

Abstract
Aging is characterized by a general decline in cellular function, which ultimately will affect whole body homeostasis. Although DNA damage and oxidative stress all contribute to aging, metabolic dysfunction is a common hallmark of aging at least in invertebrates. Since a comprehensive overview of metabolic changes in otherwise healthy aging mammals is lacking, we here compared metabolic parameters of young and 2 year old mice. We systemically integrated in vivo phenotyping with gene expression, biochemical analysis, and metabolomics, thereby identifying a distinguishing metabolic footprint of aging. Among the affected pathways in both liver and muscle we found glucose and fatty acid metabolism, and redox homeostasis. These alterations translated in decreased long chain acylcarnitines and increased free fatty acid levels and a marked reduction in various amino acids in the plasma of aged mice. As such, these metabolites serve as biomarkers for aging and healthspan.
Aging (and ultimately death) is an inevitable part of life and comes with all sorts of physical and mental ailments, including common metabolic, inflammatory, cardiovascular and neurodegenerative diseases, which will reduce healthspan. Examples of such diseases include Parkinson’s and Alzheimer’s disease, and from a cardiometabolic perspective, obesity, type 2 diabetes, and atherosclerosis. Even though these clinical manifestations are well known, the complex biomolecular networks contributing to the aging process are only beginning to be uncovered1. Several common processes are suggested to cause or at least contribute to aging, including DNA damage, accumulation of reactive oxygen species (ROS), and general metabolic dysfunction. So far, these were mostly seen as independent events, but evidence suggests that some of these pathways are interconnected, as recently highlighted by the link between DNA damage and metabolic control2,3.
Regardless of the mechanism, a common feature of aging-related disease is the involvement of metabolic systems in general, and the mitochondria in particular1,4,5,6. The best-characterized metabolic pathway implicated in aging is the insulin/IGF1 signaling pathway7. Both the C. elegans and D. melanogaster mutants of insulin receptor showed increased lifespan and the effect is mediated through the FOXO1 transcription factor, the heat-shock factor HSF1, and SKN1 (reviewed in5). Involvement of the insulin/IGF1 pathway in mammalian lifespan regulation is debated but seems likely7. A second longevity pathway is centered on the mammalian target of rapamycin (mTOR), which integrates insulin signaling with sensing of other nutrients —most notably amino acids— and as such regulates protein translation and autophagy8,9. In line with this, mTOR was shown to be involved in the aging-associated decrease in ketone body production10 and inhibition of mTOR by administration of rapamycin increased mouse lifespan11. Opposing these nutrient excess systems are the nutrient restriction pathways, such as the sirtuin and AMP-activated protein kinase (AMPK) pathways12,13. Being activated by energy stress, for instance after prolonged fasting or exercise, they inhibit energy-demanding processes in favor of energy-production14. Both sirtuins and AMPK have been suggested as positive regulators of longevity in lower organisms (discussed in1), but as for the insulin/IGF1 pathway, convincing involvement in mammalian longevity needs to be confirmed. In C. elegans, AMPK was shown to directly phosphorylate the CREB-regulated transcriptional coactivator-1, which then becomes unable to coactivate the CREB transcription factor to decrease lifespan15, indicating an aging pathway that could be conserved in mammals. Additionally, in mouse, AMPK has been connected with SIRT116,17 and PGC-1α signaling17,18 consolidating the link of this system with mitochondrial metabolism, notably mitochondrial respiration. It is still debated, however, how mitochondrial function impacts aging, as both inhibiting and stimulating mitochondrial metabolism seems to enhance lifespan (discussed in1,5). Most likely, the physiological context and the nature of the manipulation itself may determine the aging response. This phenotype seems at least partly mediated by the mitochondrial unfolded protein response and acts in a cell-non-autonomous fashion involving a yet unidentified “mitokine”19.
Several studies have reported the molecular effects of aging, for instance at the biochemical20, genomic21, or metabolomic level22. The limitation of these studies, however, lies in the problems with the multilevel integration of these different datasets. We therefore aimed to obtain an unbiased map and understand the metabolic decline during aging that predisposes to aging-related diseases in mammals. We characterized clinical, molecular and metabolomic aspects of aging in wild type C57BL/6J mice—the most commonly used strain for metabolic studies. This enabled us to define a consensus phenotype of aging by identifying metabolite sets and molecular changes that can be used as healthspan biomarkers and thereby constitute the detailed metabolic footprint of aging.
Results
Metabolic characterization and physical performance
To characterize metabolic changes during mammalian aging, we employed C57BL/6J mice of 3 months (“young”) and 22 months (“old”). We validated our aging model through an extensive clinical phenotyping effort. As expected, aged mice showed signs of physiological decline. Old mice are significantly heavier than young mice (Fig 1A), mostly because of an increase in fat mass (Fig 1B). Furthermore, old mice developed discrete glucose intolerance, reflecting potentially the development of insulin resistance (Supplementary Fig S1 online). In indirect calorimetry old mice had a tendency to decreased energy expenditure when corrected for lean body mass (Fig 1C) or when compared with multiple regression (p = 0.128), likely in part due to decreased physical activity (Supplementary Fig S1 online). The respiratory exchange ratio (RER) in old mice was lowered compared to young mice, indicating that aging mice have a proportional substrate preference towards fat (Fig 1C). When challenged with a physical exercise, maximum VO2 was significantly decreased (Fig 1D)
Figure 1. Clinical and molecular phenotyping of aging mice.
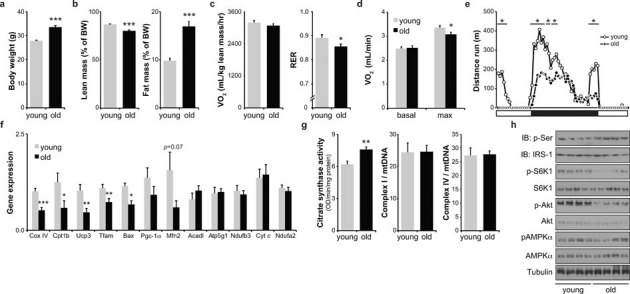
(A) Body weight. (B) Body composition. (C) Indirect calorimetry shows oxygen consumption (VO2) and respiratory exchange ratio (RER) during 24h recording. (D) Basal and maximum VO2 on a metabolic treadmill. (E) Twenty-four hours voluntary exercise recording. (F) Gene expression of young and old mice in gastrocnemius muscle (G) Enzymatic activity of citrate synthase (CS), and complex I and complex IV of oxidative phosphorylation in gastrocnemius muscle. (H) Western blot analysis of relevant metabolic signaling pathways in gastrocnemius muscle. pAMPKα represents phosphorylation/activation of the α-subunit of AMPK; Immunoprecipitated IRS1 was used to measure its phosphorylation on serine residues (p-Ser); p-S6K1 reflects S6K1 phosporylation/activation. Tubulin is used as a loading control. Values are expressed as mean±SEM; n = 6–10 (Fig 1A, n = 20). * p≤0.05; ** p≤0.01; *** p≤0.001.
The decreased physical activity in old mice during indirect calorimetry, was confirmed in a home-cage monitoring, during which mice had free access to a running wheel (Fig 1E). Old mice ran less both during the initial discovery phase and at later stages during the night phase. This hypodynamic phenotype in the old mice was also observed in a square open field test, which showed the old mice to be less agile than the young mice (Supplementary Fig S1 online). Catwalk analysis —used to determine gait disturbances in mice— revealed that the time standing on one paw before taking the next step is significantly increased with age (Supplementary Fig S1 online).
Molecular and biochemical alterations in aging tissues
Molecular characterization of tissues of the aged mice confirmed the view of metabolic —notably mitochondrial— decline that is thought to underlie part of the aging process. In gastrocnemius muscle, brown adipose tissue (BAT) and liver, we observed a drop in expression of a variety of transcripts encoding proteins involved in oxidative phosphorylation (Ndufa2, Ndufb3, Cyt c, Cox IV, Atp5g1, Ucp3), fatty acid oxidation (FAO) (Cpt1b, Acadm, Acadl), mitochondrial biogenesis (Tfam), oxidative stress defense (Sod2), and apoptosis (Bax) (Fig 1F, Supplementary Fig S1 online). Further validating the fact that our study captured mainly aging-associated changes was the fact that the gene expression data were fully consistent with a previously published survey of C57BL/6N gastrocnemius muscle gene expression comparing 5 and 25-month-old mice21 (Supplementary Fig S1 online). Interestingly, in this dataset the decreased expression of many genes involved in oxidative phosphorylation (e.g. Atpaf2, Cox4i2), FAO (e.g. Cpt1b, Acads), and oxidative stress defense (Sod2) was reverted by caloric restriction (CR), the only physiological intervention increasing lifespan known to date (Supplementary Fig S1 online). In contrast, expression of various antioxidants (Gpx3, Gpx4, Gpx6) was increased in old mice and normalized in CR mice (Supplementary Fig S1 online).
Although the expression of nuclear genes encoding mitochondrial proteins was generally decreased, some were unchanged (e.g. Acadl and Atp5g1 in gastrocnemius, and Mfn2 and Cpt1a in liver), and in BAT the expression of several genes increased with age (Tfam, Mfn2, Pgc-1α, Cox IV) (Fig 1F, Supplementary Fig S1 online). We hypothesized that this is due to compensatory mechanisms that are activated in old mice to maintain proper mitochondrial function. Another line of evidence for possible compensatory mechanisms to maintain mitochondrial capacity comes from enzyme activity analysis. Both in muscle and liver, citrate synthase (CS) activity was increased in the old mice, indicating increased mitochondrial activity or number (Fig 1G, Supplementary Fig S1 online). In contrast, relative activity of complex I and IV was equal between the two age groups (Fig 1G, Supplementary Fig S1 online). The increased CS activity in muscle and liver is not derived from mitochondrial biogenesis, as mitochondrial DNA (mtDNA) quantification in these tissues revealed no changes (Supplementary Fig S1 online). In BAT, however, an increase in mtDNA was observed (Supplementary Fig S1 online) following the increased gene expression of among others Tfam and Pgc-1α (Supplementary Fig S1 online). Deterioration of mitochondrial function is often reflected in increased levels of ROS. We therefore measured ROS damage, by virtue of 4-hydroxynonenal (4-HNE) —a marker for lipid peroxidation. As expected, old mice had increased levels of 4-HNE in muscle as well as liver (Supplementary Fig S1 online).
Signaling pathways in aging
To complement our gene expression and enzyme activity data, we also assessed metabolic signaling pathways, focusing on established metabolic regulation pathways linked to aging. In the old livers and muscles, insulin receptor substrate 1 (IRS-1) serine phosphorylation, which is a marker for insulin resistance, was increased, although to a different extent in the two tissues (Fig 1H, Supplementary Fig S1 online). In both liver and muscle, p70 S6 kinase phosphorylation was markedly decreased (Fig 1H, Supplementary Fig S1 online), reflecting decreased mTOR activity, possibly consequent to impaired insulin signaling, as suggested by the higher IRS-1 serine phosphorylation and reduced Akt phosphorylation. In addition to these nutrient excess pathways, we also analyzed the expression and activity of the nutrient restriction sensor AMPK and found that its phosphorylation, indicative for energy stress, was not changed in liver and muscle of old mice (Fig 1H, Supplementary Fig S1 online).
Metabolic disturbances in blood
To further characterize the metabolic changes upon aging, we performed clinical chemistry analysis in plasma. Various changes in plasma markers were noted, such as increased free fatty acid and decreased triglyceride levels (Fig 2A, C). To expand our characterization of blood metabolites in aging mice and to identify possible novel aging biomarkers we took a less targeted approach and generated an extensive metabolomics profile in the blood of young and old mice. Over 75 analytes including profiles of erythrocyte fatty acids and plasma acylcarnitines, amino acids and ketone bodies were measured and these data were clustered in order to identify groups of metabolites that respond similarly during aging (Fig 2B–C). Using this profile of metabolites, the mice were separated out according to age and several interesting metabolite clusters became apparent. One of these clusters contained long chain as well as medium chain acylcarnitines. These metabolites, which are intermediates of FAO, were decreased in the old mice (Fig 2C). The erythrocyte fatty acid profile reflects long-term changes in fatty acid homeostasis and is routinely performed for the diagnosis of poly-unsaturated fatty acid (PUFA) deficiency. For the saturated fatty acids, C16 was slightly decreased whereas the elongated C18 and C20 were increased. In addition, various intermediates and products of PUFA biosynthesis such as C20:4ω6 were increased, whereas the substrates C18:2ω6 and C18:3ω3 were decreased. Overall levels of PUFAs as well as plasmalogens, which both have been implicated in protection against ROS, were not decreased (Fig 2B).
Figure 2. Plasma biochemistry and targeted metabolomics.
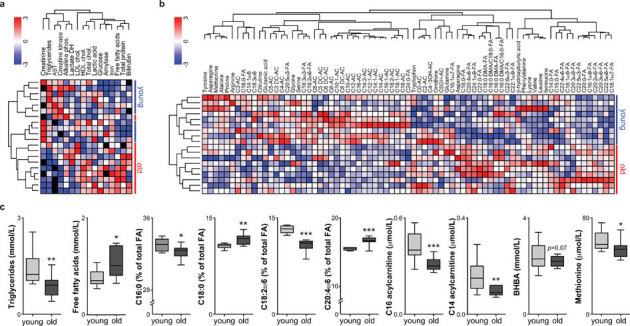
(A) Two-way hierarchical clustering of plasma clinical biochemistry. (B) Two-way hierarchical clustering of targeted metabolomics data. Metabolites described by FA are erythrocyte fatty acids, whereas ACs denote plasma acylcarnitines. (C) Plasma levels of selected metabolites. Values are expressed as box-and-whisker plots; n = 10; * p≤0.05; ** p≤0.01; *** p≤0.001.
In addition to fat metabolism, amino acid metabolism also alters significantly during aging. It was shown before that supplementation of branched-chain amino acids (BCAAs)23 or alteration in methionine content in the diet24,25 impacts lifespan. Our characterization of the plasma amino acids associated a clear reduction in the levels of proline, alanine, serine, tyrosine and methionine with aging but not in the levels of the BCAAs valine, leucine and isoleucine (Fig 2B–C). Similarly, it was previously shown that the mTOR pathway inhibits ketogenesis and that ketogenic capacity is reduced in aging10. In line with decreased ketogenesis, our analysis found that β-hydroxybutyrate (BHBA) levels, which reflect total ketone body levels, tended to decrease with age (Fig 2C).
Novel biomarkers of aging
The clear differences in plasma metabolite levels between young and old mice led us to test whether these can be used to discriminate the two groups reliably, a prerequisite for biomarker development.
Selection of relevant variables for sample classification is a common task with high dimensional data sets such as metabolomics and gene expression studies where one tries to identify the smallest possible set of metabolites or genes (i.e. biomarkers) that can still achieve good predictive performance. Random Forest (RF) is one such learning algorithm that can do this by estimating the importance of each variable to the classes. RF analysis generates many classification trees using a random training set of mice and a random small selection of metabolites. Mice that are left out of the tree are used to estimate the classification error providing an internal cross validation. As such, the “mean decrease accuracy” indicates how much a certain metabolite contributes to separation of the two test groups, and the overall “predictive accuracy” indicates the accuracy with which a set of metabolites combined can separate the groups. RF analysis of blood targeted metabolomics data defined a set of 10 metabolites that constitute the best predictors of aging, with a 100% predictive accuracy (Fig 3A). In particular, decreased erythrocyte linoleic acid (C18:2ω6) and decreased plasma C16-acylcarnitine were strong predictors (Fig 3B).
Figure 3. Metabolite and pathway enrichment of targeted metabolomics and microarray.
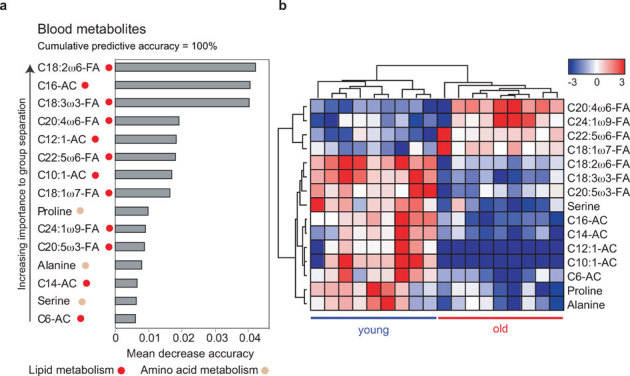
(A) To relate aging status to the blood metabolite expression data, we used random forest predictors. A random forest importance measure was used to rank metabolites according to their prognostic importance for aging status. We find that the 10 most important metabolites lead to an apparent predictive accuracy of 100%. (B) Supervised hierarchical clustering plot for visualizing the expression of the RF predictors (z-score normalized). The RF plot and the supervised hierarchical clustering plot show that the 15 most important metabolites stratify the samples according to their aging status. Metabolites described by FA are erythrocyte fatty acids, whereas ACs denote plasma acylcarnitines. A color code describes the pathway to which the respective metabolite belongs.
Following these clear metabolic changes in our blood metabolite analysis and microarray data, we also charted metabolic changes in both liver and quadriceps muscle of young and old mice using global metabolomics. We detected 473 and 263 metabolites in liver and muscle, respectively, of which 94 and 65 were significantly changed between the groups (see Supplementary tables S1 and S2 online for relative metabolite levels). We again performed RF analysis to classify these metabolic changes (Fig 4–5). In liver, the metabolite changes could 93% accurately predict the age groups (Fig 4A) and hierarchical clustering of the top 25 metabolites confirmed the separation of the two age groups (Fig 4B). Following RF analysis, which was aimed to identify relative contribution of individual metabolites from the high-dimensional metabolomics data, we performed metabolite set enrichment analysis (MSEA)26 to establish which pathways are affected. In liver, the best metabolites to predict age were in glucose metabolism, phospholipid metabolism, and redox homeostasis (Fig 4C). Additionally, we performed a metabolome view analysis to demonstrate whether a metabolite ‘hub’ node was affected, which could have more biological consequences than a ‘deadend’ metabolite node, confirming that mainly carbohydrate and glycerolipid metabolism, and redox balance were disturbed (Fig 4C). With respect to redox homeostasis, for example, various metabolites were changed in old mice, notably the metabolite hub reduced glutathione (Supplementary Fig S2 online).
Figure 4. Aging biomarker identification and pathway analysis by global metabolomics in liver.
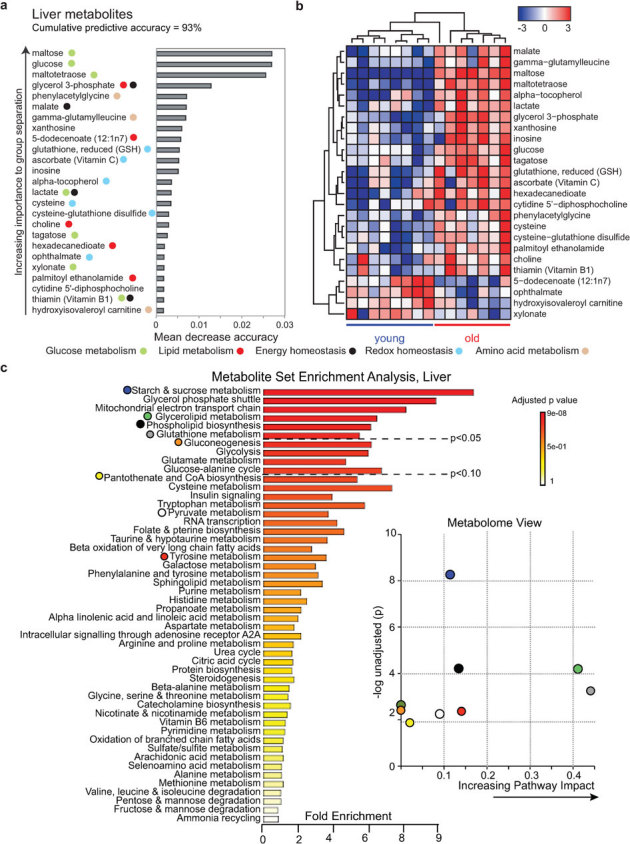
To relate aging status to the liver metabolite expression data, we used random forest predictors (A). Metabolites were ranked according to their increasing importance to group separation according to age. We find that the 25 and most important probesets lead to an apparent predictive accuracy of 93%. Colored symbols are used to indicate the pathways in which the metabolites play a role. Supervised hierarchical clustering plot (B) for visualizing the expression of the RF predictors (z-score normalized). The RF plot and the supervised hierarchical clustering plot show that the 25 most important metabolites stratify the samples according to their aging status. (C) Summary plot for metabolite set enrichment analysis (MSEA) (left panel) where metabolite sets are ranked according to Holm p-value with hatched lines showing the cut off of Holm p-value. Metabolome view (right panel) reflects on the x-axis increasing metabolic pathway impact according to the betweenness centrality measure, which reflects key nodes in metabolic pathways that have been significantly altered with aging.
Figure 5. Aging biomarker identification and pathway analysis by global metabolomics in muscle.
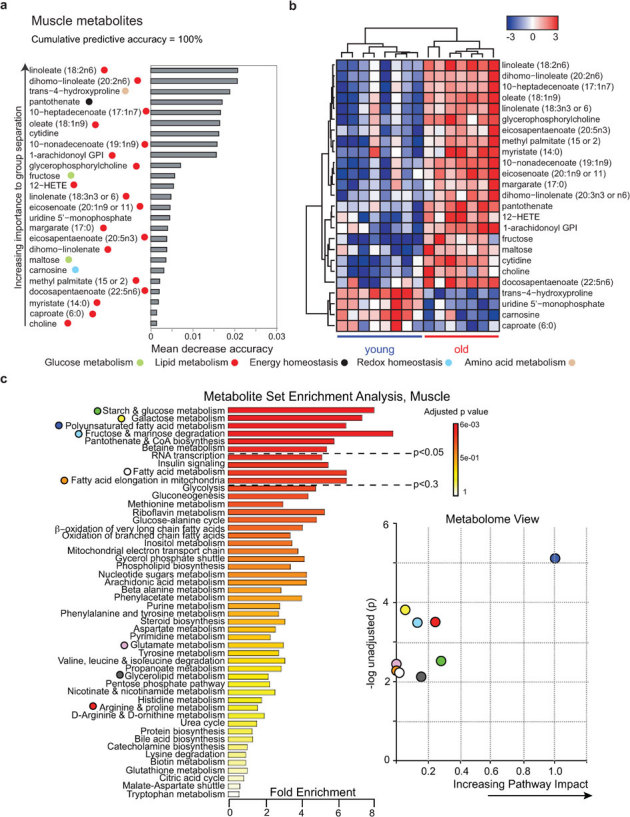
To relate aging status to the muscle metabolite expression data, we used random forest predictors (A). Metabolites were ranked according to their increasing importance to group separation according to age. We find that the 25 and most important probesets lead to an apparent predictive accuracy of 100%. Colored symbols are used to indicate the pathways in which the metabolites play a role. Supervised hierarchical clustering plot (B) for visualizing the expression of the RF predictors (z-score normalized). The RF plot and the supervised hierarchical clustering plot show that the 25 most important metabolites stratify the samples according to their aging status. (C) Summary plot for metabolite set enrichment analysis (MSEA) (left panel) and metabolome view (right panel) as discussed in Figure 4C.
In muscle, the top 25 is dominated by fatty acid metabolites (Fig 5A), with the top 20 set already showing a predictive accuracy of 100% and clear group separation (Fig 5B). MSEA and metabolome view confirmed involvement of fatty acid metabolism, but also highlighted a strong contribution of glucose metabolism, with metabolism of the poly-unsaturated fatty acids linolenic and linoleic acid being the best predictors of age in muscle (Fig 5A, Supplementary Fig S3 online). These metabolites serve as hubs in poly-unsaturated fatty acid metabolism and therefore have a high impact on predictive power (Supplementary Fig S3 online).
Discussion
Aging and the diseases associated with it are a heavy burden on society. The current increase in life expectancy not only impacts on our social systems, but also goes hand in hand with the emergence of common chronic diseases, including those of the nervous, immune, and cardio-metabolic systems, which often reach epidemic proportions. In this respect it is important to understand the natural aging process and to elucidate where lifestyle and/or pharmacological interventions can have an impact. In recent years, many novel therapeutic options have been suggested to prevent aging-associated disease. Although some of these pharmaceutical interventions were shown to extend lifespan, even in mammals11,27, there is no good readout for improved healthspan as aging biomarkers have not been identified.
Here, we characterized clinical as well as molecular, biochemical, and metabolomic alterations in young versus old mice, thereby defining a consensus phenotype of aging. Besides metabolomics in blood and plasma, we also performed an unbiased approach to determine metabolic changes in muscle and liver. We used C57BL/6J mice, as these are particularly suitable for studying metabolic disease28,29. We realize that our study could be biased by the changes in body composition that accompany aging, but this phenomenon is inherent to aging in all species. Although it could be argued that an aging model devoid of changes in fat mass or glucose homeostasis would be optimal, such a model does not exist, as physiological decline is a hallmark of aging.
The compendium that we generated of the changes occurring in naturally aging mice highlighted a wide variety of molecular and metabolic changes. Our data suggest strong involvement of impaired mitochondrial function. Not only did we observe a marked decrease in expression of various transcripts involved in mitochondrial function —e.g. oxidative phosphorylation, FAO, apoptosis—, but also we measured changes in enzymatic activity, altogether possibly culminating in the accumulation of oxidative damage, as evidenced by elevated 4-HNE levels. This coincided with the identification of redox homeostasis and its hub glutathione as affected pathways in our liver and muscle metabolomic analysis, suggesting that reactive oxygen species play a role in the initiation or progression of the aging phenotype. Interestingly, the lowered gene expression and oxidative phosphorylation activity in various tissues seems to be counteracted by a compensatory increase in mitochondrial biogenesis and/or activity, as CS in muscle and mtDNA abundance in BAT are increased in old mice.
From a signaling perspective, the insulin and mTOR pathways were affected. Increased IRS-1 serine phosphorylation is linked to insulin resistance30, a signature of aging that was also observed in our study (Supplementary Fig S1 online). IRS-1 serine phosphorylation impedes its proper association with the insulin receptor and, therefore, the ability of insulin to promote IRS-1 tyrosine phosphorylation and activate Akt and PI3-kinase and its downstream pathways31,32,33. Consistent with this, S6K1 phosphorylation was reduced in tissues from old mice. S6K1 phosphorylation depends on mTOR, which is coordinately activated by nutrients and PI3-kinase34. Serine phosphorylation of IRS-1, hence, provides a plausible explanation for decreased mTOR signaling. Several kinases can induce IRS-1 serine phosphorylation, including JNK35, IKK36, and well as novel PKCs36,37. The activation of JNK and IKK is linked to chronic oxidative and inflammatory stress, as observed with aging. Novel PKCs, such as PKC-θ, are activated by diacylglycerols, which accumulate when serum free fatty acids increase and mitochondrial function is defective, both conditions again being met with aging. In agreement with previous observations38,39, basal AMPK phosphorylation, reflective of AMPK activation, was not changed with aging, indicating that the defective mitochondrial function observed in aged tissues does not compromise ATP generation in the basal state.
One of the main goals of our study was to identify novel aging biomarkers. Therefore, we performed extensive blood and plasma metabolomics, mainly focusing on nutrient pathways. In this approach we identified marked changes in amino acid and fatty acid metabolite levels that could accurately predict aging. Besides blood metabolomics we also performed an unbiased approach to determine metabolic changes in aging tissues. Both in muscle and liver, we identified metabolite sets that are changed upon aging—including fatty acid and glucose metabolism.
We found glucose and intermediates of glycolysis and glycogen metabolism, such as maltose and maltotetraose, as biomarkers for aging in liver as well as muscle (Fig 4–5, 6A). In both liver and muscle, accumulation of glycogen intermediates suggests altered glycogen metabolism in aged mice, whereas the increased lactate and reduced glycolytic intermediates suggest elevated anaerobic glycolysis. In muscle, the elevated levels of glucose, glucose-6-phosphate and maltose, also suggest changes in glucose and glycogen metabolism (Fig 6A). Indeed, the increase in muscle maltose levels is consistent with increased glycogenolysis. Importantly, these metabolite data go in pair with the clinical —impaired glucose tolerance— and signaling —increased IRS-1 serine phosphorylation— data. Elevated maltose levels were also detected in a recent metabolomics survey of blood samples from diabetes patients40, again supporting that some of the metabolic changes we found are linked to aging-related insulin resistance.
Figure 6. Models of aging-related changes in glucose and fat metabolism.
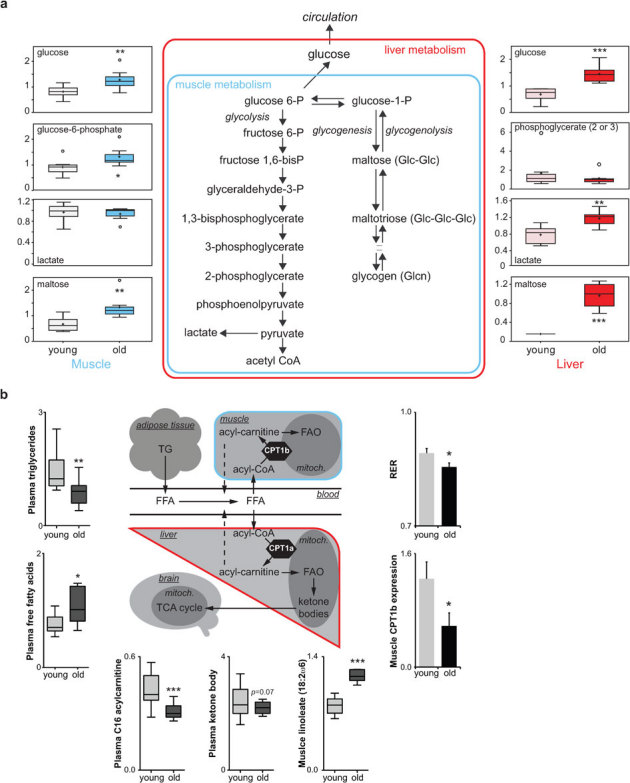
(A) A summary of the biochemical pathways of glucose metabolism affected in muscle and liver. Metabolites that changed between young and old are indicated in bold; representative metabolites data from the muscle and liver are shown in the left and right margin respectively. In summary, accumulation of the glycogen intermediates maltose and maltotetraose suggests increased liver glycogen breakdown in old mice. On the other hand, increased lactate and reduced glycolysis intermediates suggest elevated glycolysis. Elevated glucose and glucose--phosphate in muscle, in combination with the unchanged lactate levels, suggest decreased glucose disposal in muscle, either by breakdown or glycogen synthesis. Indeed, the increase in muscle maltose levels is consistent with increased glycogenolysis. (B) A schematic representation of fat metabolism, showing representative changes. Plasma triglycerides were decreased, whereas free fatty acids were increased in old mice, suggesting enhanced triglyceride deposition in tissues, as well as enhanced liberation and/or decreased breakdown of fatty acids. Decreased FAO is supported by accumulation fatty acid in the muscle, decreased leak of acylcarnitines into plasma (dashed line) and reduced ketogenesis. Decreased Cpt1b expression in muscle likely contributes to the decreased FAO, also explaining lower plasma acylcarnitine levels. The fact that RER is lower in old mice —indicative for relative reliance on fat oxidation— suggests that glucose oxidation is also hampered and fat metabolism is still the primary source for ATP generation. Values are expressed as mean±SEM or as box-and-whisker plots. * p≤0.05; ** p≤0.01; *** p≤0.001.
A second pathway that was clearly affected in old mice is fatty acid metabolism (Fig 6B). Not only did we observe a decrease in gene expression of genes involved in FAO (Cpt1b in muscle, Acadm in liver, Acadl in BAT), but also metabolites derived from this pathway were perturbed. We found plasma free fatty acids to be increased, whereas plasma medium and long chain acylcarnitines were lower in old mice. In contrast, it is well known that during fasting both free fatty acids as well as medium and long chain acylcarnitines in plasma increase, reflecting increased FAO flux41. Therefore, our data fit with reduced entry in and flux through the mitochondrial FAO pathway42,43, causing an increase in FA levels and a concomitant reduced “leak” of acylcarnitines back into the plasma compartment (dashed line in Fig 6B). It is important to note that in aged humans, several fatty acids were also more abundant in plasma22, although no information is available for acylcarnitines. Our hypotheses also fit with the accumulation of fatty acids in muscle and with the decrease in Cpt1b expression in muscle, suggesting a reduced carnitine esterification at the mitochondrial membrane and hence reduced entry into the mitochondria for FAO. This seems in apparent contradiction with the fact that RER is reduced in old mice —indicative of increased reliance on fat for energy generation. Indirect calorimetry, however, measures whole body metabolism and reflects the combination of relative changes in all metabolic pathways (e.g. glucose and fat oxidation) and all organs (e.g. liver, muscle, etc) that contribute to energy expenditure. Although we cannot exclude tissue-specific differences in FAO, our data suggest fat metabolism is still the primary source for ATP generation.
We identified molecular and biochemical profiles of aging in various tissues with high predictive accuracy. The cross validation of these pathways between our various approaches —for instance FAO metabolites in both blood and tissue metabolomics, and FAO genes in gene expression— strengthens their potential value as a biomarker. For biomarker qualification, however, we suggest to analyze metabolite levels in plasma, as it is easily obtained without the need of sacrificing the animal or being too invasive in humans. As discussed above, the changes in plasma free fatty acid and acylcarnitine levels have a high predictive value for separation of young and old mice, and could prove a strong candidate biomarker for aging and healthspan. It is also interesting to note that the biosynthetic pathway for poly-unsaturated fatty acids was strongly affected in aged muscle, but similar changes were found in erythrocyte membranes, again proving the value of blood and plasma as a source for biomarkers. Additionally, measurement of selected plasma amino acids adds predictive value. Importantly, the relevance of amino acids as a biomarker was recently corroborated as a combination of several amino acids, including the BCAAs, was associated with insulin resistance44 and predictive of future diabetes45. We found some changes in BCAA metabolites in aged liver and muscle, but not in plasma, making them less suitable as aging biomarkers.
Even though perhaps less practical from a clinical point of view, a few additional pathways turned out to be useful to monitor the aging process and to target by preventive and therapeutic strategies to delay aging and/or aging-associated pathologies, as they were proven equally predictive for aging. These include increased tissue oxidative damage and the accumulation of poly-unsaturated fatty acid intermediates, particularly in the muscle. Additionally, mRNA expression of a selected set of genes (Cox IV and Tfam) was predictive of aging as it was changed in all tissues tested. In contrast to these biomarkers that are altered in most of the analyzed tissues, some changes are indicative of aging in specific tissues, as exemplified by the increased mitochondrial gene expression and mtDNA content in BAT, perhaps enabling a better adaptation of this tissue to the metabolic challenges of aging.
Altogether, our data provide a footprint of metabolic aging in a well-controlled mammalian system. The pathways that we identified by our integrative approach also opens up a new avenue of research, aimed to identify the individual contributions of these molecular and metabolomic systems to the aging process. Although some of these pathways have already been investigated in relation to aging, further research is needed to fully establish their relevance, also in man. Future studies will also have to address how physiological challenges, such as fasting/feeding, exercise, or high-fat feeding in aged mice impact on these biomarkers. Additionally, we hope that the identification of these biomarkers of aging may spur the rational design of strategies that can prevent or reverse the decline in certain of the affected networks and as such could extend healthspan and prevent accelerated aging.
Methods
Mice
C57BL/6J mice of either 13 weeks old (“young”) or 93 weeks old (“old”) were purchased from Janvier (St. Berthevin, France). Mice were group housed and received standard chow (#2018, containing 18% protein, 50% carbohydrate and 6.0% fat; Harlan Laboratories, Madison, WI, USA). After in vivo phenotyping, mice were sacrificed after overnight fasting, reaching the age of 24 or 103 weeks. Heparinized plasma was taken and tissues were frozen in liquid nitrogen for biochemical and molecular analyses.
In vivo phenotyping
Most clinical tests were carried out according to rigorous SOPs established and validated within the Eumorphia program28,46. Body composition was determined by Echo-MRI (Echo Medical Systems, Houston, TX, USA) and oxygen consumption (VO2), respiratory exchange ratios (RER), activity levels and activity were monitored by indirect calorimetry using the comprehensive laboratory animal monitoring system (CLAMS) (Columbus Instruments, Columbus, OH, USA)47. Glucose tolerance was analyzed by measuring blood glucose and insulin following intraperitoneal injection of 2 g/kg glucose after an overnight fast48. Maximum respiration potential (VO2max) was determined in a metabolic treadmill (Columbus instruments, Columbus, OH, USA) with an incremental speed protocol. During the run, VO2 and VCO2 are measured. The experiment is stopped when mice are exhausted, RER is above 1 for more than one interval, or the VO2 reaches a clear plateau. Voluntary exercise is recorded with a recording running wheel placed in the home cage for 24h. During that period the mice are individually caged. Gait analysis was performed using the CatWalk system (Noldus Technology, Wageningen, The Netherlands)49. Mice were placed for 30 min in a square open field and their movement was recorded using video tracking (Ethovision XT, Noldus Technology, Wageningen, The Netherlands)50.
Histology and biochemical assays
Mitochondrial complex I and IV activity (Mitosciences, Eugene, OR, USA), citrate synthase (Sigma) were determined according to the manufacturer’s protocols. 4-HNE was determined in PBS tissue lysates using an ELISA-based method (Cell Biolabs, San Diego, CA, USA). Western blotting was performed as described before2. Additional antibodies include Tubulin (Santa Cruz Biotechnology), p-Ser (Sigma), p-ACC (Millipore), IRS-1, p-AMPKα, AMPKα, S6K1 and p-S6K1 (Cell Signaling Technology)51.
Gene expression and mitochondrial DNA abundance
RNA was extracted from tissues using Trizol. cDNA was synthesized using the QuantiTect RT kit (Qiagen), followed by qPCR on a Roche Lightcycler 480 and using Roche SYBR-green mastermix. qPCR primers are described in the Extended Experimental Procedures. Mitochondrial DNA abundance (mtDNA) was quantified as described before47. In short, DNA was extracted using a standard phenol extraction, and the resulting genomic DNA (gDNA) was used for qPCR using the primers in the Extended Experimental Procedures. Microarray data from gastrocnemius muscle of C57BL/6N mice of either 5 months (young), 25 months (old) or CR diet21, were retrieved from the public Gene Aging Nexus database (http://gan.usc.edu/public/index.jsp)52.
Plasma biochemistry and targeted metabolomics
Basic plasma clinical chemistry analyses were performed according to Eumorphia SOPs28,46. Plasma acylcarnitines and amino acids were determined by tandem mass spectrometry (MSn) and LC-MSn respectively53,54. Fatty acids in erythrocytes were directly transesterified and analyzed by gas chromatography with flame ionization detection55. Beta-hydroxybutyric acid was measured in perchloric acid deproteinized plasma using established procedures56
Global metabolomics
Metabolomics of quadriceps muscle and liver of young and old mice (8 and 7 mice per group, respectively) was performed by Metabolon (Durham, NC, USA), according to published methods57. In brief, sample preparation was conducted using a proprietary series of organic and aqueous extractions to remove the protein fraction while allowing maximum recovery of small molecules. The extracted samples were split into equal parts for analysis on the GC/MS and LC-MS/MS platforms. For LC-MS/MS, samples were split in two aliquots that were either analyzed in positive (acidic solvent) or negative (basic solvent) ionization mode. GC-MS was performed on bistrimethyl-silyl-triflouroacetamide derivatized samples in a 5% phenyl GC column.
Bioinformatics and statistics
Data in figures is presented as mean±SEM or as box-and-whisker plots indicating the sample minimum, lower quartile, median, upper quartile, and sample maximum with outliers represented as small circles. A Students T-test or Welch’s T-test (liver and muscle metabolomics data) was performed for statistical comparison between young and old mice. A p-value of 0.05 or less was considered significant.
Unsupervised hierarchical clustering was performed using complete linkage and Pearson rank correlation distance on the normalized metabolites using software implemented in Genepattern (http://www.broadinstitute.org/cancer/software/genepattern/)58,59. The z-score was calculated by subtracting the mean expression value for each metabolite from each of the values and then dividing the resulting values by the standard deviation. Color in the heat-maps reflects the relative metabolite abundance level with red being higher and blue lower than the mean metabolite abundance value. Metabolite and animal ordering is determined as in hierarchical clustering using the distance function 1-correlation.
The web-based metabolomic data processing tool, MetaboAnalyst60 was used for tissue metabolite data analysis. See http://www.metaboanalyst.ca for detailed methodology. Briefly, for the RF analysis, the metabolite data was log transformed (log 2) and the number of predictors to try for each node was set to the square root of the total number of variables per tissue and the number of trees to grow was set to 10.000. RF is performed using the random forest package and the resulting confusion matrix, out of bag (OOB) error and variable importance (as determined by the mean decrease in accuracy when data is permuted) are presented. For MSEA, metabolite data was mapped according to HMDB and the ‘Metabolite pathway associated metabolite set’ library (currently 88 entries) was chosen for the enrichment analysis, which is performed using package global test. The metabolome view displays the pathway topological analysis, a method that takes into account the pathway structure when determining which pathways are more likely to be of significance. The parameters chosen for this analysis included global test and the node centrality measure ‘betweenness centrality’ to estimate node importance. This measure considers the global network structure such that nodes that occur between two dense clusters will have a high betweenness centrality even if its degree centrality is not high. The node importance values calculated from centrality measures are further normalized by the sum of the importance of the pathway for comparison among different pathways. Therefore, the total/maximum importance of each pathway is 1; the importance measure of each metabolite node is actually the percentage with respect to the total pathway importance, and the pathway impact is the cumulative percentage from the matched metabolite nodes.
Author Contributions
RHH and JA conceived and designed the project, with input from CA, SMH, CC and KS. RH, CA, SMH, CC, EHJ, and PAA performed experiments. CT and RD planned and supervised mouse experiments. RHH, CA, SMH and JA wrote the manuscript, with contributions from all authors.
Supplementary Material
Supplementary info
Table S1
Table S2
Acknowledgments
We thank team members of the Auwerx lab and the mouse phenotyping unit for technical support and helpful discussions. RHH is supported by a Rubicon fellowship of the Netherlands Organization for Scientific Research. JA is the Nestlé Chair in Energy Metabolism. The work in the laboratories of the authors is supported by grants of the Ecole Polytechnique Fédérale de Lausanne, the EU Ideas program (ERC-2008-AdG-), the Velux Stiftung, the Swiss National Science Foundation (31003A-124713 and 31003A-125487) and the Netherlands Organization for Scientific Research (NWO VIDI No. 016.086.336 to SMH).
References
- Houtkooper R. H., Williams R. W. & Auwerx J. Metabolic networks of longevity. Cell 142, 9–14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P. et al.. PARP-1 Inhibition Increases Mitochondrial Metabolism through SIRT1 Activation. Cell Metab 13, 461–468 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin E. et al.. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470, 359–365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L., Partridge L. & Longo V. D. Extending healthy life span–from yeast to humans. Science 328, 321–326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C. J. The genetics of ageing. Nature 464, 504–512 (2010). [DOI] [PubMed] [Google Scholar]
- Lin M. T. & Beal M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 (2006). [DOI] [PubMed] [Google Scholar]
- Russell S. J. & Kahn C. R. Endocrine regulation of ageing. Nat Rev Mol Cell Biol 8, 681–691 (2007). [DOI] [PubMed] [Google Scholar]
- Wullschleger S., Loewith R. & Hall M. N. TOR signaling in growth and metabolism. Cell 124, 471–484 (2006). [DOI] [PubMed] [Google Scholar]
- Zoncu R., Efeyan A. & Sabatini D. M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12, 21–35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S., Peterson T. R., Laplante M., Oh S. & Sabatini D. M. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468, 1100–1104 (2010). [DOI] [PubMed] [Google Scholar]
- Harrison D. E. et al.. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis M. C. & Sinclair D. A. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5, 253–295 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Mitochondria–a nexus for aging, calorie restriction, and sirtuins? Cell 132, 171–176 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C. & Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 20, 98–105 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair W. et al.. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 470, 404–408 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco M. et al.. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell 14, 661–673 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C. et al.. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C. et al.. Interdependence of AMPK and SIRT1 for Metabolic Adaptation to Fasting and Exercise in Skeletal Muscle. Cell Metab 11, 213–219 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J., Wolff S. & Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux H., Vazquez E. J., Fujioka H. & Hoppel C. L. Decrease in mitochondrial function in rat cardiac permeabilized fibers correlates with the aging phenotype. J Gerontol A Biol Sci Med Sci 65, 1157–1164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M. G. et al.. Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genomics 8, 80 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton K. A. et al.. Analysis of the adult human plasma metabolome. Pharmacogenomics 9, 383–397 (2008). [DOI] [PubMed] [Google Scholar]
- D'Antona G. et al.. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab 12, 362–372 (2010). [DOI] [PubMed] [Google Scholar]
- Grandison R. C., Piper M. D. & Partridge L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 462, 1061–1064 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. A. et al.. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 4, 119–125 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J. & Wishart D. S. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res 38, W71–77 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson K. J. et al.. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab 8, 157–168 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champy M. F. et al.. Genetic background determines metabolic phenotypes in the mouse. Mamm Genome 19, 318–331 (2008). [DOI] [PubMed] [Google Scholar]
- Biddinger S. B. et al.. Effects of diet and genetic background on sterol regulatory element-binding protein-1c, stearoyl-CoA desaturase 1, and the development of the metabolic syndrome. Diabetes 54, 1314–1323 (2005). [DOI] [PubMed] [Google Scholar]
- Morino K., Petersen K. F. & Shulman G. I. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 55 Suppl 2, S9–S15 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz K. et al.. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem 272, 29911–29918 (1997). [DOI] [PubMed] [Google Scholar]
- Aguirre V. et al.. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem 277, 1531–1537 (2002). [DOI] [PubMed] [Google Scholar]
- Moeschel K. et al.. Protein kinase C-zeta-induced phosphorylation of Ser318 in insulin receptor substrate-1 (IRS-1) attenuates the interaction with the insulin receptor and the tyrosine phosphorylation of IRS-1. J Biol Chem 279, 25157–25163 (2004). [DOI] [PubMed] [Google Scholar]
- Pearce L. R., Komander D. & Alessi D. R. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11, 9–22 (2010). [DOI] [PubMed] [Google Scholar]
- Rui L. et al.. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest 107, 181–189 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. et al.. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem 277, 48115–48121 (2002). [DOI] [PubMed] [Google Scholar]
- Li Y. et al.. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J Biol Chem 279, 45304–45307 (2004). [DOI] [PubMed] [Google Scholar]
- Reznick R. M. et al.. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab 5, 151–156 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A. A., Kumar R., Mulligan J. D., Davis A. J. & Saupe K. W. Effects of aging on cardiac and skeletal muscle AMPK activity: basal activity, allosteric activation, and response to in vivo hypoxemia in mice. Am J Physiol Regul Integr Comp Physiol 287, R1270–1275 (2004). [DOI] [PubMed] [Google Scholar]
- Suhre K. et al.. Metabolic footprint of diabetes: a multiplatform metabolomics study in an epidemiological setting. PLoS ONE 5, e13953 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C. C., de Almeida I. T., Jakobs C., Poll-The B. T. & Duran M. Dynamic changes of plasma acylcarnitine levels induced by fasting and sunflower oil challenge test in children. Pediatr Res 46, 440–444 (1999). [DOI] [PubMed] [Google Scholar]
- Tucker M. Z. & Turcotte L. P. Impaired fatty acid oxidation in muscle of aging rats perfused under basal conditions. Am J Physiol Endocrinol Metab 282, E1102–1109 (2002). [DOI] [PubMed] [Google Scholar]
- Hyyti O. M., Ledee D., Ning X. H., Ge M. & Portman M. A. Aging impairs myocardial fatty acid and ketone oxidation and modifies cardiac functional and metabolic responses to insulin in mice. Am J Physiol Heart Circ Physiol 299, H868–875 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard C. B. et al.. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9, 311–326 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. J. et al.. Metabolite profiles and the risk of developing diabetes. Nat Med 17, 448–453 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champy M. F. et al.. Mouse functional genomics requires standardization of mouse handling and housing conditions. Mamm Genome 15, 768–783 (2004). [DOI] [PubMed] [Google Scholar]
- Lagouge M. et al.. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127, 1109–1122 (2006). [DOI] [PubMed] [Google Scholar]
- Heikkinen S., Argmann C. A., Champy M. F. & Auwerx J. Evaluation of glucose homeostasis. Curr Protoc Mol Biol Chapter 29, Unit 29B 23 (2007). [DOI] [PubMed] [Google Scholar]
- Angeby-Moller K., Berge O. G. & Hamers F. P. Using the CatWalk method to assess weight-bearing and pain behaviour in walking rats with ankle joint monoarthritis induced by carrageenan: effects of morphine and rofecoxib. J Neurosci Methods 174, 1–9 (2008). [DOI] [PubMed] [Google Scholar]
- Mandillo S. et al.. Reliability, robustness, and reproducibility in mouse behavioral phenotyping: a cross-laboratory study. Physiol Genomics 34, 243–255 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um S. H. et al.. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205 (2004). [DOI] [PubMed] [Google Scholar]
- Pan F. et al.. Gene Aging Nexus: a web database and data mining platform for microarray data on aging. Nucleic Acids Res 35, D756–759 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chace D. H., Kalas T. A. & Naylor E. W. Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin Chem 49, 1797–1817 (2003). [DOI] [PubMed] [Google Scholar]
- Piraud M. et al.. ESI-MS/MS analysis of underivatised amino acids: a new tool for the diagnosis of inherited disorders of amino acid metabolism. Fragmentation study of 79 molecules of biological interest in positive and negative ionisation mode. Rapid Commun Mass Spectrom 17, 1297–1311 (2003). [DOI] [PubMed] [Google Scholar]
- Dacremont G. & Vincent G. Assay of plasmalogens and polyunsaturated fatty acids (PUFA) in erythrocytes and fibroblasts. J Inherit Metab Dis 18 Suppl 1, 84–89 (1995). [DOI] [PubMed] [Google Scholar]
- Bergmeyer H. U., Bergmeyer J. & Grassl M. Methods of enzymatic analysis. (VCH Verlagsgesellschaft mbH, 1986). [Google Scholar]
- Gall W. E. et al.. alpha-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PLoS ONE 5, e10883 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M. et al.. GenePattern 2.0. Nat Genet 38, 500–501 (2006). [DOI] [PubMed] [Google Scholar]
- de Hoon M. J., Imoto S., Nolan J. & Miyano S. Open source clustering software. Bioinformatics 20, 1453–1454 (2004). [DOI] [PubMed] [Google Scholar]
- Xia J., Psychogios N., Young N. & Wishart D. S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res 37, W652–660 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary info
Table S1
Table S2