damage after fluid percussion injury, centered at the site of the craniotomy (Figure 1). Parenchymal hemorrhage is evident throughout the ipsilateral cortex, with a smaller area of ischemia. There is also edema involving both the ipsilateral and contralateral cortex.
Figure 1.

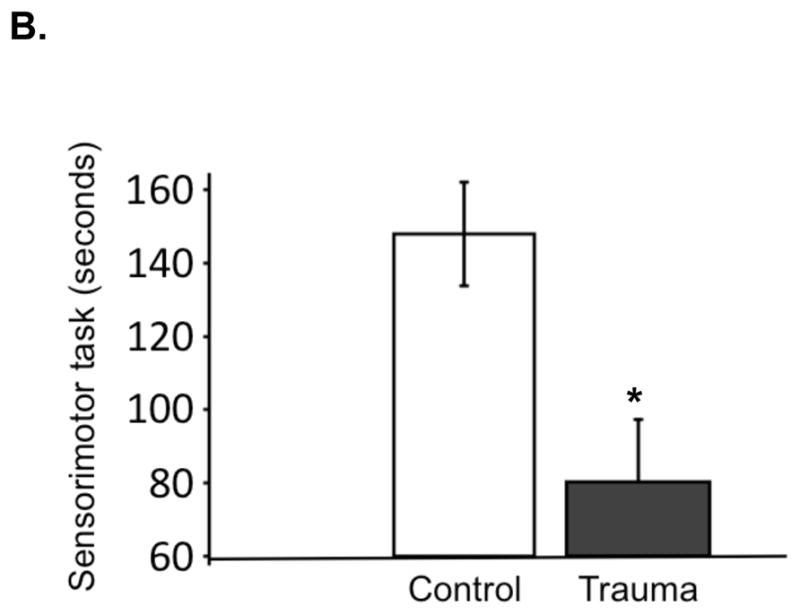
A. Neuroimaging with Achieva 3.0T TX magnet after TBI surgery (trauma) or sham surgery (control). Imaging obtained from animals under isofluorane anesthesia 36–48 hours following surgery. Representative sequences are shown with enhancement indicating structural injury (T2-weighted gradient and spin echo [GRaSE]), surrounding edema (fluid-attenuated inversion recovery [FLAIR]) and larger ischemic penumbra (diffusion weighted images [DWI]). B. Neurological function assessment after TBI surgery or sham surgery. Animals were pre-acclimated to the rotarod test, subjected to surgery, and allowed to recover. On the third day after surgery, animals were tested in five consecutive trials to measure length of time on the rotating rod. Values are expressed as mean +/− SEM (control, n = 17; trauma, n=16). *Different from control, two-tailed t test (P < 0.05).
We also measured reactive gliosis in cortical tissue after TBI. In response to insult, astrocytes undergo reactive gliosis, characterized by increased expression of GFAP19, 20. We observed increased gliosis in cortical tissue ipsilateral to site of fluid percussion injury, as measured by total GFAP fluorescence in the brains of animals receiving TBI vs. control surgery (Figure 2).
Figure 2.

A. Immunohistochemistry for reactive gliosis after TBI surgery (trauma) or sham surgery (control). Rats were euthanized 7–10 days after surgery for acquisition of ipsilateral cortical tissue adjacent to the area of injury. Confocal microscopy was used to detect fluorescence targeted by an antibody specific to glial fibrillary acidic protein (GFAP) in frozen tissue. Representative slides are shown. B. Graph depicts GFAP fluorescence from cortical tissue sections. At least 3 microscope fields per animal were normalized to background and averaged to determine total area of fluorescence per animal. Values are expressed as mean +/− SEM (control, n = 9; trauma, n=9). *Different from control, two-tailed t test (P < 0.05).
Additionally, we were able to quantify a significant difference in our research animals’ ability to perform a sensorimotor coordination task three days after injury. This task evaluated balance, coordination, and capacity for physical activity17. In our experiments, the mean duration of time a rat remained on the rod was 80 seconds vs. 145 seconds, for TBI (n=8) vs. control (n=10), respectively (Figure 1).
We measured the short term and long term blood pressure response to TBI. Using arterial monitoring of blood pressure during TBI surgery (n=3), we noted that blood pressure peaked within one minute (147mmHg +/− 11 mmHg, from baseline 105 +/− 18 mmHg) and normalized within 5 minutes. We acclimated 20 additional animals to tail cuff measurement of blood pressure, and collected serial measurements of blood pressure before and after TBI (n=10) or sham surgery (n=10). Two TBI animals had hypotension after surgery, and did not survive. There were no differences in blood pressure between TBI and sham animals on the first day after surgery. At 48 hours post-injury, TBI animals showed a significant increase in systolic blood pressure (Figure 3). Differences between TBI and sham were no longer significant by 5 days post-injury; some TBI animals had persistent hypertension but others had normalized their blood pressure. At 2 days after surgery, cardiac MRI demonstrated that TBI animals were hypercontractile with significantly elevated ejection fraction (Figure 4). Heart rates during left ventricle ejection fraction (LVEF) measurement were 162 +/− 12 bpm, with no significant differences between TBI and sham animals.
Figure 3.

A. Fluid percussion injury measured using high frequency pressure transducer in line with fluid column during procedure. The maximum pressure is shown (49 PSI) over the 500 msec time period. B. Representative tracing of systolic blood pressure after fluid percussion injury (60 second tracing). Intra-operative blood pressure was transduced from the tail artery using an indwelling line placed under anesthesia. The blood pressure increased from 100 mmHg to 153 mmHg over 3 seconds, and then returned to baseline within 30 seconds.
B. Blood pressure after TBI surgery (trauma) or sham surgery (control) measured with a non-invasive tail cuff. Representative cuff pressure tracings are shown for control and trauma animals. B. Summary blood pressure data at 2 days after injury. Five to ten readings were taken and averaged for each animal. Values are expressed as mean +/− SEM (control, n=8; trauma, n=8). *Different from control, two-tailed t test (P < 0.05).
Figure 4.

A. Cardiac MRI to measure left ventricular ejection fraction (LVEF) after TBI surgery (trauma) or sham surgery (control). Rats recovered from surgery for 2 days and were then placed under isofluorane anesthesia for respiratory and ECG-gated cardiac MRI. LVEF was estimated by determining the average ratio of biplane area of short-axis images of the ventricle in end-systole and end-diastole at three different LV sections (base, mid-LV, and apex) for each animal. Representative images from diastole and systole are shown with the LV cavity highlighted. B. Summary LVEF data. Values are expressed as mean +/− SEM (control, n=4; trauma, n=6). *Different from control, two-tailed t test (P < 0.05).
We measured cardiac ROS in 24 TBI animals who survived surgery and received two weeks of treatment with either propranolol (n=12) or no propranolol (n=12), and compared these groups to animals receiving the sham surgery (n=12). ROS in myocardial tissue was recorded as fluorescence of DHE (total counts/ area) and revealed a significant increase in TBI as compared with control (Figure 5a). We found that the addition of propranolol to the drinking water of TBI animals caused normalization of ROS levels to that of control animals (Figure 5b). As a control of the DHE assay, we added of superoxide dismutase to TBI tissue (n=9) and demonstrated that the signal drops, which confirms that the fluorescence is due to ROS.
Figure 5.

A. Reactive oxygen species (ROS) in LV myocardium after sham surgery (control, n=12), TBI surgery (trauma, n=12), or TBI surgery followed by treatment with propranolol (trauma + propranolol, n=12). Rats were euthanized 7– 10 days after surgery for acquisition of myocardial tissue and fresh frozen sections were incubated with dihydroethidium (DHE) to determine levels of ROS detected by fluorescence using Zeiss LSM 510 META Laser Scanning Microscope. Negative control slides for each animal were incubated with super oxide dismutase (SOD) immediately prior to addition of DHE (trauma + SOD, n=9). Representative images are shown. B. Graph depicts DHE fluorescence from myocardial tissue sections. Values are expressed as mean +/− SEM *Different from control, ANOVA (P < 0.05).
Discussion
We provide new insight into the patterns of traumatic brain injury in an animal model visualized on MRI in vivo, with demonstration of reactive gliosis by immunohistochemistry in vitro. We have pioneered a novel technology for collecting images in rodent models using a 3.0 tesla magnet and an inexpensive, single loop disposable receive coil. We were surprised by the extent of injury seen on MRI, because animals appeared generally normal and were able to recover from surgery to an extent it would not be expected in a human with a similar pattern of injury; however, when we tested sensorimotor coordination, we measured significant deficits in neurological function in TBI compared to sham surgery animals.
Our observations of hypertension and increased cardiac contractility after TBI, are consistent with reports of a hyperdynamic cardiac state during recovery from TBI in both animal models and humans.3–5 The hyperadrenergic state observed in moderate to severe TBI patients contributes to increased mortality and may guide surgical therapy 22.
In our study, animals with hypotension immediately after surgery did not survive (<20%). Cardiovascular responses to TBI have been well described, and sympathetic hyperactivity following TBI in its most severe form, paroxysmal sympathetic storm, is characterized by tachycardia, hypertension, tachypnea, mydriasis, and diaphoresis. Blood pressure elevation occurs not only when intracranial pressure is elevated (Cushing response) 23 but also occurs in models of brain trauma, with or without elevation in intracranial pressure24. The hypertension of a Cushing response is due sympathetic neuron activation, including both direct cardiac stimulation and adrenal release of circulating catecholamines 25, 26. Severe head injury has been associated with a greater than 500 fold increase in plasma epinephrine and a 100 fold increase in plasma norepinephrine in experimental models.27 The increase in catecholamines correlates with the severity of brain injury and has a direct correlation with the increase in systolic blood pressure. Elevations in plasma catecholamines following human head injury occur immediately but may peak at 10–15 days.28 The increased ejection fraction we observed is consistent with the known effects of elevated catecholmines, and with prior research showing that head injury causes a hyperdynamic cardiac state.3–5
Prior research has also shown damage to the myocardium after severe brain injury29. Pathologic studies showed that the myofibrillar degeneration associated with head injury was similar to the histological findings in patients with pheochromocytoma or in experimental animals following exogenous catecholamine administration, suggesting that high levels of catecholamines exerted a direct toxic effect on the myocardium30. The pattern of injury associated with catecholamine toxicity, distinct from the inflammatory pattern characteristic of infarction, has been called contraction band necrosis30. In traumatic brain injury, signs of cardiac dysfunction including ECG changes, arrhythmias, or necrosis, develop in 20% of patients, and cardiac complications have been identified in one third of TBI cases that result in mortality.12 The ECG abnormalities that have been described following TBI include ST-T changes, ventricular arrhythmias, and reduced heart rate variability. 31, 32 Elevated creatinine kinase and tropinin I levels are found in trauma patients with brain injury, even without mechanical chest injury.33–35 Transient, reversible left ventricular dysfunction has been described in patients with acute brain injury36.
One mechanism that might explain this cardiac damage would be oxidative stress. Recent studies have shown oxidative damage in the central nervous system after traumatic brain injury37. There is evidence that catecholamines generate free radicals leading to oxidative stress, and altered redox balance reduces nitric oxide bioavailability and impairs endothelial reactivity 6, 7. We now show that myocardial oxidative stress after TBI can be mitigated by administration of a beta-adrenergic receptor antagonist.
Cardioprotective strategies including beta-adrenergic blockade are common in the surgical setting and there is evidence to support beta-adrenergic blockade in trauma, even in those patients with cardiac risk factors.38,11, 12,8–10 It may be that the increased contractility in TBI is not pathologic, per se, but rather a physiologic response to the increased catecholamines after TBI. This response may be necessary for homeostasis, in the short term, but it may also become deleterious particularly in patients with pre-existing cardiovascular risk factors, or possibly even in otherwise healthy individuals who experience a prolonged or extended cathecholaminergic response. Otherwise healthy individuals with adequate cardiac reserve may be able to tolerate the cardiac stimulation and oxidative stress without complications; however, in those patients with pre-existing cardiac disease or decreased cardiac reserve due to their injury, the stress of TBI may result in cardiac complications through the mechanisms we have described. Alternatively, the mechanism by which beta-adrenergic blockade may be cardioprotective in trauma may be multifactorial and could involve central effects such as inhibition of the Bezold-Jarish reflex 39. Further research is required before our results can be extrapolated to a clinical scenario. Our results suggest that prevention of deleterious cardiovascular ROS may be a mechanism of the beneficial effect of beta-adrenergic blockade in trauma. The implications of these findings are that adrenergic blockade or antioxidant administration following TBI may protect against cardiovascular stress.
Acknowledgments
The project described was supported by Grant Number K08GM098795 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
The MRI was supported in part by US Department of Energy SC 0001753.
MRI technician: Jay Gonyea
ECG gating/ LVEF analysis: Tim Christian, MD
Surgery technician: Sheila Russell
Students: Angus Beal, Chary Loadholt, Peter Andriakos, Tabitha Trahan
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Faul MXL, Wald MM, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; Atlanta (GA): 2010. [Google Scholar]
- 2.Bybee KA, Prasad A. Stress-related cardiomyopathy syndromes. Circulation. 2008 Jul 22;118(4):397–409. doi: 10.1161/CIRCULATIONAHA.106.677625. [DOI] [PubMed] [Google Scholar]
- 3.Clifton GL, Robertson CS, Kyper K, Taylor AA, Dhekne RD, Grossman RG. Cardiovascular response to severe head injury. Journal of neurosurgery. 1983 Sep;59(3):447–454. doi: 10.3171/jns.1983.59.3.0447. [DOI] [PubMed] [Google Scholar]
- 4.Clifton GL, Ziegler MG, Grossman RG. Circulating catecholamines and sympathetic activity after head injury. Neurosurgery. 1981 Jan;8(1):10–14. doi: 10.1227/00006123-198101000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Robertson CS, Clifton GL, Grossman RG. Oxygen utilization and cardiovascular function in head-injured patients. Neurosurgery. 1984 Sep;15(3):307–314. doi: 10.1227/00006123-198409000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Kojda G, Harrison D. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovascular research. 1999 Aug 15;43(3):562–571. doi: 10.1016/s0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 7.Singal PK, Beamish RE, Dhalla NS. Potential oxidative pathways of catecholamines in the formation of lipid peroxides and genesis of heart disease. Advances in experimental medicine and biology. 1983;161:391–401. doi: 10.1007/978-1-4684-4472-8_22. [DOI] [PubMed] [Google Scholar]
- 8.Cruickshank JM, Dwyer GN. Proceedings: Electrocardiographic changes in subarachnoid haemorrhage: role of catecholamines and effects of beta-blockade. British heart journal. 1974 Apr;36(4):395. [PubMed] [Google Scholar]
- 9.Cruickshank JM, Neil-Dwyer G, Degaute JP, et al. Reduction of stress/catecholamine-induced cardiac necrosis by beta 1-selective blockade. Lancet. 1987 Sep 12;2(8559):585–589. doi: 10.1016/s0140-6736(87)92984-9. [DOI] [PubMed] [Google Scholar]
- 10.Neil-Dwyer G, Walter P, Cruickshank JM, Doshi B, O’Gorman P. Effect of propranolol and phentolamine on myocardial necrosis after subarachnoid haemorrhage. British medical journal. 1978 Oct 7;2(6143):990–992. doi: 10.1136/bmj.2.6143.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arbabi S, Campion EM, Hemmila MR, et al. Beta-blocker use is associated with improved outcomes in adult trauma patients. The Journal of trauma. 2007 Jan;62(1):56–61. doi: 10.1097/TA.0b013e31802d972b. discussion 61–52. [DOI] [PubMed] [Google Scholar]
- 12.Cotton BA, Snodgrass KB, Fleming SB, et al. Beta-blocker exposure is associated with improved survival after severe traumatic brain injury. The Journal of trauma. 2007 Jan;62(1):26–33. doi: 10.1097/TA.0b013e31802d02d0. discussion 33–25. [DOI] [PubMed] [Google Scholar]
- 13.DeRoss AL, Adams JE, Vane DW, Russell SJ, Terella AM, Wald SL. Multiple head injuries in rats: effects on behavior. The Journal of trauma. 2002 Apr;52(4):708–714. doi: 10.1097/00005373-200204000-00017. [DOI] [PubMed] [Google Scholar]
- 14.Horinouchi T, Morishima S, Tanaka T, et al. Different changes of plasma membrane beta-adrenoceptors in rat heart after chronic administration of propranolol, atenolol and bevantolol. Life Sci. 2007 Jul 12;81(5):399–404. doi: 10.1016/j.lfs.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Takita M, Oda Y, Kigoshi S, Muramatsu I. Effects of propranolol and atenolol on immobilization stress-induced hypertension and down-regulation of central beta-adrenoceptors in rats. Pharmacol Biochem Behav. 1995 Feb;50(2):225–232. doi: 10.1016/0091-3057(94)00301-x. [DOI] [PubMed] [Google Scholar]
- 16.Marano G, Palazzesi S, Fadda A, Vergari A, Ferrari AU. Attenuation of aortic banding-induced cardiac hypertrophy by propranolol is independent of beta-adrenoceptor blockade. J Hypertens. 2002 Apr;20(4):763–769. doi: 10.1097/00004872-200204000-00036. [DOI] [PubMed] [Google Scholar]
- 17.Rozas G, Guerra MJ, Labandeira-Garcia JL. An automated rotarod method for quantitative drug-free evaluation of overall motor deficits in rat models of parkinsonism. Brain Res Brain Res Protoc. 1997 Dec 1;2(1):75–84. doi: 10.1016/s1385-299x(97)00034-2. [DOI] [PubMed] [Google Scholar]
- 18.Kurtz TW, Griffin KA, Bidani AK, Davisson RL, Hall JE Association SoPaPEotAH. Recommendations for blood pressure measurement in humans and experimental animals. Part 2: Blood pressure measurement in experimental animals: a statement for professionals from the subcommittee of professional and public education of the American Heart Association council on high blood pressure research. Hypertension. 2005 Feb 1;45(2):299–310. doi: 10.1161/01.HYP.0000150857.39919.cb. [DOI] [PubMed] [Google Scholar]
- 19.Kakinuma Y, Hama H, Sugiyama F, et al. Impaired blood-brain barrier function in angiotensinogen-deficient mice. Nat Med. 1998 Sep;4(9):1078–1080. doi: 10.1038/2070. [DOI] [PubMed] [Google Scholar]
- 20.Murakami K, Koide M, Dumont TM, Russell SR, Tranmer BI, Wellman GC. Subarachnoid Hemorrhage Induces Gliosis and Increased Expression of the Pro-inflammatory Cytokine High Mobility Group Box 1 Protein. Transl Stroke Res. 2011 Mar 1;2(1):72–79. doi: 10.1007/s12975-010-0052-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Troncoso Brindeiro CM, da Silva AQ, Allahdadi KJ, Youngblood V, Kanagy NL. Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. American journal of physiology. 2007 Nov;293(5):H2971–2976. doi: 10.1152/ajpheart.00219.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ley EJ, Berry C, Mirocha J, Salim A. Mortality is reduced for heart rate 80 to 89 after traumatic brain injury. The Journal of surgical research. 2010 Sep;163(1):142–145. doi: 10.1016/j.jss.2010.04.046. [DOI] [PubMed] [Google Scholar]
- 23.Cushing H. Concerning a Definite Regulatory Mechanism of the Vaso-Motor Centre Which Controls Blood Pressure During Cerebral Compression. Bull Johns Hopkins Hosp. 1901;12:3. [Google Scholar]
- 24.Walker A. The Physiological Basis of Concussion. Journal of neurosurgery. 1944;1:14. [Google Scholar]
- 25.Freeman N. Effect of Progressive Sympathectomy on Hypertension Produced by Increased Intracranial Pressure. The American journal of physiology. 1939;128:10. [Google Scholar]
- 26.Grimson K. The Early and Remote Effects of Total and Partial Paravertebral Sympathetomy on Blood Pressure. Annals of surgery. 1937;106:25. doi: 10.1097/00000658-193711000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosner MJ, Newsome HH, Becker DP. Mechanical brain injury: the sympathoadrenal response. Journal of neurosurgery. 1984 Jul;61(1):76–86. doi: 10.3171/jns.1984.61.1.0076. [DOI] [PubMed] [Google Scholar]
- 28.Hortnagl H, Hammerle AF, Hackl JM, Brucke T, Rumpl E, Hortnagl H. The activity of the sympathetic nervous system following severe head injury. Intensive care medicine. 1980 May;6(3):169–167. doi: 10.1007/BF01757299. [DOI] [PubMed] [Google Scholar]
- 29.Connor RC. Myocardial damage secondary to brain lesions. American heart journal. 1969 Aug;78(2):145–148. doi: 10.1016/0002-8703(69)90001-5. [DOI] [PubMed] [Google Scholar]
- 30.Reichenbach DB, EP Catecholamines and Cardiomyopathy. Human Pathology. 1970;1:26. [Google Scholar]
- 31.Morris JA, Jr, Norris PR, Ozdas A, et al. Reduced heart rate variability: an indicator of cardiac uncoupling and diminished physiologic reserve in 1,425 trauma patients. The Journal of trauma. 2006 Jun;60(6):1165–1173. doi: 10.1097/01.ta.0000220384.04978.3b. discussion 1173–1164. [DOI] [PubMed] [Google Scholar]
- 32.Norris PR, Ozdas A, Cao H, et al. Cardiac uncoupling and heart rate variability stratify ICU patients by mortality: a study of 2088 trauma patients. Annals of surgery. 2006 Jun;243(6):804–812. doi: 10.1097/01.sla.0000219642.92637.fd. discussion 812–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaste M, Hernesniemi J, Somer H, Hillbom M, Konttinen A. Creatine kinase isoenzymes in acute brain injury. Journal of neurosurgery. 1981 Oct;55(4):511–515. doi: 10.3171/jns.1981.55.4.0511. [DOI] [PubMed] [Google Scholar]
- 34.Maramattom BV, Manno EM, Fulgham JR, Jaffe AS, Wijdicks EF. Clinical importance of cardiac troponin release and cardiac abnormalities in patients with supratentorial cerebral hemorrhages. Mayo Clinic proceedings. 2006 Feb;81(2):192–196. doi: 10.4065/81.2.192. [DOI] [PubMed] [Google Scholar]
- 35.Martin M, Mullenix P, Rhee P, Belzberg H, Demetriades D, Salim A. Troponin increases in the critically injured patient: mechanical trauma or physiologic stress? The Journal of trauma. 2005 Nov;59(5):1086–1091. doi: 10.1097/01.ta.0000190249.19668.37. [DOI] [PubMed] [Google Scholar]
- 36.Kono T, Morita H, Kuroiwa T, Onaka H, Takatsuka H, Fujiwara A. Left ventricular wall motion abnormalities in patients with subarachnoid hemorrhage: neurogenic stunned myocardium. Journal of the American College of Cardiology. 1994 Sep;24(3):636–640. doi: 10.1016/0735-1097(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 37.Petronilho F, Feier G, de Souza B, et al. Oxidative stress in brain according to traumatic brain injury intensity. The Journal of surgical research. 2010 Dec;164(2):316–320. doi: 10.1016/j.jss.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 38.Jacobs DG, Plaisier BR, Barie PS, et al. Practice management guidelines for geriatric trauma: the EAST Practice Management Guidelines Work Group. The Journal of trauma. 2003 Feb;54(2):391–416. doi: 10.1097/01.TA.0000042015.54022.BE. [DOI] [PubMed] [Google Scholar]
- 39.Wisbach G, Tobias S, Woodman R, Spalding A, Lockette W. Preserving cardiac output with beta-adrenergic receptor blockade and inhibiting the Bezold-Jarisch reflex during resuscitation from hemorrhage. The Journal of trauma. 2007 Jul;63(1):26–32. doi: 10.1097/TA.0b013e31806864e2. [DOI] [PubMed] [Google Scholar]