

Abstract
Mitotic homologous recombination promotes genome stability through the precise repair of DNA double-strand breaks and other lesions that are encountered during normal cellular metabolism and from exogenous insults. As a result, homologous recombination repair is essential during proliferative stages in development and during somatic cell renewal in adults to protect against cell death and mutagenic outcomes from DNA damage. Mutations in mammalian genes encoding homologous recombination proteins, including BRCA1, BRCA2 and PALB2, are associated with developmental abnormalities and tumorigenesis. Recent advances have provided a clearer understanding of the connections between these proteins and of the key steps of homologous recombination and DNA strand exchange.
Cells have evolved various strategies to contend with the multitude of DNA lesions, including DNA strand breaks, that the genome incurs on a continuous basis1. The importance of DNA repair is evident, as deficiencies in several repair pathways are associated with human diseases, including cancer2, and with ageing. In the past decade, homologous recombination (HR) has emerged as a crucial DNA repair pathway in mammalian cells3. HR has a role in the repair of several types of DNA lesions that pose a threat to genome integrity, including double-strand breaks (DSBs), damage encountered during DNA replication and DNA interstrand cross links (ICLs). HR is a key pathway during late S phase to G2 phase of the mammalian cell cycle, as it leads to precise repair of DNA damage using the sister chromatid as the repair template. HR deficiency directs cells along more error-prone repair pathways, including non-homologous end joining (NHEJ) and single-strand annealing (SSA). Error-prone repair contributes to genome instability through the accumulation of spontaneous and damage-induced chromosomal aberrations as well as more subtle mutations such as small deletions. Not surprisingly then, crucial HR proteins, such as breast and ovarian cancer type 1 susceptibility protein (BRCA1) and BRCA2 (also known as FANCD1), suppress genome instability and are also tumour suppressors. Defects in HR are manifested when repair itself is defective and when upstream DNA damage signalling is hindered.
This Review provides a detailed overview of HR, including homologous partners and HR outcomes as related to genome stability, noting the relationship between HR and other DSB repair pathways. Recent cellular and structural advances that clarify the roles of key HR proteins are emphasized. We also provide a discussion of the distinct human tissue- and age-specific phenotypes associated with inherited monoallelic and biallelic mutated HR genes.
The HR pathway
Our understanding of HR is based on several decades of research in bacteria, yeast and other model systems4. Although too extensive to summarize here, this prior research provides a framework for investigating HR in mammalian systems. HR repair of DNA damage such as a DSB requires a second, homologous DNA that can act as a template for the repair reaction5–8 (FIG. 1). In this process, frequently called gene conversion, the information from the homologous ‘donor’ sequence is copied into the damaged site, making the repaired locus the recipient of genetic information. If the donor is identical to the recipient at the region surrounding the DSB, repair is precise and restores the DNA to the sequence that was present before breakage. This precision in repair presumably occurs even when the DNA ends incur extensive damage, because the donor sequence acts as a template for repair.
Figure 1. Pathways of DNA DSB repair.

Double-strand breaks (DSBs) are efficiently repaired in mammalian cells by homologous recombination (HR) and non-homologous end joining (NHEJ). HR initiates with end resection, which produces a 3′ single-stranded end that can invade a homologous template to initiate repair. Alternative HR pathways can ensue from the displacement loop (D-loop) intermediate: synthesis-dependent strand annealing (SDSA) and DSB repair (DSBR). In SDSA, the newly synthesized strand is displaced to anneal to the other DNA end, resulting in a non-crossover outcome with no change to the template DNA. In DSBR, the second DNA end is ‘captured’ by the D-loop to form a double Holliday junction, which in principle can result in a non-crossover (cleavage at black or grey arrowheads) or a crossover (cleavage at black arrowheads on one side and grey arrowheads) outcome. NHEJ involves the joining of non-homologous DNA ends. It can be imprecise and lead to deletions and other mutations through numerous end-processing steps (not shown). Single-strand annealing takes place when end resection occurs at sequence repeats (arrowheads) to provide complementary single strands that anneal, giving rise to a product with a single copy of the repeat and a deletion of intervening sequences.
DSBs can also be repaired by NHEJ, a distinct but efficient pathway in which non-homologous DNA ends are joined9 (FIG. 1). In contrast to HR, repair by NHEJ is often imprecise because the DNA ends are modified before joining, leading to deletions or insertions at the break site. SSA, a pathway specific to homologous repeats, leads to deletion of sequences between the repeats (FIG. 1).
The importance of mitotic HR in DNA repair in mammalian cells emerged in the mid1990s following the development of reporters to introduce site-specific damage into the genome to detect and quantify HR repair3,10,11 (BOX 1) and the discovery that RAD51 is essential in mice and maintains genome integrity in mice12,13. RAD51 catalyses the defining biochemical step of HR, strand exchange14,15, during which single-stranded DNA (ssDNA) invades homologous duplex DNA, displacing the identical strand of the duplex and forming a displacement loop (D-loop) (FIG. 1). RAD51, similarly to its bacterial homologue, RecA, is a DNA-dependent ATPase that forms helical nucleoprotein filaments with ssDNA16,17.
Box 1. Assaying HR repair of a DSB in mammalian cells.
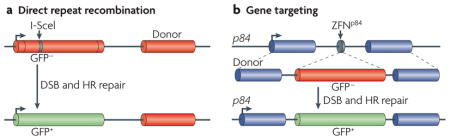
Reporters to assess repair of a double-strand break (DSB) have been instrumental for deciphering the importance of homologous recombination (HR) in mammalian cells. Most estimates of HR rely on reporters that consist of homologous repeats located close by on the same chromosome, with one repeat targeted for DSB formation by the rare-cutting I-SceI endonuclease and the other repeat acting as a template for repair (see the figure, part a). With this approach, HR repair of the DSB results in a scoreable phenotype such as fluorescence of green fluorescent protein (GFP) or drug resistance3,45,143,144. A commonly used reporter is DR-GFP, which consists of direct repeats of mutated GFP genes: a full-length GFP mutated to contain an I-SceI site and a 5′ and 3′-truncated GFP143. Repair of the I-SceI-generated DSB by HR results in GFP-positive cells that are quantified by flow cytometry (see the figure, part a). DR-GFP has been integrated into the genome of a range of wild-type and HR repair-defective mammalian cell lines.
Tandem repeat reporters such as DR-GFP are used as a surrogate to measure inter-sister HR, given that inter-sister HR is genetically silent, although in principle the repeat on the same chromatid can also participate in HR. In some reporters, repeat triplication can definitively identify a portion of inter-sister HR events6,42,144. Inter-chromosomal HR has also been analysed using a similar approach, but instead of being present in tandem on the same chromosome, repeats are on homologous (inter-homologue HR) or heterologous (inter-heterologue HR) chromosomes. Because inter-chromosomal HR is much less efficient than tandem repeat HR, drug selection is used to identify recombinants.
As I-SceI endonuclease recognizes an 18-bp sequence, sites are rare or non-existent in mammalian genomes. Recent alternatives to I-SceI include endonucleases directed towards endogenous genomic sites145. For example, in human cells, the p84 locus (PPP1R12C; protein phosphatase 1, regulatory (inhibitor) subunit 12C) on chromosome 19 can be cleaved by a zinc finger nuclease (ZFN) (see the figure, part b). In this case, HR is assayed with a transfected donor fragment that is homologous to the p84 locus and contains a promoterless GFP146. DSB repair by homologous gene targeting leads to expression of GFP.
The central role of DNA strand exchange
The mechanism of strand exchange has recently been illuminated by crystal structure determinations of RecA filaments in complex with ssDNA and double-stranded DNA (dsDNA)18, which provided insight into how RecA and presumably RAD51 function (FIG. 2). As expected from previous studies, ssDNA within the RecA filament is stretched approximately 50% relative to B-form DNA; unexpectedly, the stretching is not uniform. Instead, the ssDNA has a repeating unit of three nucleotides, which maintains a B-form structure, whereas the DNA between every triplet is greatly stretched. The B-form structure of the triplets in the ssDNA is therefore well suited to pair through canonical Watson–Crick hydrogen bonds with complementary triplets in the donor duplex DNA. Although the structure of a RecA–ssDNA–dsDNA complex is not available, it is thought that the binding of duplex DNA to RecA disrupts duplex base stacking and base pairing to promote interaction with the invading ssDNA. Correct base pairing between the invading ssDNA and the complementary DNA provides the stable interaction between the two DNA strands, as RecA has few contacts with the complementary DNA to stabilize the interaction. Furthermore, the constrained B-form DNA in the filament excludes non-standard structures from forming with mismatched bases. This reliance on DNA–DNA interactions, rather than on protein–DNA interactions, works to ensure the fidelity of strand exchange. Finally, dissociation of the newly formed heteroduplex DNA and the displaced single strand is promoted by ATP hydrolysis.
Figure 2. Mechanism of Hr revealed by recA–ssDNA and recA–dsDNA structures.

In these structures18, five RecA molecules (grey) bind to ATP (yellow) and single-stranded DNA (ssDNA; red) to form a nucleoprotein filament in which ssDNA adopts a helical conformation and is stretched relative to B-form DNA. Triplets within the ssDNA form a repeating unit with a B-form DNA-like structure, in which the bases can pair through canonical Watson–Crick hydrogen bonds. Engagement of the double-stranded DNA (magenta and blue) forms a synaptic complex that allows homology sampling, presumably by destabilizing the duplex through disruption of base stacking and pairing. Fidelity is achieved by base pairing of the invading strand (red) with the complementary strand (magenta), as RecA forms few contacts with the complementary strand. In addition, the RecA-imposed B-form DNA-like structure of the invading strand allows only canonical Watson–Crick base pairing, which does not allow pairing to mismatched bases. The new duplex (red and magenta) and the displaced strand (blue; not present in the crystal structures) are released following ATP hydrolysis. Image courtesy of Nikola Pavletich, Memorial Sloan–Kettering Cancer Center, USA.
End resection: a key first step in HR
The active HR intermediate for strand invasion is a RAD51–ssDNA nucleoprotein filament. The ssDNA used for strand invasion by RAD51 is generated by 5′ to 3′ DNA end resection, resulting in 3′ single-stranded tails (FIG. 1). In yeast, end resection occurs by a two-step mechanism: initial limited resection followed by processive resection. Limited resection involves the Mre11 complex and Sae2; more extensive resection involves either exodeoxyribonuclease 1 (Exo1; a 5′ to 3′ exonuclease) or the Sgs1 helicase (which unwinds duplex DNA) together with a nuclease to digest the 5′ strand19,20. Experiments support a role for homologues of these proteins in end resection in mammalian cells as well. Human EXO1 resects DNA ends in vitro, and its activity is stimulated by Bloom’s syndrome protein (BLM), the human Sgs1 homologue; the resected ends can then be used by RAD51 in strand exchange reactions21. In addition, the nuclease activity of MRE11 promotes recruitment of the ssDNA-binding protein replication protein A (RPA) to sites of DSBs, presumably by promoting end resection to generate ssDNA22. Similarly, CtBP-interacting protein (CtIP; also known as RBBP8), which has limited homology to Sae2 and a binding partner of BRCA1 (REF. 23), also promotes RPA recruitment to DSBs24. Studies in yeast have shown that DNA end resection is regulated during the cell cycle by cyclin-dependent kinase 1 (Cdk1; also known as Cdc28) (REFS 25,26). Sae2 and CtIP seem to be key targets for CDK phosphorylation in yeast and mammalian cells, respectively, limiting end resection to the S and G2 phases of the cell cycle27,28.
RAD51 filament formation and beyond
Once DNA ends are resected, RPA efficiently binds ssDNA to melt DNA secondary structures that can interfere with the formation of RAD51 filaments. However, RPA also impedes RAD51 binding to ssDNA, such that RAD51 needs accessory factors — sometimes termed mediators — for filament formation29. Several factors, including BRCA2 (see below), are involved in this process7. Once RAD51 nucleoprotein filaments form and strand invasion ensues, repair synthesis occurs from the invading DNA end, using the donor duplex as a template (FIG. 1). In the synthesis-dependent strand annealing (SDSA) pathway, the newly synthesized strand is displaced and anneals to the other DNA end to form a non-crossover. In the canonical DSB repair (DSBR) pathway, the second DNA end is ‘captured’ to form two Holliday junctions, which can either be dissolved to form a non-crossover (for example, by the BLM helicase30) or resolved to form a crossover, for which several proteins have been implicated8. Crossovers are crucial in meiotic cells to generate haploid germ cells31; however, non-crossovers seem to be the more beneficial outcome of mitotic HR (see below).
DSB pathway choice: HR, NHEJ and SSA
HR is restricted to the S and G2 phases of the cell cycle by several factors, including the availability of sister chromatids, transcription of HR genes and CDK-mediated phosphorylation of HR proteins28,32, whereas NHEJ functions throughout the cell cycle33. A crucial determinant of DSB repair pathway choice during S and G2 phases is the requirement of a resected DNA end for HR. Binding of NHEJ components to DNA ends interferes with end resection34; as a result of this competition for DNA ends, HR is increased in NHEJ mutants35–37.
Efficient HR during S phase may have evolved to repair damage encountered during DNA replication38,39. For example, replication of a nicked template gives rise to a DSB with just one end; this end is a substrate for repair by HR from the replicated unbroken strand. Because NHEJ requires two ends for joining, faithful repair of one-ended DSBs by NHEJ is not possible; instead, NHEJ of two one-ended DSBs would give rise to a genomic rearrangement. Thus, NHEJ has an important role only in maintaining genomic integrity in response to two-ended DSBs. Not surprisingly, HR and NHEJ are in competition with each other for the repair of two-ended, but not one-ended, DSBs35.
In addition to HR and NHEJ, SSA provides a third DSB repair pathway in the context of sequence repeats. In this pathway, ssDNA that was formed after end resection at homologous repeats anneals, leading to the deletion of the intervening sequence (FIG. 1). Because SSA requires resected ends, it is also inhibited by NHEJ components40,41. Unlike HR, SSA is a RAD51-independent pathway, and in fact is inhibited by HR components that work downstream of resection because the two pathways compete for the resected DNA ends40.
HR: partners and outcomes
The fidelity of HR makes it a relatively non-mutagenic pathway of repair when compared with NHEJ, but the availability of different homologous donors raises the question of whether (or how frequently) HR leads to genetic change. For any genomic locus in cycling cells (except on the non-pseudoautosomal region on the XY pair in males), at least two possible homologous donors are available, the sister chromatid and the homologous chromosome (FIG. 3a,b). Sister chromatids are present after DNA replication until cell division, whereas the homologue is present throughout the cell cycle. Inter-sister HR restores the sequence to how it was before DNA breakage; by contrast, inter-homologue HR has the potential to lead to loss of heterozygosity (LOH) of parental markers.
Figure 3. Homologous templates and repair outcomes of Hr.

A homologous sequence that can act as a template to repair a double-strand break (DSB) can be found on the sister chromatid, the homologous chromosome and, in the case of repeated sequences, a sequence on the same (not shown) or a different chromosome. a | Inter-sister repair is genetically silent regardless of whether the outcome is a crossover or a non-crossover. b | Inter-homologue repair can lead to local regions of loss of heterozygosity (LOH) when the outcome is a non-crossover or to LOH of entire distal regions of chromosomes when the outcome is a crossover and recombinant sister chromatids segregate in anaphase to different daughter cells. If recombinant sister chromatids end up in the same daughter cell, the chromosomes have undergone an exchange, but there is no loss of parental information (not shown). c | Inter-heterologue repair involving a crossover would in principle lead to reciprocal translocations, but they have rarely been observed. Instead, oncogenic translocations typically involve non-homologous end joining.
Inter-sister versus inter-homologue HR
Accurately comparing the frequency of inter-sister and inter-homologue HR is challenging because inter-sister HR is genetically silent. Most estimates of HR rely on reporters consisting of homologous repeats that are located close by on the same chromosome (for inter-sister HR) or on the homologous chromosome (for inter-homologue HR), in which one repeat is targeted for DSB formation (BOX 1). Generally, HR between repeats on the same chromosome is efficient in both rodent and human cells, and can be as efficient as NHEJ42–44. In mouse embryonic stem cells, HR between repeats on homologous chromosomes is approximately two orders of magnitude less efficient than that seen with tandem repeats45,46. A similar 100-fold difference is also seen in a human lymphoblastoid cell line47,48. It is perhaps not surprising that inter-homologue HR is so much less efficient, as sister chromatids are held in proximity by cohesion, whereas homologues are more distant from each other in the nuclear volume.
Non-crossover versus crossover HR
Inter-homologue HR does not lead to genomic rearrangement, but it has the potential to lead to genetic loss (that is, LOH) because information from one of the parental chromosomes is duplicated and information on the other parental chromosome is lost (FIG. 3b). The extent of genetic loss is minimal if HR results in a non-crossover gene conversion46,49. By contrast, gene conversion associated with a crossover leads to LOH of the entire region of the chromosome that is distal to the HR event if recombinant sister chromatids segregate away from each other in mitosis.
The question that arises is what fraction of inter-homologue HR events is resolved as crossovers. In both mouse embryonic stem cells and human lymphoblastoid cells, non-crossover inter-homologue HR events predominate over crossovers, although the ratio of non-crossovers to crossovers ranges from 30 to 1 (REF. 46; J. Stark and M.J., unpublished observations) to 6 to1 (REF. 48), respectively. Thus, crossing over seems to be an infrequent outcome of mitotic HR. The low overall frequency of inter-homologue HR (1 in 100 HR events) coupled to a non-crossover bias reduces the probability that inter-homologue HR leads to genetic loss during DSB repair. Nonetheless, inter-homologue HR has an important role during the development of some tumours, the classic case being hereditary retinoblastoma, during which it is estimated that inter-homologue HR leads to loss of the wild-type retinoblastoma 1 (RB1) in about 40% of tumours50. The contribution of inter-homologue HR to LOH of other tumour suppressor genes has not been studied in as much detail and is difficult to gauge in genetically unstable tumours.
HR in repeat-laden genomes
Repetitive elements and low copy number repeats that are present in different locations on the same chromosome or on different chromosomes can participate in non-allelic HR events (also known as ectopic HR events), with the potential to give rise to deletions, inversions and reciprocal translocations (FIG. 3c). Owing to the highly repetitive nature of mammalian genomes and the potential for genome rearrangements, investigators had initially considered it unlikely that HR could have an important role in DNA repair without scrambling the genome. However, sequence repeats have often highly diverged from each other, and even low levels of divergence substantially suppresses HR51. Moreover, when HR does occur between dispersed repeats, it rarely leads to genomic rearrangements because crossing over is a rare outcome of mitotic HR52,53. More commonly, genomic rearrangements that arise from DSBs in the vicinity of two homologous repeats are repaired by SSA if the repeats are not highly divergent or by NHEJ if they are highly divergent53 rather than HR. These mechanistic studies confirm the generally genome-stabilizing nature of HR. Ongoing genome instability, as would be found in HR mutants, may paradoxically lead to genetic reversion (BOX 2) in addition to deleterious mutations.
Box 2. Reversions restore gene function: an outcome of ongoing somatic instability.
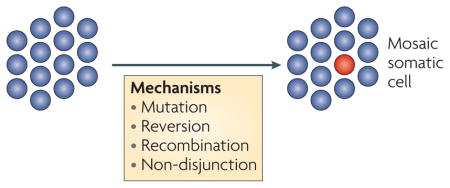
Somatic mosaicism (see the figure) of lymphocytes in patients with Fanconi anaemia with no, partial or complete protein function restoration is not uncommon147,148. In one report, revertant lymphocytes were found in patients with FANCA and FANCC mutations, whereas skin fibroblasts from these patients retained two non-functional FANC alleles149. As a result, a portion of lymphocytes was no longer sensitive to DNA interstrand cross link (ICL)-inducing agents. DNA sequencing identified mutations in a second site close to the inherited mutation that restored the open reading frame of FANC. Mechanisms of reversion seem to include replication slippage at short sequence repeats150. Restoration of partner and localizer of BRCA2 (PALB2) function in a Fanconi anaemia N lymphoblast cell line has also been reported, in this case by Alu–Alu recombination, which also restores the reading frame92. The first description of breast and ovarian cancer type 2 susceptibility protein (BRCA2) correction was noted in a cell line propagated from leukaemic blasts derived from a patient with Fanconi anaemia-associated leukaemia151, although the mechanism of reversion was not identified. Experimentally induced BRCA2 genetic reversion was achieved with chronic exposure to either an ICL-inducing agent152 or a PARP (poly (ADP-ribose) polymerase) inhibitor153. Three features of the PARP inhibitor-resistant clones are notable: restoration of double-strand break (DSB)-induced homologous recombination (HR) repair by revertant BRCA2 peptides missing the DNA-binding domain, large BRCA2 deletions at regions of homology (which suggests that the increased single-strand annealing (SSA) associated with BRCA2 loss drives genetic reversion) and, importantly, observation of similar deletions in human BRCA2 tumours that are resistant to ICL-inducing agents152,153. Of note, only half of the ICL-resistant clones showed restored BRCA2 function through genetic reversion, indicating that there are other routes to resistance, and few revertants generated deletions that would be compatible with an SSA event, which is indicative of other mechanisms of reversion152. For further information on Fanconi anaemia see REFS 154,155.
Studies of cancer genomes support the conclusion that NHEJ is more prone to giving rise to genomic rearrangements than HR54,55. Recurrent chromosomal translocations are the initiating events in several tumour types, including several leukaemias, lymphomas, sarcomas and even prostate cancer. Some type of NHEJ is responsible for joining the two chromosome ends because translocation breakpoints have little or no sequence homology54. More recent global analysis of rearrangements in lung cancer genomes using paired-end sequencing has also shown a predominance of non-homologous events55.
Thus, restrictions on HR, including sequence heterology and crossover suppression, attenuate potentially deleterious HR events in the genomes of somatic cells, emphasizing the key role that HR has in promoting precise repair in lieu of mutagenic repair by other pathways. The question arises as to what benefit NHEJ has if it can be promiscuous in joining DNA ends. The canonical NHEJ pathway has an important role in maintaining genome integrity, especially in G1 or G0, when HR does not operate. Thus, loss of canonical NHEJ proteins, similarly to loss of HR proteins, results in genomic rearrangements56. However, these rearrangements involve joining at non-homologous sequences, pointing to a non-canonical type of NHEJ for their formation57–59, the normal role of which remains unclear.
HR and tumour suppression
The connection between HR proteins and tumour suppressor genes followed soon after the discovery that RAD51 is important in maintaining genomic integrity in mammals. Similarly to RAD51, BRCA1 and BRCA2 form DNA damage-induced nuclear foci, and, importantly, interact with RAD51 (REFS 60–62). These key observations led investigators to directly test the importance of BRCA1 and BRCA2 in HR. Brca1-mutant mouse embryonic stem cells were found to have reduced HR63, and targeted correction of the Brca1 mutation reversed cellular phenotypes such as genome instability64. Subsequently, Brca2-mutant mouse cells and human cancer cells were found to have HR defects65.
Overall, Brca1- and Brca2-mutants fall into the classic paradigm of HR mutants: spontaneous and damage-induced chromosomal instability, impaired RAD51 focus formation, mild ionizing radiation sensitivity, severe ICL sensitivity and centrosome abnormalities66–68. As with Rad51, Brca1 or Brca2 disruption in mice leads to embryonic lethality69,70. Despite these similarities, differences have been noted between the roles of BRCA1 and BRCA2 in the other DNA repair pathway assessments: Brca1-mutant cells have reduced SSA40,63, whereas Brca2-mutant cells have increased SSA40,71; Brca1-mutant cells have reduced gene targeting compared with Brca2-mutant cells, despite a similar magnitude of the HR defect63,65; and Brca1-mutant cells have a small increase in NHEJ63, whereas Brca2-mutant cells do not40,72. These results point to different roles in DSB repair: BRCA1 is involved early, perhaps at an end processing step, whereas BRCA2 is clearly central to the strand exchange step (see below). Of note, in addition to differences in repair phenotypes, human tumour suppression by BRCA1 and BRCA2 is also different. Inherited mono-allelic mutations predispose to tumorigenesis when the wild-type BRCA1 or BRCA2 allele is lost, but there are differences in the tissues at risk, gender specificity, disease penetrance and tumour pathology69,73. Although both proteins suppress tumours of the female breast and ovary, BRCA2 is also important for the suppression of male breast, pancreas and prostate cancer. And although an inherited BRCA1 biallelic mutation has not been observed, inherited BRCA2 biallelic mutation leads to a severe Fanconi anaemia phenotype with early tumours of other types (see below). Additional genes that encode BRCA1- and BRCA2-interacting proteins — BRCA1-interacting protein 1 (BRIP1; also known as BACH1 and FANCJ) and partner and localizer of BRCA2 (PALB2; also known as FANCN) — have also been identified as tumour suppressors; their loss results in moderate penetrance breast cancer susceptibility with monoallelic mutations and Fanconi anaemia with biallelic mutations, as detailed below.
BRCA2: requirement for RAD51 function
Human BRCA2 is a large, ~ 410 kDa protein comprising several domains that function to bind RAD51 and DNA, which are features thought to be required to facilitate HR74. Of note, the central region of the protein contains a series of eight short repeats, termed BRC repeats, which bind RAD51 (FIG. 4). The BRC repeats are divergent from each other and thus may not all bind to RAD51 identically75. An unrelated RAD51 interaction domain is found at the carboxyl terminus of BRCA2, which can bind and stabilize RAD51–DNA filaments from disruption by BRC repeats76,77. Recently, biochemical studies have shown that one or multiple BRC repeats stimulate the formation of RAD51 nucleoprotein filaments on short ssDNA in the presence of ATP78. Furthermore, the repeats maintain the active ATP-bound form of RAD51–ssDNA filaments, stimulating strand exchange and preventing the formation of non-productive RAD51–dsDNA filaments. How all of the RAD51 interactions are managed in the context of full-length BRCA2 remains to be determined.
Figure 4. Hr protein interactions and domains.

The homologous recombination proteins breast and ovarian cancer type 1 susceptibility protein (BRCA1), partner and localizer of BRCA2 (PALB2) and BRCA2 form complexes with RAD51 and are tumour suppressors. BRCA1-interacting protein 1 (BRIP1) also promotes homologous recombination and may be a tumour suppressor. Abraxas and CtBP-interacting protein (CtIP) have key roles in the repair functions of BRCA1, although they have not been identified as tumour suppressors. These proteins are involved in various steps of DNA repair, which includes damage recognition, end processing, repair protein localization (at damage-induced nuclear foci) and DNA strand exchange. BRIP1, abraxas and CtIP interact with BRCA1 through its BRCT domain. Other relevant interactions and functional domains are indicated. Arrows show protein–protein interactions. Mutually exclusive binding of the BRCA1 BRCT domains to either BRIP1, abraxas or CtIP through their phosphorylated SerXXPhe (pSerXXPhe) residues are shown (dashed arrows). Structures of defined domains with interacting peptides are shown on the right: the BRCA1 BRCT repeats with a pSerXXPhe-containing peptide (1), the PALB2 carboxy-terminal β-propeller with a small amino-terminal fragment of BRCA2 (magenta; 2), and the BRCA2 BRC4 peptide with RAD51 (3). In the BRCA1 BRCT domains, Met1775 forms the base of the recognition pocket for the Phe residue in the pSerXXPhe peptide and has been found to be mutated to Arg in breast cancers; this mutation abrogates the ability of the BRCA1 BRCT to bind pSerXXPhe peptides in vitro104. In the PALB2 interaction with the BRCA2 peptide, the BRCA2 residue Trp31 is highlighted because mutations of this residue that abrogate the interaction with PALB2 have been found in breast cancers87. The asterisks in PALB2 highlight the interaction of the N- and C-terminal residues of the WD40 structure; deletion of the last four amino acids (Tyr1183X; in which X denotes a stop codon), which has been found in patients, disrupts the structure of the protein to destabilize it88. In the BRCA2 BRC4 peptide, the aromatic ring of Phe1524 is buried within a hydrophobic pocket of RAD51, probably mimicking the self interaction of Phe86 of RAD51 with this RAD51 pocket79,80. BRC4 contains two modules that interact with RAD51; the N terminus is shown in magenta and the C terminus in blue. Image in part 1 is reproduced, with permission, from REF. 104 © (2004) Elsevier. Image in part 2 is reproduced, with permission, from EMBO Reports REF. 88 © (2009) Macmillan Publishers Ltd. All rights reserved. Image in part 3 is reproduced from REF. 80. BARD1, BRCA1-associated RING domain protein 1; DSS1, deleted in split hand/split foot protein 1; OB, oligonucleotide–oligosaccharide binding.
Structure determinations of one repeat, BRC4, have identified two distinct modules within a 33-amino acid peptide that contribute to RAD51 binding79,80 (FIG. 4). The BRC4 amino terminus adopts a hairpin structure that mimics the oligomerization motif of RAD51, occupying hydrophobic pockets that would normally be occupied by an adjacent RAD51 monomer in a filament79. This mimicry provides a structure-based explanation of how BRC repeats interfere with HR when overexpressed in cells81, although it presumably promotes RAD51 function in its normal context. The BRC4 C terminus forms an α-helix and contributes to RAD51 binding by occupying a distinct hydrophobic pocket80. Therefore, complexity of binding exists even within a single BRC repeat.
Results with BRC4, which makes up just 1% of BRCA2, raise the question regarding the role of the remaining portion of the protein. The structure of a well-conserved ~ 800-amino acid region from the C terminus showed that BRCA2 is a ssDNA-binding protein82. This region of BRCA2 consists of four globular domains arranged in a linear manner and a fifth domain, which has a coiled-coil region extending out like a tower. Three globular domains are oligonucleotide–oligosaccharide-binding (OB) folds found in ssDNA-binding proteins such as RPA, whereas the tower domain has a three-helix bundle at the apex that is similar in structure to some dsDNA-binding domains. The presence of ssDNA and possible dsDNA binding led to the suggestion that BRCA2 delivers RAD51 to ssDNA–dsDNA junctions that are formed by end resection; experiments carried out with a BRCA2 orthologue from the fungus Ustilago maydis (the agent of corn smut) supports this interpretation83. Nonetheless, dsDNA binding may not be essential, as RPA fused to a BRC repeat corrects the HR defects of BRCA2-deficient cells81. More recently, the absolute requirement for the DNA binding domain has been called into question by genetic reversion of BRCA2 mutations that are missing this domain (BOX 2). Furthermore, mice in which BRCA2 lacks this domain, but retains at least some BRC repeats, survive much longer than BRCA2-null mice69,70.
In hereditary breast cancers, BRCA2 mutations are found throughout the length of the protein, most often as truncating mutations but also as missense mutations (see the breast cancer information core database). In contrast to the monoallelic inheritance of mutations found in adults with tumours, biallelic inheritance of BRCA2 mutations results in Fanconi anaemia of the D1 subtype (Fanconi anaemia D1)84. Because BRCA2 mutations are found in patients with Fanconi anaemia D1, it is also alternatively referred to as FANCD1. At least one of the two mutated BRCA2 alleles in patients with Fanconi anaemia D1 has a partial loss of function mutation, explaining how children with Fanconi anaemia D1 survive to term. However, children with Fanconi anaemia D1 manifest more severe and earlier clinical phenotypes than patients with other types of Fanconi anaemia. A distinguishing feature of patients with Fanconi anaemia D1 is the frequent occurrence of medulloblastoma, other brain tumours and Wilms’ tumour (a pediatric malignancy of the kidney), in addition to the haematological malignancies that are common to most Fanconi anaemia groups85,86. As a result, they have a short life expectancy and consequently do not develop bone marrow failure, which is a characteristic of other Fanconi anaemia groups.
PALB2: a link between BRCA1 and BRCA2
PALB2 was first identified as a BRCA2-interacting protein by mass spectrometry of proteins that immunoprecipitate with BRCA2 (REF. 87). A substantial fraction of cellular BRCA2 associates with PALB2, and a large fraction of PALB2 associates with BRCA2. So far, PALB2 has been identified only in higher eukaryotes, despite the presence of BRCA2 in several model organisms74. The N terminus of BRCA2 interacts with the C terminus of PALB2, which forms a WD40 β-propeller domain that is commonly involved in protein–protein interactions87,88 (FIG. 4). The BRCA2 N terminus is conserved among vertebrates, but not among species that do not have PALB2, consistent with its function being tied to that of PALB2. A crystal structure of the BRCA2–PALB2 interaction has recently been reported and it shows the BRCA2 peptide forming a short α-helix binding to an outer pocket of the PALB2 β-propeller domain88.
Importantly, PALB2 deficiency produces similar cellular phenotypes to those seen with BRCA2 deficiency. Notably, HR is reduced by knockdown of PALB2 or by expression of N-terminal BRCA2 peptides that would interfere with their interaction87. As expected by the HR defect, PALB2 disruption sensitizes cells to ICLs. BRCA2 and PALB2 colocalize in nuclear foci during S phase and after DNA damage87, and both colocalize with BRCA1 and RAD51 (REFS 89,90). Evidence supports a hierarchy of recruitment to DNA damage sites: BRCA1 does not depend on any of these proteins for nuclear focus formation, but PALB2 shows some dependence on BRCA1. BRCA2 nuclear focus formation requires PALB2, whereas RAD51 focus formation requires all three proteins.
A longstanding question in the field has been how BRCA1 and BRCA2 interact. Recent work has provided strong evidence that PALB2 serves as the link between BRCA1 and BRCA2 (REFS 89–91). Specifically, immunoprecipitation of PALB2 brings down both BRCA1 and BRCA2, and disruption of PALB2 expression abolishes the interaction between BRCA1 and BRCA2 (REF. 90). BRCA1 and PALB2 interact at coiled-coil regions found in both proteins, at residues upstream of the BRCT repeats in BRCA1 and at the N-terminal residues of PALB2 (FIG. 4). Importantly, abolishing the interaction of BRCA1 with PALB2 impairs HR, linking the function of these three proteins89,90.
PALB2 mutations and human disease
Several reports have provided evidence that PALB2 is a Fanconi anaemia protein and a tumour suppressor, although the frequency of PALB2 familial mutations is much lower than that of BRCA1 and BRCA2 (REFS 92–94). In studies of diverse populations, monoallelic truncating mutations of PALB2 have been identified in ~ 1% of familial breast cancer cases that do not have BRCA1 or BRCA2 mutations94–96, and a founder mutation of PALB2 in the Finnish population occurs at a frequency of ~ 4% (REF. 97). Cancer risk is estimated to be increased fourfold in individuals with the Finnish founder mutation and approximately twofold in other populations with PALB2 mutations94,96,98. The number of tumours analysed so far is too small to draw conclusions as to whether PALB2 mutations cause tumours that are similar to those seen in patients with BRCA2 mutations94,97,99. However, surprisingly, out of the handful of tumours analysed only one PALB2 tumour has been reported with LOH of the wild-type PALB2 allele94,97,100. PALB2 mutations have also been reported in male breast cancer and familial pancreatic cancer96,100,101. Children with Fanconi anaemia N (which have a mutation in PALB2) have clinical similarities with children with Fanconi anaemia D1 in that they are at high risk for developing Wilms’ tumour and medulloblastoma within the first few years of life93.
Truncating mutations found in tumours and in children with Fanconi anaemia N are located throughout the PALB2 coding sequence92,93. The crystal structure of the BRCA2–PALB2 complex provides an explanation as to why even small deletions in the PALB2 C terminus are deleterious: they disrupt the β-propeller, rendering the protein susceptible to proteolysis88. For example, the shortest deletion Tyr1183X (in which X denotes a stop codon) deletes only the last four amino acids, but this deletion disrupts the interaction between two β-strands that seal the last blade of the β-propeller (red–blue strand interface in FIG. 4) and prevents closure of the ring. Tumour-promoting missense mutations in BRCA1 and BRCA2 that abrogate PALB2 binding have been identified87,102. Mutation of BRCA2 Trp31 disrupts the interaction of BRCA2 with a hydrophobic pocket on the outside of the β-propeller of PALB2 (REF. 88) (FIG. 4). Similarly, mutations in the coiled-coil domain of BRCA1 (Met1400Val, Leu1407Pro and Met1411Thr) abolish its interaction with PALB2 (REF. 102).
BRCA1 BRCT domains and genome stability
Although the BRCA1 tumour suppressor was linked to DNA repair a decade ago, its mechanistic role in HR is not fully elucidated. Several key BRCA1 functional domains, including the N-terminal RING domain and the C-terminal BRCT tandem repeats, promote HR and are necessary for tumour suppression. Interestingly, the RING domain has E3 ubiquitin ligase activity, but this function is not required for HR103. BRCT domains at the C terminus of BRCA1 are sequence repeats of approximately 90 amino acids that mediate interactions with phosphorylated proteins which are involved in the DNA damage response104. In BRCA1, the tandem BRCT repeats pack close together in a head to tail fashion, forming a hydrophobic interface105, and bind the phosphorylated Ser motif pSerXXPhe106. The phosphorylated Ser binds to a pocket in the N-terminal BRCT domain and the Phe binds to the interface created by the tandem BRCT structure107 (FIG. 4).
Importantly, binding to the BRCT region is mutually exclusive: only one protein with the pSerXXPhe motif can occupy the site on BRCA1. As a result, the complexes formed between BRCA1 and BRCT-interacting proteins are functionally distinct108; these are described below. Recent work has identified new roles for BRCT-interacting proteins, and several cancer-associated missense mutations located at the BRCA1 BRCT domains abrogate binding to these proteins. Disturbance of the binding of the phosphorylated Ser to the N-terminal BRCT repeat or the hydrophobic interface of the tandem repeats has been shown biochemically and structurally, highlighting the importance of the tandem BRCT structure to BRCA1 tumour suppression107,109,110.
Abraxas — localization of BRCA1 to DNA damage
Post-translational modification of proteins by ubiquitylation of Lys residues is a dynamic regulatory process in various biological pathways, similar to phosphorylation. In DNA damage and repair, monoubiquitylation and Lys63 polyubiquitylation conjugates predominate111. In a search for additional BRCA1 BRCT-interacting proteins, several groups identified new proteins, the binding of which depends on ubiquitylation. Abraxas binds directly to the BRCT domain, whereas RAP80 interacts with BRCA1 indirectly through abraxas108. RAP80 binds to polyubiquitylated histone H2AX, modifications of which are important in the DNA damage response112, thereby bringing BRCA1 to damaged DNA108,113,114. The heterodimer UBC13–RNF8 (ubiquitin-conjugating enzyme 13–RING finger protein 8) carries out Lys63 polyubiquitylation of H2AX115–118. UBC13-deficient cells have severe HR defects119, whereas cells depleted of abraxas or RAP80 exhibit mild HR defects108, implicating additional functions of ubiquitylation in HR120. Therefore, abraxas-mediated localization of BRCA1 has a role in the DNA damage response, although the less severe HR phenotype associated with abraxas knockdown suggests it is not essential for this pathway.
BRIP1 — a DNA helicase
BRIP1, a DNA helicase originally identified as BACH1 and later as FANCJ, binds to the BRCA1 BRCT repeats in S phase following its phosphorylation121–123. Human BRIP1 interacts with BRCA1 at its C terminus, whereas the ATP-dependent helicase domain is in the N-terminal two thirds of the protein124. BRIP1 depletion in human cells results in defective HR and hypersensitivity and chromosome instability following exposure to ICL-inducing agents122. Notably, in contrast to depletion of BRCA1, RAD51 focus formation is intact following exposure to both hydroxyurea and ICL-inducing agents102,122. Interestingly, BRIP1-depleted cells have a greater degree of ICL hypersensitivity compared with BRCA1-depleted cells. BRIP1 is also required for timely progression through S phase; this and other functions of BRIP1 depend on its helicase activity121,125,126.
Surprisingly, the worm and avian homologues of BRIP1 lack the C-terminal domain or the C-terminal pSerXXPhe residues, respectively, that are required for BRCA1 BRCT binding125,127. Consistent with this, a physical interaction between the worm BRIP1 and BRCA1 orthologues is not detected. Despite the notable divergence, the worm and chicken BRIP1 mutants are hypersensitive to ICL-inducing agents, incur genetic instability after damage and monoubiquitylate FANCD2 (REFS 122,127). However, unlike human cells, chicken BRIP1 mutants show no decrease in HR and arrest in G2, a phenotype that is similar to that of Fanconi anaemia core complex mutants after ICL damage125.
The worm BRIP1 homologue is known as DOG-1 (deletion of guanine-rich DNA 1) based on the observation that its loss results in deletions at poly-G tracts that have the ability to form non-canonical DNA structures which can impede replication128. HR (and translesion synthesis) reduce the number of deletions, which supports the genome stabilizing effect of HR when unstable DNA structures are encountered129. Importantly, resolution of four-stranded structures (G quadraplexes or G4 DNA) by purified BRIP1 was identified in human cell extracts130. More recently, additional genome destabilizing phenotypes have been observed in worms, including chromosome rearrangements, large complex deletions, duplications and translocations131. In humans, array comparative genomic hybridization have identified large deletions with a bias towards adjacent G4 DNA regions in Fanconi anaemia J cells compared with Fanconi anaemia D2 and control cells132. BRIP1 may function to regulate HR by both DNA unwinding and displacement of proteins at the damaged site. Preliminary biochemical data suggest that excess BRIP1 may inhibit RAD51-mediated D-loop formation133. The S phase-dependent, BRCA1-mediated association of BRIP1 with chromatin along with its DNA helicase substrate specificity for secondary DNA structures, including ICL damage, supports a role for BRIP1 in maintaining genome stability during replication-dependent HR.
BRIP1 has been identified as the deficient protein in Fanconi anaemia J subtype122,125,134,135. Unlike the markedly severe clinical syndromes of patients with Fanconi anaemia D1 and Fanconi anaemia N described above, patients with Fanconi anaemia J typically present with growth and developmental anomalies and bone marrow failure occurring with an average latency of 4.5 years (ranging between 2–6 years)135. One patient developed leukaemia at 13.5 years; however, no solid tumours have been observed. Notably, several early deaths occurred in utero and within the first days and weeks of life135. Both truncating and missense mutations are identified in patients with Fanconi anaemia J, including the recurrent homozygous Arg798X truncation, which deletes part of the helicase domain and the BRCA1 BRCT-interacting domain. Clinical phenotypic variation is observed even with the recurrent homozygous Arg798X truncation. Similar to PALB2, BRIP1 has been implicated as a human breast tumour suppressor, and inheritance of a mutated BRIP1 allele is estimated to result in a twofold increased risk of breast cancer136. So far, BRIP1 mutations have been identified in 12 patients with breast cancer. Because the mutation frequency is low (0.4% for the recurring Arg798X mutation in selected populations and 0.05% in unselected populations), there are limited data confirming the role of BRIP1 in tumour suppression136,137.
CtIP — DNA end resection promotes HR
A key determinant for repair pathway choice between HR and NHEJ is the requirement for RAD51 to bind ssDNA, as discussed above. Resection of the DNA strand is regulated during the S and G2 phases of the cell cycle by CDK phosphorylation, which then allows binding of phosphorylated CtIP to the BRCA1 BRCT domain and subsequent 5′ to 3′ end resection of a DNA strand23,24. This reaction also involves the MRE11–RAD50–NBS1 (MRN) complex24,138. CtIP genetic loss is associated with early embryonic lethality in mice139, and cells deficient in CtIP show HR defects102,138,140. Further analysis of repair defects with single and combined RNA interference depletion experiments confirmed a mild HR repair defect with loss of BRIP1 or CtIP alone, moderately severe HR defects with depletion of BRCA1 or PALB2, and markedly severe defect with depletion of RAD51. Notably, combined knockdown of any two proteins resulted in severe HR defects, including knockdown of CtIP and BRIP1 (REF. 102). No inactivating CTIP mutations have been reported in human diseases.
Perspectives
HR has evolved to be tightly regulated to promote precise repair and limit genomic aberrations and genetic loss. This is achieved through cell cycle phase coordination, post-translational modifications and many accessory factors that either catalyse or inhibit interactions. Although BRCA1 (REF. 141) and to a lesser extent BRCA2 have been implicated in additional functions, specific tumour susceptibility missense mutations that interrupt recently identified HR protein–protein interactions, such as BRCA1–PALB2 and PALB2–BRCA2, emphasize the central role of HR in tumour suppression in the human breast. Most heritable breast cancer predisposition results from defects in genes that are involved in DNA damage signalling and repair. BRCA1 and BRCA2, the loss of which confers the highest inherited risk of breast cancer, are also the most crucial for HR repair. Factors with more peripheral roles in HR, such as BRIP1, or with roles in DNA damage signalling, such as ataxia telangiectasia mutated (ATM) and checkpoint kinase 2 (CHEK2), confer a more modest risk of breast cancer when mutated. The current outlier to this classification is PALB2, as PALB2 mutations are estimated to confer modest breast cancer risk but severe Fanconi anaemia phenotypes; the severe Fanconi anaemia phenotypes are consistent with experimental evidence demonstrating a central repair function. It is possible that additional factors that modify HR proficiency can predispose to tumorigenesis when mutated. However, it is difficult to ascertain whether subtle defects in HR owing to alterations in peripheral modifying factors lead to clinically deleterious phenotypes. As defective HR represents a target for emerging therapies in cancer therapeutics142, this question is immediately relevant.
Acknowledgments
The authors thank past and present members of the laboratory, especially E. Kass, and colleagues in the field for discussions. Work in the authors’ laboratory is supported the Hecksher Foundation for Children (M.E.M.) and National Institutes for Health grants P01CA94060 (M.E.M and M.J.) and R01GM54668 (M.J.).
Glossary
- B-form DNA
The most common helical DNA structure, also called canonical DNA, comprising two aligned strands of DNA in opposite polarity forming a right-handed helix
- Non-crossover
Homologous recombination in which DNA sequences are copied from the donor strand to the recipient strand without an exchange of genetic information with the recipient strand flanking DNA
- Holliday junction
A structural intermediate formed between four DNA strands during homologous recombination
- Crossover
Resolution of homologous recombination resulting in an exchange of DNA sequences between the donor and recipient
- Loss of heterozygosity
Reduction of genetic information from both maternal and paternal alleles to genetic information from a single parent
- Centrosome
A cytoplasmic organelle that organizes microtubules. Preceding mitosis, the centrosome doubles and then is involved in the generation of the mitotic spindle for subsequent chromosome segregation during mitosis
- Fanconi anaemia
A genetic disorder arising from biallelic mutations in one of 13 different genes, characterized by chromosome instability that typically presents early in life, with developmental disorders, anaemia, bone marrow failure and solid and haematologic malignancy. There is a high degree of clinical variation that depends on both the gene defect and mutation type
- Somatic mosaicism
The existence of more than one genetically distinct population of somatic cells in an organism. This can arise by DNA mutation, chromosome non-disjunction, recombination or the spontaneous reversion of inherited mutations
- E3 ubiquitin ligase
A ubiquitin ligase that, in combination with an e2 ubiquitin-conjugating enzyme, adds ubiquitin (a 76-amino acid protein) to a Lys on a target protein
Footnotes
Competing interests statement
The authors declare no competing financial interests.
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/gene
BRIP1|PALB2
uniProtKb: http://www.uniprot.org
abraxas | BRCA1 | BRCA2 | Cdk1 | CtIP | RAD51|
FURTHER INFORMATION
Maria Jasin’s homepage: http://www.mskcc.org/mskcc/html/10566.cfm
Breast cancer information core database: http://research.nhgri.nih.gov/bic/
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 2.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability — an evolving hallmark of cancer. Nature Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 3.Liang F, Han M, Romanienko PJ, Jasin M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc Natl Acad Sci USA. 1998;95:5172–5177. doi: 10.1073/pnas.95.9.5172. Compares HR and NHEJ repair of a genomic DSB in mammalian cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aquilera A, Rothstein R. Molecular Genetics of Recombination. Springer; Berlin Germany: 2007. [Google Scholar]
- 5.Haber JE. Partners and pathways repairing a double-strand break. Trends Genet. 2000;16:259–264. doi: 10.1016/s0168-9525(00)02022-9. [DOI] [PubMed] [Google Scholar]
- 6.Johnson RD, Jasin M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans. 2001;29:196–201. doi: 10.1042/0300-5127:0290196. [DOI] [PubMed] [Google Scholar]
- 7.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nature Rev Mol Cell Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 8.Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci. 2009;34:264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 9.Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 10.Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14:8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang F, Jasin M. Ku80 deficient cells exhibit excess degradation of extrachromosomal DNA. J Biol Chem. 1996;271:14405–14411. doi: 10.1074/jbc.271.24.14405. [DOI] [PubMed] [Google Scholar]
- 12.Tsuzuki T, et al. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci USA. 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim D-S, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. References 12 and 13 provided two of the first indications of a crucial role for HR in mammals. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265:1241–1243. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- 15.Baumann P, Benson FE, West SC. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87:757–766. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- 16.Sung P, Robberson DL. DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell. 1995;82:453–461. doi: 10.1016/0092-8674(95)90434-4. [DOI] [PubMed] [Google Scholar]
- 17.Benson FE, Stasiak A, West SC. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J. 1994;13:5764–5771. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Z, Yang H, Pavletich NP. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453:489–494. doi: 10.1038/nature06971. Crystal structures of RecA with ssDNA and dsDNA, leading to the discovery that, although stretched, DNA adopts a local B-form DNA-like structure, which restricts the homology search to sequences that can form Watson–Crick base pairs. [DOI] [PubMed] [Google Scholar]
- 19.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. References 19 and 20 provide evidence for a two-step mechanism for DNA end resection in yeast to initiate HR: initial limited resection by the Mre11 complex and Sae2, followed by processive resection involving either Exo1 or Sgs1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA. 2008;105:16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buis J, et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem. 1998;273:25388–25392. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 24.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. Discovery that CtIP promotes HR and is related to the yeast protein Sae2, which is involved in DNA end-resection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ira G, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004;23:4868–4875. doi: 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sung P, Krejci L, Van Komen S, Sehorn MG. Rad51 recombinase and recombination mediators. J Biol Chem. 2003;278:42729–42732. doi: 10.1074/jbc.R300027200. [DOI] [PubMed] [Google Scholar]
- 30.Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 31.Baudat F, de Massy B. Regulating double-stranded DNA break repair towards crossover or non-crossover during mammalian meiosis. Chromosome Res. 2007;15:565–577. doi: 10.1007/s10577-007-1140-3. [DOI] [PubMed] [Google Scholar]
- 32.Esashi F, et al. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi: 10.1038/nature03404. [DOI] [PubMed] [Google Scholar]
- 33.Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. Using phosphorylated histone H2AX (γH2AX) as a marker for the repair of DSBs generated by low doses of ionizing radiation, this study shows that NHEJ is important for DSB repair in all cell cycle phases, and HR is important in late S and G2. By contrast, DSBs produced by a replication inhibitor are predominately repaired by HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- 35.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–3242. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allen C, Kurimasa A, Brenneman MA, Chen DJ, Nickoloff JA. DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proc Natl Acad Sci USA. 2002;99:3758–3763. doi: 10.1073/pnas.052545899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinstock DM, Jasin M. Alternative pathways for the repair of RAG-induced DNA breaks. Mol Cell Biol. 2006;26:131–139. doi: 10.1128/MCB.26.1.131-139.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cox MM, et al. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 39.Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu Rev Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 40.Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bennardo N, Gunn A, Cheng A, Hasty P, Stark JM. Limiting the persistence of a chromosome break diminishes its mutagenic potential. PLoS Genet. 2009;5:e1000683. doi: 10.1371/journal.pgen.1000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000;19:3398–3407. doi: 10.1093/emboj/19.13.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delacote F, Han M, Stamato TD, Jasin M, Lopez BS. An xrcc4 defect or wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res. 2002;30:3454–3463. doi: 10.1093/nar/gkf452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakanishi K, et al. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci USA. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dronkert ML, et al. Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol Cell Biol. 2000;20:3147–3156. doi: 10.1128/mcb.20.9.3147-3156.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stark JM, Jasin M. Extensive loss of heterozygosity is suppressed during homologous repair of chromosomal breaks. Mol Cell Biol. 2003;23:733–743. doi: 10.1128/MCB.23.2.733-743.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiese C, Pierce AJ, Gauny SS, Jasin M, Kronenberg A. Gene conversion is strongly induced in human cells by double-strand breaks and is modulated by the expression of BCL-xL. Cancer Res. 2002;62:1279–1283. [PubMed] [Google Scholar]
- 48.Neuwirth EA, Honma M, Grosovsky AJ. Interchromosomal crossover in human cells is associated with long gene conversion tracts. Mol Cell Biol. 2007;27:5261–5274. doi: 10.1128/MCB.01852-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moynahan ME, Jasin M. Loss of heterozygosity induced by a chromosomal double-strand break. Proc Natl Acad Sci USA. 1997;94:8988–8993. doi: 10.1073/pnas.94.17.8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hagstrom SA, Dryja TP. Mitotic recombination map of 13cen-13q14 derived from an investigation of loss of heterozygosity in retinoblastomas. Proc Natl Acad Sci USA. 1999;96:2952–2957. doi: 10.1073/pnas.96.6.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elliott B, Jasin M. Repair of double-strand breaks by homologous recombination in mismatch repair-defective mammalian cells. Mol Cell Biol. 2001;21:2671–2682. doi: 10.1128/MCB.21.8.2671-2682.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richardson C, Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 2000;405:697–700. doi: 10.1038/35015097. [DOI] [PubMed] [Google Scholar]
- 53.Elliott B, Richardson C, Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell. 2005;17:885–894. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 54.Lieber MR, Yu K, Raghavan SC. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair (Amst) 2006;5:1234–1245. doi: 10.1016/j.dnarep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 55.Campbell PJ, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nature Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferguson DO, et al. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci USA. 2000;97:6630–6633. doi: 10.1073/pnas.110152897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weinstock DM, Brunet E, Jasin M. Formation of NHEJ-derived reciprocal chromosomal translocations does not require Ku70. Nature Cell Biol. 2007;9:978–981. doi: 10.1038/ncb1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang JH, et al. Mechanisms promoting translocations in editing and switching peripheral B cells. Nature. 2009;460:231–236. doi: 10.1038/nature08159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4/ligase IV during chromosomal translocation formation. Nature Struct Mol Biol. doi: 10.1038/nsmb.1773. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haaf T, Golub EI, Reddy G, Radding CM, Ward DC. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci USA. 1995;92:2298–2302. doi: 10.1073/pnas.92.6.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scully R, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. First report to link BRCA1 to RAD51: co-localization of the two proteins is observed in nuclear foci. [DOI] [PubMed] [Google Scholar]
- 62.Sharan SK, et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386:804–810. doi: 10.1038/386804a0. First report to link BRCA2 to RAD51 function; early embryonic lethality of Brca2-mutant mice and radiation sensitivity of Brca2-mutant embryos are observed. [DOI] [PubMed] [Google Scholar]
- 63.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. Shows that BRCA1 deficiency results in HR defects. [DOI] [PubMed] [Google Scholar]
- 64.Moynahan ME, Cui TX, Jasin M. Homology-directed DNA repair, mitomycin-C resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61:4842–4850. [PubMed] [Google Scholar]
- 65.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. Similarly to reference 63, this study shows that BRCA2 deficiency results in HR defects, although phenotypic differences between BRCA1 and BRCA2 mutants are noted. [DOI] [PubMed] [Google Scholar]
- 66.Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 2002;21:8981–8993. doi: 10.1038/sj.onc.1206176. [DOI] [PubMed] [Google Scholar]
- 67.Pellegrini L, Venkitaraman A. Emerging functions of BRCA2 in DNA recombination. Trends Biochem Sci. 2004;29:310–316. doi: 10.1016/j.tibs.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 68.Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864–5874. doi: 10.1038/sj.onc.1209874. [DOI] [PubMed] [Google Scholar]
- 69.Moynahan ME. The cancer connection: BRCA1 and BRCA2 tumor suppression in mice and humans. Oncogene. 2002;21:8994–9007. doi: 10.1038/sj.onc.1206177. [DOI] [PubMed] [Google Scholar]
- 70.Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006;25:5885–5897. doi: 10.1038/sj.onc.1209871. [DOI] [PubMed] [Google Scholar]
- 71.Tutt A, et al. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20:4704–4716. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xia F, et al. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc Natl Acad Sci USA. 2001;98:8644–8649. doi: 10.1073/pnas.151253498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Honrado E, Osorio A, Palacios J, Benitez J. Pathology and gene expression of hereditary breast tumors associated with BRCA1, BRCA2 and CHEK2 gene mutations. Oncogene. 2006;25:5837–5845. doi: 10.1038/sj.onc.1209875. [DOI] [PubMed] [Google Scholar]
- 74.Christ N, Moynahan ME, Jasin M. In: Molecular Genetics of Recombination. Rothstein R, editor. Springer; Berlin: 2007. pp. 363–380. [Google Scholar]
- 75.Galkin VE, et al. BRCA2 BRC motifs bind RAD51-DNA filaments. Proc Natl Acad Sci USA. 2005;102:8537–8542. doi: 10.1073/pnas.0407266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Davies OR, Pellegrini L. Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nature Struct Mol Biol. 2007;14:475–483. doi: 10.1038/nsmb1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Esashi F, Galkin VE, Yu X, Egelman EH, West SC. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nature Struct Mol Biol. 2007;14:468–474. doi: 10.1038/nsmb1245. [DOI] [PubMed] [Google Scholar]
- 78.Carreira A, et al. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell. 2009;136:1032–1043. doi: 10.1016/j.cell.2009.02.019. Proposes that BRC repeats of BRCA2 control RAD51–DNA interactions by directing active RAD51 to ssDNA and inhibiting RAD51 nucleation on dsDNA to promote HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pellegrini L, et al. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. Crystal structure of a fusion between the BRC4 repeat from BRCA2 with the core domain of RAD51, demonstrating that BRC4 can adopt a structure that mimics a RAD51 self-interaction motif. [DOI] [PubMed] [Google Scholar]
- 80.Rajendra E, Venkitaraman AR. Two modules in the BRC repeats of BRCA2 mediate structural and functional interactions with the RAD51 recombinase. Nucleic Acids Res. 2009;38:82–96. doi: 10.1093/nar/gkp873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saeki H, et al. Suppression of the DNA repair defects of BRCA2-deficient cells with heterologous protein fusions. Proc Natl Acad Sci USA. 2006;103:8768–8773. doi: 10.1073/pnas.0600298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang H, et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. Crystal structure of an 800-amino acid domain of BRCA2, showing that BRCA2 is a ssDNA-binding protein. [DOI] [PubMed] [Google Scholar]
- 83.Yang H, Li Q, Fan J, Holloman WK, Pavletich NP. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature. 2005;433:653–657. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- 84.Howlett NG, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. Discovery of biallelic mutations in BRCA2, with a link to developmental defects and tumours in children, similar to mutations in genes encoding Fanconi anaemia proteins. [DOI] [PubMed] [Google Scholar]
- 85.Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. 2007;44:1–9. doi: 10.1136/jmg.2006.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reid S, et al. Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet. 2005;42:147–151. doi: 10.1136/jmg.2004.022673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xia B, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. Identification of PALB2 as a BRCA2-interacting protein with a role in HR. [DOI] [PubMed] [Google Scholar]
- 88.Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009;10:990–996. doi: 10.1038/embor.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sy SM, Huen MS, Zhu Y, Chen J. PALB2 regulates recombinational repair through chromatin association and oligomerization. J Biol Chem. 2009;284:18302–18310. doi: 10.1074/jbc.M109.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang F, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19:524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res. 2009;7:1110–1118. doi: 10.1158/1541-7786.MCR-09-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xia B, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nature Genet. 2007;39:159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 93.Reid S, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nature Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 94.Tischkowitz M, et al. Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci USA. 2007;104:6788–6793. doi: 10.1073/pnas.0701724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Foulkes WD, et al. Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007;9:R83. doi: 10.1186/bcr1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rahman N, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nature Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Erkko H, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–319. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 98.Erkko H, et al. Penetrance analysis of the PALB2 c. 1592delT founder mutation. Clin Cancer Res. 2008;14:4667–4671. doi: 10.1158/1078-0432.CCR-08-0210. [DOI] [PubMed] [Google Scholar]
- 99.Heikkinen T, et al. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res. 2009;15:3214–3222. doi: 10.1158/1078-0432.CCR-08-3128. [DOI] [PubMed] [Google Scholar]
- 100.Garcia MJ, et al. Analysis of FANCB and FANCN/PALB2 fanconi anemia genes in BRCA1/2-negative Spanish breast cancer families. Breast Cancer Res Treat. 2009;113:545–551. doi: 10.1007/s10549-008-9945-0. [DOI] [PubMed] [Google Scholar]
- 101.Jones S, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA. 2009;106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reid LJ, et al. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc Natl Acad Sci USA. 2008;105:20876–20881. doi: 10.1073/pnas.0811203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Glover JN, Williams RS, Lee MS. Interactions between BRCT repeats and phosphoproteins: tangled up in two. Trends Biochem Sci. 2004;29:579–585. doi: 10.1016/j.tibs.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 105.Williams RS, Green R, Glover JN. Crystal structure of the BRCT repeat region from the breast cancer- associated protein BRCA1. Nature Struct Biol. 2001;8:838–842. doi: 10.1038/nsb1001-838. [DOI] [PubMed] [Google Scholar]
- 106.Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 107.Williams RS, Lee MS, Hau DD, Glover JN. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nature Struct Mol Biol. 2004;11:519–525. doi: 10.1038/nsmb776. [DOI] [PubMed] [Google Scholar]
- 108.Wang B, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Clapperton JA, et al. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nature Struct Mol Biol. 2004;11:512–518. doi: 10.1038/nsmb775. [DOI] [PubMed] [Google Scholar]
- 110.Tischkowitz M, et al. Pathogenicity of the BRCA1 missense variant M1775K is determined by the disruption of the BRCT phosphopeptide-binding pocket: a multi-modal approach. Eur J Hum Genet. 2008;16:1820–1832. doi: 10.1038/ejhg.2008.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Panier S, Durocher D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair (Amst) 2009;8:436–443. doi: 10.1016/j.dnarep.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 112.Ayoub N, et al. The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr Biol. 2009;19:1075–1085. doi: 10.1016/j.cub.2009.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- 114.Sobhian B, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huen MS, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kolas NK, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mailand N, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 118.Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci USA. 2007;104:20759–20763. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao GY, et al. A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol Cell. 2007;25:663–675. doi: 10.1016/j.molcel.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 120.Huang J, et al. RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nature Cell Biol. 2009;11:592–603. doi: 10.1038/ncb1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cantor SB, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 122.Litman R, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 123.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phosphoprotein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 124.Cantor S, et al. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci USA. 2004;101:2357–2362. doi: 10.1073/pnas.0308717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nature Genet. 2005;37:953–957. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- 126.Kumaraswamy E, Shiekhattar R. Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol Cell Biol. 2007;27:6733–6741. doi: 10.1128/MCB.00961-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Youds JL, et al. DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross-link repair. Mol Cell Biol. 2008;28:1470–1479. doi: 10.1128/MCB.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cheung I, Schertzer M, Rose A, Lansdorp PM. Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nature Genet. 2002;31:405–409. doi: 10.1038/ng928. [DOI] [PubMed] [Google Scholar]
- 129.Youds JL, O’Neil NJ, Rose AM. Homologous recombination is required for genome stability in the absence of DOG-1 in Caenorhabditis elegans. Genetics. 2006;173:697–708. doi: 10.1534/genetics.106.056879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wu Y, Shin-ya K, Brosh RM., Jr FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhao Y, Tarailo-Graovac M, O’Neil NJ, Rose AM. Spectrum of mutational events in the absence of DOG-1/FANCJ in Caenorhabditis elegans. DNA Repair (Amst) 2008;7:1846–1854. doi: 10.1016/j.dnarep.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 132.London TB, et al. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J Biol Chem. 2008;283:36132–36139. doi: 10.1074/jbc.M808152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sommers JA, et al. FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J Biol Chem. 2009;284:7505–7517. doi: 10.1074/jbc.M809019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Levitus M, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J Nature Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 135.Levran O, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nature Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 136.Seal S, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nature Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 137.De Nicolo A, et al. A novel breast cancer-associated BRIP1 (FANCJ/BACH1) germ-line mutation impairs protein stability and function. Clin Cancer Res. 2008;14:4672–4680. doi: 10.1158/1078-0432.CCR-08-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1. CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 139.Chen PL, et al. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol Cell Biol. 2005;25:3535–3542. doi: 10.1128/MCB.25.9.3535-3542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Huen MS, Sy SM, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nature Rev Mol Cell Biol. 2010;11:138–148. doi: 10.1038/nrm2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 143.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Johnson RD, Liu N, Jasin M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature. 1999;401:397–399. doi: 10.1038/43932. Shows the importance of sister chromatid recombination for DSB repair in mammalian cells and provides direct evidence for a factor involved in HR repair in mammalian cells. [DOI] [PubMed] [Google Scholar]
- 145.Carroll D. Progress and prospects: zinc-finger nucleases as gene therapy agents. Gene Ther. 2008;15:1463–1468. doi: 10.1038/gt.2008.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brunet E, et al. Chromosomal translocations induced at specified loci in human stem cells. Proc Natl Acad Sci USA. 2009;106:10620–10625. doi: 10.1073/pnas.0902076106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kalb R, et al. Hypomorphic mutations in the gene encoding a key Fanconi anemia protein, FANCD2, sustain a significant group of FA-D2 patients with severe phenotype. Am J Hum Genet. 2007;80:895–910. doi: 10.1086/517616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lo Ten Foe JR, et al. Somatic mosaicism in Fanconi anemia: molecular basis and clinical significance. Eur J Hum Genet. 1997;5:137–148. [PubMed] [Google Scholar]
- 149.Waisfisz Q, et al. Spontaneous functional correction of homozygous fanconi anaemia alleles reveals novel mechanistic basis for reverse mosaicism. Nature Genet. 1999;22:379–383. doi: 10.1038/11956. [DOI] [PubMed] [Google Scholar]
- 150.DeMarini DM, Shelton ML, Abu-Shakra A, Szakmary A, Levine JG. Spectra of spontaneous frameshift mutations at the hisD3052 allele of Salmonella typhimurium in four DNA repair backgrounds. Genetics. 1998;149:17–36. doi: 10.1093/genetics/149.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ikeda H, et al. Genetic reversion in an acute myelogenous leukemia cell line from a Fanconi anemia patient with biallelic mutations in BRCA2. Cancer Res. 2003;63:2688–2694. [PubMed] [Google Scholar]
- 152.Sakai W, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Edwards SL, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. References 152 and 153 identify genetic reversion of BRCA2, which leads to resistance to therapeutic agents. [DOI] [PubMed] [Google Scholar]
- 154.Auerbach AD. Fanconi anaemia and its diagnosis. Mutat Res. 2009;668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang W. Emergence of DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nature Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]