

Abstract
Sexual transmission of human immunodeficiency virus type 1 (HIV-1) across mucosal barriers is responsible for the vast majority of new infections. This relatively inefficient process results in the transmission of a single transmitted/founder (T/F) virus, from a diverse viral swarm in the donor, in approximately 80% of cases. Here we compared the biological activities of 24 clade B T/F envelopes (Envs) with those from 17 chronic controls to determine whether the genetic bottleneck that occurs during transmission is linked to a particular Env phenotype. To maximize the likelihood of an intact mucosal barrier in the recipients and to enhance the sensitivity of detecting phenotypic differences, only T/F Envs from individuals infected with a single T/F variant were selected. Using pseudotyping to assess Env function in single-round infectivity assays, we compared coreceptor tropism, CCR5 utilization efficiencies, primary CD4+ T cell subset tropism, dendritic cell trans-infections, fusion kinetics, and neutralization sensitivities. T/F and chronic Envs were phenotypically equivalent in most assays; however, T/F Envs were modestly more sensitive to CD4 binding site antibodies b12 and VRC01, as well as pooled human HIV Ig. This finding was independently validated with a panel of 14 additional chronic HIV-1 Env controls. Moreover, the enhanced neutralization sensitivity was associated with more efficient binding of b12 and VRC01 to T/F Env trimers. These data suggest that there are subtle but significant structural differences between T/F and chronic clade B Envs that may have implications for HIV-1 transmission and the design of effective vaccines.
INTRODUCTION
Sexual transmission of HIV-1 across mucosal barriers is a relatively inefficient process and is most often due to the transmission of a single transmitted/founder (T/F) virus from the swarm of viral variants present in the donor, resulting in a profound genetic bottleneck (19, 36, 49, 52, 71, 72, 75, 79). A question of central importance is whether T/F viruses have particular phenotypic properties which favor their transmission. If so, viruses with these properties should logically be targets of vaccination and microbicide efforts. The viral envelope (Env) protein is a likely candidate for transmission-related signatures. For example, viruses expressing Envs that utilize the CCR5 coreceptor (R5-tropic) are transmitted far more frequently than those expressing Envs that utilize CXCR4 (X4-tropic) (36, 56, 63, 79). Variations in Env have also been linked to differences in the utilization of CD4 and coreceptor, the rate and efficiency of membrane fusion, and binding to C-type lectins such as DC-SIGN that are expressed on dendritic cells (DCs) and can function as virus attachment factors (25, 53, 54, 67).
Studies to characterize the properties of transmitted HIV-1 strains face several challenges. First, it is difficult to identify individuals during the acute phase of HIV-1 infection, particularly before the onset of immune responses (that is, at early Fiebig stages [23]), thus limiting sample sizes. Second, individual viruses cloned from the peripheral blood or plasma of acutely infected individuals within weeks of transmission may have already evolved away from the actual T/F virus and may thus have acquired phenotypic changes (8). Third, in the absence of extensive sampling of the early viral quasispecies by single-genome amplification (SGA), it is impossible to know if one or more virus strains established the clinical infection, making it difficult to assess the integrity of the mucosal barrier (36). Infection with multiple T/F viruses may reflect a different mechanism of transmission, with these T/F Envs likely facing different or reduced transmission selection pressure (30, 39). Nonetheless, small numbers of Envs cloned from acutely infected individuals have been obtained and compared to Envs cloned from corresponding donors or from other chronically infected individuals. Derdeyn et al. examined clade C Envs from eight heterosexual transmission pairs and concluded that transmitted Envs have fewer putative N-linked glycosylation sites (PNGs), more compact variable loops, and enhanced neutralization sensitivity to donor plasma (19), although subsequent phenotypic studies of a subset of viruses bearing these Envs did not reveal functional differences (2, 35). Analysis of clade A and D transmission pairs also identified shorter recipient Envs with a lower V3 charge, although no differences in the number of PNGs were noted (58). For clade B Envs, initial studies suggested that transmission was independent of variable-loop length and PNGs (13, 15, 24); however, more recent comparisons of thousands of clade B T/F and chronic env sequences confirmed significantly fewer total PNGs and a trend toward fewer in the V1/V2 loops of transmitted Envs (S. Gnanakaran et al., submitted for publication). Finally, several studies have investigated neutralization sensitivities of acute or T/F Envs compared to chronic control Envs, but conflicting results were reported (24, 36, 57, 64). These discrepancies may have resulted from differences in sample size, demographic characteristics of acutely infected individuals and chronic controls, cloning strategy, and whether the Envs under investigation represented true T/F viruses.
The use of SGA of plasma viral RNA during the earliest stages of infection has allowed the inference of the nucleotide sequences of T/F viruses from an increasingly large number of individuals (1, 36, 59, 60). Recent analyses of a large number of clade B T/F Env sequences led to the identification of transmission signatures in the CCR5 binding site, certain PNGs, and sites in the signal peptide and gp41 cytoplasmic domain that could affect Env processing and localization (Gnanakaran et al., submitted). These results suggested that T/F Envs might differ in some phenotypic properties from chronic Envs. To examine this, we conducted a comprehensive phenotypic analysis of T/F and chronic clade B HIV-1 Envs in the context of viral pseudotypes. Specifically, we assessed coreceptor tropism, CCR5 utilization efficiency, CD4+ T cell subset tropism, efficiency of DC-mediated trans-infection of T cells, and membrane fusion kinetics. In addition, we examined the sensitivities of T/F and chronic Envs to neutralization by purified immunoglobulin from infected patients (HIV Ig) and a panel of broadly neutralizing monoclonal antibodies (MAbs) and assessed the efficiencies of binding of these MAbs to trimeric Env on the cell surface. Our results failed to identify a major transmission phenotype but uncovered subtle functional differences between T/F and chronic Envs that may be of biological significance.
MATERIALS AND METHODS
Pseudovirus production.
Pseudotyped virus was produced by calcium phosphate cotransfection of 6 μg of pcDNA3.1+ containing env with 10 μg of HIV-1 core (pNL43-ΔEnv-vpr+-luc+ or pNL43-ΔEnv-vpr+-eGFP) into 293T17 cells. Virus was harvested at 72 h posttransfection, filtered through a 0.45-μm filter, aliquoted, and stored at −80°C. For primary CD4+ T cell infections, pseudovirus was concentrated by ultracentrifugation through a 20% sucrose cushion. Pelleted pseudovirus was then resuspended in phosphate-buffered saline (PBS). All luciferase-encoding pseudoviral stocks were serially diluted and used to infect NP2 cells to define the linear range of the assay. A viral dilution was chosen in the middle of the 5-fold linear range of the assay to maximize sensitivity.
env cloning and sequence analysis.
The derivations of most T/F Env clones used in this study have been described previously (36). THRO.F4.2026, SUMAd5.B2.1713, 9010-09.A1.4924, and PRB959-02.A7.4345 were cloned from SGA amplicons known to contain the nucleotide sequence of the corresponding T/F env sequence into pcDNA3.1 according to the manufacturer's instructions (Invitrogen). The AD17.1 env gene was subcloned from a full-length infectious molecular T/F clone described elsewhere (39). Chronic Envs HEMA.A4.2125 and HEMA.A23.2143 were also cloned in pcDNA3.1. Briefly, viral RNA was extracted from plasma samples from chronically infected patients and amplified using SGA methods. Individual env genes were then either cloned at random or selected to maximize within-patient env sequence diversity. Env clones were sequenced to confirm that they did not contain Taq polymerase errors but represented env genes of viruses circulating in the patient. The nucleotide sequences of all T/F and chronic Envs have previously been reported (Gnanakaran et al., submitted). PNGs were determined with N-glycosite (hiv.lanl.org) (76). To assess lengths of the V1/2, V3, V4, V5, and V1-4 regions, sequences were aligned to HXB2, and then boundaries were identified for each region and nongap residues were counted.
Coreceptor tropism testing and cell line infections.
NP2 cells stably expressing CD4 and either CCR5 (NP2/CD4/CCR5) or CXCR4 (NP2/CD4/CXCR4) were infected with HIV-1 pseudoviruses expressing luciferase by spinoculation in 96-well plates at 450 × g for 90 min at 25°C. Cells were lysed with Brite-Glo (Promega) at 72 h postinfection and analyzed on a Luminoskan Ascent luminometer. Coreceptor tropism was arbitrarily defined by mean relative light units (RLUs) greater than 1 (approximately 100-fold over background). To assess sensitivity to coreceptor inhibitors, NP2/CD4/CCR5 or NP2/CD4/CXCR4 cells were preincubated for 30 min with saturating concentrations of the CCR5 inhibitor maraviroc (MVC) (1 μM), the CXCR4 inhibitor AMD3100 (2 μM), or the fusion inhibitor enfuvirtide (10 μg/ml) prior to infection. To assess sensitivity to broadly neutralizing MAbs, viral pseudotypes were preincubated with 10 μg/ml of antibody for 30 min at 37°C. Virus and antibody mixes were then used to infect NP2/CD4/CCR5 or NP2/CD4/CXCR4 cells. All NP2 cell line infections were done in at least triplicate in at least three independent experiments using R5-tropic JRFL as a positive control and Env-deficient pseudotypes as a negative control.
The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: pNL4-3-deltaE-eGFP (catalog no. 11100) from Haili Zhang, Yan Zhou, and Robert Siliciano (74); bicyclam JM-2987 (hydrobromide salt of AMD-3100; catalog no. 8128) (9, 18, 34); maraviroc (catalog no. 11580) (6, 22, 65), and HIV-1 gp120 MAb IgG1 b12 (catalog no. 2640) from Dennis Burton and Carlos Barbas (5, 11, 12, 55).
Primary human CD4+ T cell tropism assay.
Primary human CD4+ T cells, purified by negative selection, were obtained from the University of Pennsylvania's Human Immunology Core. A total of 2 × 106 cells per virus were stimulated with plate-bound anti-CD3 (clone OKT3) (eBiosciences) and anti-CD28 (clone 28.2) (BD Biosciences) and 20 U/ml recombinant interleukin-2 (IL-2) in RPMI containing 10% fetal bovine serum (FBS). At 3 days poststimulation cells were transferred to 96-well V-bottom plates prior to infection. Five microliters/well of concentrated HIV-green fluorescent protein (GFP) was used to infect cells in triplicate. Plates were then spinoculated at 1,200 × g for 2 h. Cells were then transferred to new 24-well plates, and new medium containing 20 U/ml IL-2 was added. At 3 days postinfection, cells were stained for flow cytometry.
Determination of alternative coreceptor use.
Primary human CD4+ T cells from two different ccr5Δ32 homozygous donors were obtained and purified as described above. Prior to infection, cells were preincubated with 50 μM AMD3100 for 30 min. Cells were infected as described above. At 2 hours after spinfection, enfuvirtide (1-μg/ml final concentration) was added to all samples to prevent additional fusion prior to transferring cells to a 24-well plate for further incubation. Samples were stained and analyzed as described above.
Flow cytometry.
A total of 1 × 106 to 2 × 106 cells were stained per tube for flow cytometry. All incubations were done at room temperature (RT) and in fluorescence-activated cell sorter (FACS) wash buffer (PBS, 2.5% FBS, 2 mM EDTA), and all antibodies were from BD Biosciences, unless otherwise noted. To stain CD4+ T cells, cells were first washed in PBS, and then, live/dead Aqua (Invitrogen) was added and incubated for 10 min. Next, anti-CCR7 IgM in FACS wash buffer was added and incubated for 30 min. Cells were then washed in FACS wash buffer before staining with anti-CD3–Qdot 655 (Invitrogen), anti-CD4–Alexa Fluor 700, anti-CD45RO–phycoerythrin (PE)-Texas Red (Beckman Coulter), and anti-IgM–PE (Invitrogen) for 30 min. Cells were then washed in FACS wash buffer and resuspended in 1% paraformaldehyde (PFA). Samples were run on an LSRII (BD) instrument and analyzed with FlowJo 8.8.6 (Treestar). Cells were gated as follows: singlets (FSC-A by FSC-H), then live cells (SSC-A by live/dead), then lymphocytes (SSC-A by FSC-A), then CD3+ cells (SSC-A by CD3), then memory markers (CCR7 by CD45RO).
DC trans-infection assay.
To differentiate DCs, freshly isolated monocytes from the University of Pennsylvania's Human Immunology Core were treated with 50 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (R&D systems) and 100 ng/ml IL-4 (R&D systems) in AIM V serum-free medium (Invitrogen). New medium containing GM-CSF and IL-4 was added on day 3. At 6 days poststimulation, DCs were washed and plated at 3 × 104 cells per well in a V-bottom 96-well plate; 3 × 104 CD4+ T cells alone at 3 days poststimulation with plate-bound anti-CD3/anti-CD28 were used as a negative control. Titers of viral stocks were first determined by RLUs on NP2/CD4/CCR5 or CXCR4 cells. Virus sufficient to generate 80 RLUs was added to DCs or a CD4+ T cell control and allowed to bind for 2 h at 37°C. Cells were washed three times with fresh medium to remove cell-free virus, and then 3 × 105 stimulated heterologous CD4+ T cells were added to each well containing 3 × 104 HIV-bound DCs or CD4+ T cells. As an additional control, an equal amount of virus was added to 3 × 105 stimulated CD4+ T cells to ensure that there was no differential infection of CD4+ T cells. For CD4+ T cell luciferase infection, cells were spinoculated at 450 × g for 90 min and then incubated without washing off virus. Cells were then transferred to a flat-bottom 96-well plate for 3 days prior to takedown with Brite-Glo. Experiments for each condition were done in triplicate, and each viral pseudotype was used in at least three independent experiments with cells from different healthy donors.
Enfuvirtide time-of-addition assay.
To assess the entry kinetics of T/F and chronic Envs, NL43vpr+luc+ pseudotypes were chilled to 4°C and added to NP2/CD4/CCR5 (or NP2/CD4/CXCR4 for the one X4-tropic Env) cells on metal blocks embedded in ice covered by a moist towel. Cells were then spinoculated at 1,300 rpm for 90 min at 4°C to enhance viral binding. Immediately postspinoculation, cold supernatant was aspirated off and all wells were flooded with 270 μl of prewarmed 37°C medium and transferred to a 37°C incubator. Thirty microliters of 10-μg/ml enfuvirtide (final concentration of 1 μg/ml) was then added at 0, 5, 10, 20, 40, 80, or 160 min postwarming. A no-drug control was also included to normalize percent infection. Cells were then incubated for 72 h and assessed for RLUs. At least three wells per virus per time point were included in each experiment, and all Envs were examined in at least three independent experiments. Data were analyzed with Prism 4.0 (GraphPad Software, Inc.) by fitting a best-fit sigmoidal line to the results from each independent experiment prior to averaging the Hill slopes and time to half-maximal fusion.
Neutralization sensitivity.
Neutralization assays were performed using both NP2 and TZMbl cells in two independent laboratories. To assess sensitivity to MAbs b12, VRC01, PG9, and PG16, viral pseudotypes were preincubated with 10 μg/ml of antibody for 1 h prior to infection of NP2 cells. To assess sensitivity to HIV Ig, pseudotypes were preincubated with 2-fold serial dilutions of clade B HIV Ig from 1,500 to 23 μg/ml. This mix was then added to NP2 cells and spinoculated as described above. For MAbs, neutralization was assessed by determining the maximum percent inhibition (MPI) compared to a no-antibody control. Clade B HIV Ig (lot 12 100158) was obtained from the AIDS Repository.
Neutralization sensitivity was assessed on TZMbl cells as previously described (17, 69). Briefly, 8 × 103 TZMbl cells were plated overnight. Five-fold dilutions of MAbs (b12, VRC01, PG9, PG16, and clade B HIV Ig) were incubated in the presence of 40 μg/ml DEAE-dextran and 2.000 infectious units (as measured on TZMbl cells) of pseudovirus for 1 h at 37°C. After the medium was removed from TZMbl cells, the virus-MAb dilutions were added to the cells and incubated for 48 h before being analyzed for luciferase expression (Promega). The highest concentration tested for b12, VRC01, PG19, and P16 was 10 μg/ml. The highest concentration of clade B HIV Ig was 1,500 μg/ml. Samples were tested in duplicate, with all experiments repeated at least two times. Fifty percent inhibitory concentrations (IC50s) were calculated as described previously (17).
CELISA.
The binding of MAbs to HIV-1 Env trimers expressed on cells was measured using a cell-based enzyme-linked immunosorbent assay (CELISA) system, as previously described (32). Briefly, COS-1 cells were seeded in 96-well plates (1.8 × 104 cells/well) and transfected the next day with 0.1 μg of a plasmid expressing Env and 0.02 μg of a Rev-expressing plasmid per well using Effectene transfection reagent. Three days later, cells were incubated with the indicated MAb suspended in blocking buffer (35 mg/ml bovine serum albumin [BSA], 10 mg/ml nonfat dry milk, 1.8 mM CaCl2, 1 mM MgCl2, 25 mM Tris [pH 7.5], and 140 mM NaCl) for 1 h at room temperature. Cells were then washed four times with blocking buffer and four times with washing buffer (140 mM NaCl, 1.8 mM CaCl2, 1 mM MgCl2, and 20 mM Tris [pH 7.5]). A horseradish peroxidase (HRP)-conjugated antibody specific for the Fc region of human IgG was then incubated with the samples for 45 min at room temperature. Cells were washed five times with blocking buffer and five times with washing buffer. HRP enzyme activity was determined after the addition of 33 μl per well of a 1:1 mix of Western Lightning oxidizing and luminol reagents (Perkin-Elmer Life Sciences) supplemented with 150 mM NaCl. Light emission was measured with a Mithras LB 940 luminometer (Berthold Technologies). To correct for the level of cell surface expression of each envelope glycoprotein, binding of the antibodies is expressed as percent binding of the CD4-Ig probe at saturating concentrations (5 μg/ml). We decided a priori to exclude all envelope glycoproteins that bound CD4-Ig at less than 20% of the binding measured for the SC05.8H2.3243 control isolate. Five of the 57 Envs were thus not analyzed, including three T/F and two chronic Envs. Measurements of antibody binding and neutralization were performed under code to prevent potential bias.
Statistical analyses.
T/F and chronic Envs were compared by using Mann-Whitney tests, and correlations were assessed by using Spearman tests. P values of less than 0.05 were considered significant. Data were analyzed with Prism 4.0 software.
Ethics statement.
All human cells used in this study were from normal healthy donors who provided written informed consent after approval by the University of Pennsylvania's institutional review board.
RESULTS
Panels of T/F and chronic Envs.
To determine if there are functional differences between T/F Envs and those that predominate during chronic infection, we assembled a panel of 24 clade B T/F Envs previously inferred and cloned from plasma viral RNAs of 24 individuals with acute HIV-1 infection as defined by the Fiebig staging system, in which patients are classified from stage I (viral RNA positive and antibody and antigen negative) to stage VI (ELISA and Western blot positive with multiple bands) (4, 36, 39) (Table 1). Twelve individuals were sampled during Fiebig stage II, five during Fiebig stage III, two during Fiebig stage IV, and five during Fiebig stage V. Acutely infected individuals were predominantly males (22 of 24) from the southeastern United States (18 of 24) with a variety of sexual risk factors, all denying intravenous drug use (IDU). All T/F Envs were inferred from SGA-derived sequences, which are devoid of PCR-induced errors and cloning bias (36). Env clones identical to this inferred T/F sequence were then chosen for phenotypic analysis. Importantly, all T/F Envs were selected from subjects with single-variant transmissions. This was done to increase the likelihood that the viruses encoding these Envs were transmitted across an intact mucosal barrier, thereby maximizing our chances of observing properties required for this process (30, 39).
Table 1.
Description of T/F and chronic Envs
| Env type | Subject | env clone(s) | Fiebig stage or minimal time since infection | Viral load (copies/ml) | Genderc | Risk behaviord | Geographic location | Sampling date (mo/day/yr) | Coreceptor tropisme |
|---|---|---|---|---|---|---|---|---|---|
| T/F | REJO4541 | REJO.D12.1972 | V | 722,349 | M | Heterosexual | Alabama | 9/28/01 | R5 |
| RHPA4259 | RHPA.A19.2000 | V | 1,458,354 | F | Heterosexual | Alabama | 12/5/00 | R5 | |
| SUMA0874 | SUMAd5.82.1713 | II | 939,260 | M | MSM | Alabama | 5/13/91 | R5 | |
| THRO4156 | THRO.F4.2026 | V | 5,413,140 | M | MSM | Alabama | 8/1/00 | R5 | |
| WEAU0575 | WEAUd15.410.5017 | II | 216,415 | M | MSM | Alabama | 5/30/90 | R5X4 | |
| WITO4160 | WITO.B10.2062 | II | 325,064 | M | Heterosexual | Alabama | 8/4/00 | R5 | |
| 700010040 | CH40.C9.4520 | V | 298,026 | M | MSM | North Carolina | 7/27/06 | R5 | |
| 700010058 | CH58.A4.4375 | III | 394,649 | M | Unknown | North Carolina | 9/8/06 | R5 | |
| 700010077 | CH77.SA2.6559 | V | 144,145 | M | Unknown | North Carolina | 9/31/06 | R5X4 | |
| 1006-11 | 1006-11.C3.1601 | III | 1,600,000 | M | SPD | North Carolina | 6/5/97 | R5 | |
| 1018-10 | 1018-10.A5.1732 | III | 270,000 | M | SPD | South Carolina | 6/20/97 | R5 | |
| 1053-07 | 1053-07.B15.1648 | III | 1,400,000 | M | SPD | South Carolina | 12/3/97 | R5 | |
| 1056-10 | 1056-10.TA11.1826 | II | 140,000 | M | SPD | South Carolina | 1/14/98 | R5 | |
| 1058-11 | 1058-11.B11.1550 | IV | 550,000 | M | SPD | South Carolina | 3/18/98 | R5X4 | |
| 9010-09 | 9010-09.A1.4924 | II | 146,954 | F | SPD | South Carolina | 11/25/97 | R5 | |
| 9015-07 | 9015-07.A1.4729 | II | 500,000 | M | SPD | South Carolina | 12/27/97 | R5 | |
| 9021-14 | 9021-14.B2.4571 | II | 143,379 | M | SPD | California | 6/10/98 | R5 | |
| 9032-08 | 9032-08.A1.4685 | III | 40,815 | M | SPD | Alabama | 7/30/98 | R5 | |
| PRB956-04 | PRB956-04.B22.4267 | II | 600,000 | SPD | Virginia | 8/19/97 | R5 | ||
| AD17 | AD17.1 | II | 47,600,000 | M | MSM | New York | 6/14/99 | R5 | |
| PRB959-02 | PRB959-02.A7.4345 | II | >2,000,000 | SPD | South Carolina | 11/17/99 | R5 | ||
| TT35P | TT35P.11H8.2874 | II | 1,849,301 | M | Heterosexual | Trinidad | 1/26/99 | R5 | |
| SC20 | SC20.8A8.2437 | IV | 2,789,313 | M | Heterosexual | Trinidad | 2/18/98 | R5 | |
| SC05 | SC05.8C11.2344 | II | 9,980,952 | M | Heterosexual | Trinidad | 6/28/93 | R5 | |
| Chronic 1a | SC05 | SC05.A10.2362, SC05.8H2.3243, SC05.8A11.2363 | 5 yr 5 mo | 19,514 | M | Heterosexual | Trinidad | 12/9/98 | R5 |
| SHKE4761 | SHKE.A26.4112, SHKE.A7.2118, SHKE.A4.2116 | 1 yr 2 mo | 544,000 | M | MSM | Alabama | 8/2/06 | R5 | |
| HEMA4284 | HEMA.A4.2125, HEMA.A23.2143 | 1 yr 10 mo | 49,755 | M | MSM | Alabama | 10/2/02 | R5 | |
| WICU4248 | WICU.B4.2973, WICU.C1.2992 | 5 yr 11 mo | 8,424 | M | MSM | Alabama | 10/6/05 | R5 | |
| CRPE4571 | CRPE.B28.4072 | 2 y | 21,917 | F | Heterosexual | Alabama | 11/30/01 | R5X4 | |
| JOTO5278 | JOTO.TA1.2247 | 1 yr 6 mo | 404,180 | M | Heterosexual | Alabama | 2/13/04 | X4 | |
| OLLA4645 | OLLA.A14.1923 | 2 yr 1 mo | 382,000 | F | Heterosexual | Alabama | 2/22/02 | R5 | |
| SAMI4303 | SAMI.A8.1863 | 3 yr 11 mo | 116,000 | M | MSM | Alabama | 8/5/04 | R5 | |
| SMRE4166 | SMRE.A13.4127 | 1 yr 4 mo | 135,858 | F | Heterosexual | Alabama | 11/16/01 | R5 | |
| TALA4022 | TALA.A2.1780 | 6 yr 11 mo | 228,200 | M | MSM | Alabama | 12/9/03 | R5 | |
| YOMI4024 | YOMI.F2.4137 | 6 yr 1 mo | 14,178 | M | MSM | Alabama | 2/15/06 | R5 | |
| Chronic 2b | 1632 | 1632.TA9, 1632.A17, 1632.A6, 1632.TA1, 1632.A7, 1632.A23 | 2 yr 5 mo | 97,800 | M | MSM | Washington | 9/14/05 | R5 |
| 1451 | 1451.D17, 1451.C16, 1451.D1, 1451.C8 | 20 yr 3 mo | 532,000 | M | MSM | Washington | 3/1/05 | R5 | |
| 1588 | 1588.TA7 | 7 yr 2 mo | 99,600 | M | MSM/IDU | Washington | 7/6/05 | R5 | |
| 1470 | 1470.D27 | 4 yr 3 mo | 492,200 | M | MSM/IDU | Washington | 3/15/05 | R5 | |
| 1599 | 1599.B11 | 6 yr 7 mo | 112,000 | M, F | IDU | Washington | 7/26/05 | R5 | |
| 1444 | 1444.A21 | 7 y | 86,300 | M | MSM | Washington | 2/22/05 | R5 |
Original panel of chronic Envs.
Chronic Envs from Washington state assessed only in neutralization assays.
M, male; F, female.
MSM, men who have sex with men; SPD, serial plasma donor; IDU, intravenous-drug user.
Coreceptor tropism was assessed on NP2 cells for the T/F and Chronic 1 Envs. Tropism for the Chronic 2 Envs was previously assessed on TZMbl cells (Gnanakaran et al., submitted).
To generate chronic clade B control Envs, we used SGA to amplify env genes from plasma viral RNAs of two groups of antiretroviral therapy-naïve individuals. The first group consisted of 11 individuals sampled at 14 to 83 months postinfection (mean, 42 months). A test set of 17 Env clones was derived from this group, consisting predominantly of males (8 of 11) from the southeastern United States (10 of 11), all denying IDU (Table 1). An additional 14 clade B control Envs were SGA amplified and cloned from six chronically infected individuals residing in the northwestern United States. This second group of chronic Envs served as a validation set to confirm differences in neutralization sensitivity observed with the first test set. A phylogenetic tree of the 31 chronic Envs is depicted in Fig. 1, along with the 24 T/F Envs. None of the Envs were from epidemiologically linked infections.
Fig. 1.

Phylogenetic relationships of T/F and chronic Envs selected for phenotypic analyses. The tree was constructed from Env amino acid sequences of T/F (red), Chronic 1 control (blue), and Chronic 2 (green) viruses (subtype B reference sequences from the database are shown in black). All sequences were derived by SGA methods; Env sequences from the same individuals form discrete subclusters. A bracket indicates epidemiologically linked infections from Trinidad and Tobago (14). The tree was inferred using maximum-likelihood methods (29). Numbers on nodes indicate posterior probabilities (only values above 0.95 are shown). The scale bar represents 0.05 amino acid substitution per site.
Previous studies noticed fewer PNGs in the gp120 region of T/F Envs than in that of chronic Envs (40; Gnanakaran et al., submitted). To determine whether our selected subset of T/F and chronic Envs differed from this much larger group, we compared the variable-loop lengths as well as the numbers and distributions of putative PNGs. There were no differences in V1/2, V3, V4, and V1-4 lengths between T/F and chronic clade B Envs. Further, the median number of gp120 PNGs in T/F Envs was 26.0, compared to 27.0 for the chronic controls (P = 0.16) and 26.0 for clade B Envs in general (74). Thus, the panel of T/F Envs selected for our functional analyses exhibited no statistically significant differences in patient demographics, virus phylogeny, variable-loop length, or PNGs relative to the panel of chronic Envs we assembled or to clade B Envs in general.
Determination of coreceptor tropism.
R5-tropic viruses represent the vast majority of transmitted viruses, with dual (R5X4)-tropic viruses being transmitted less frequently (36, 56, 63, 79). On rare occasions, X4-tropic viruses can be transmitted (3, 47, 66). To determine the coreceptor usage in our panel, we characterized the CCR5 and CXCR4 utilization of the 24 T/F and 17 chronic Envs by producing viral pseudotypes and using these to infect NP2 cell lines expressing CD4 and either CCR5 (NP2/CD4/CCR5) or CXCR4 (NP2/CD4/CXCR4), as well as primary human CD4+ T cells. NP2 cells were selected because they provide a 5- to 6-log linear range of infection, approximately 2 to 3 logs greater than that of the TZMbl assay. We found that of the 24 T/F Envs, 21 were R5-tropic and three were R5X4-tropic, while of the 17 chronic Envs, 15 were R5-tropic, one was R5X4-tropic, and one was X4-tropic (Table 1). This is consistent with previous results, with the exception of T/F Envs 1058-11.B11.1550 and CH77.SA2.6559, which were R5X4-tropic on NP2 cells and R5-tropic on TZMbl cells (36) (Gnanakaran et al., submitted). This discrepancy is likely due to differences in CXCR4 expression, as the NP2 cells used stably express high levels of CXCR4 compared to the HeLa-derived TZMbl cells, which express lower endogenous CXCR4 levels. All four R5X4-tropic Envs utilized CCR5 and CXCR4 with approximately equivalent efficiencies as assessed by a less-than-2-fold difference in RLUs between the NP2/CD4/CCR5 and NP2/CD4/CXCR4 cells. To assess coreceptor use on human CD4+ T cells, we infected ccr5Δ32 or ccr5wt CD4+ T cells in the presence or absence of saturating concentrations of the CXCR4 inhibitor AMD3100. The results paralleled those obtained with the NP2 cell lines. R5-tropic Envs mediated infection of ccr5wt but not ccr5Δ32 CD4+ T cells, while R5X4 Envs mediated infection of both cell types. Infection of ccr5Δ32 CD4+ T cells by three of the R5X4 Envs was completely inhibited by AMD3100, while Env CRPE.B28.4072 could infect ccr5Δ32 cells in the presence of AMD3100, though with reduced efficiency (data not shown). However, we found that AMD3100 inhibited infection of NP2/CD4/CXCR4 cells by viruses bearing the CRPE.B28.4072 Env by only 50%. In addition, this Env was unable to infect NP2 cells expressing CD4 alone or in combination with any of 17 different putative alternative coreceptors, indicating that this Env can utilize the drug-bound conformation of CXCR4 (data not shown). Several other HIV-1 Env proteins have been shown to exhibit this property (33). In summary, all T/F Envs utilized CCR5, while three were R5X4-tropic. Thus, there were no differences in coreceptor tropism between the T/F and chronic Envs, with the exception of the one X4-tropic chronic Env, and there was no evidence for utilization of coreceptors other than CCR5 or CXCR4 to infect human CD4+ T cells.
Sensitivity to coreceptor antagonists and CCR5 utilization efficiency.
Mucosal transmission of HIV-1 is dependent upon CCR5. Hypothesizing that the ability to use low levels of CCR5 may confer a selective advantage to viruses at the moment of transmission, we determined the sensitivity of each Env to the CCR5 inhibitor maraviroc (MVC) as a surrogate for CCR5 utilization efficiency. High MVC IC50s indicate that an Env can mediate infection at low levels of CCR5, while low IC50s suggest that an Env requires high CCR5 expression for viral entry. We found no significant difference in median MVC IC50s between the T/F (2.4 nM) and chronic (2.3 nM) Envs (P = 0.79 by Mann-Whitney test) (Fig. 2A). In addition, we determined the maximal percent inhibition (MPI) of infection by MVC. While they are uncommon, several in vivo-derived MVC-resistant R5-tropic viruses that can utilize the drug-bound form of CCR5 have been identified (67, 70). Such viruses engage the coreceptor differently, relying predominantly upon the N terminus for entry whereas most viruses require the N terminus as well as the extracellular loops of CCR5. Furthermore, determining the MVC sensitivities of T/F viruses has implications for microbicides and preexposure prophylaxis. All 41 Envs examined had MPIs of greater than 85%, with the vast majority greater than 95%. There were no significant differences (P = 0.17 by Mann-Whitney test) between the T/F (median = 99.1%) and chronic (median = 98.3%) Envs (Fig. 2B). Together, these data indicate that the HIV-1 transmission bottleneck does not impose a selection pressure for viruses capable of using low concentrations of CCR5.
Fig. 2.

CCR5 utilization efficiency. (A) Viral pseudotypes were used to infect NP2/CD4/CCR5 cells in the presence of serial dilutions of the CCR5 antagonist maraviroc (MVC). Higher IC50s correspond to Envs that can utilize CCR5 more efficiently, and vice versa. T/F and chronic clade B Envs have similar MVC IC50s (P = 0.79), suggesting that they engage CCR5 comparably. (B) Since some Envs can utilize the MVC-bound conformation of CCR5 and since MVC is a candidate microbicide, we assessed the maximal percent inhibition (MPI) of MVC for each Env. All Envs were sensitive to MVC, and there was no difference in MPI between the T/F and chronic Envs (P = 0.17). All infections were done in at least triplicate in each of at least three independent experiments. Data were analyzed by a Mann-Whitney test.
Primary CD4+ T cell tropism.
CD4+ T cells, the major target and source of HIV-1 in vivo (37, 77), can be broadly divided into four subsets: naïve (CCR7+ CD45RO−), central memory (TCM) (CCR7+ CD45RO+), effector memory (TEM) (CCR7-CD45RO+), and CD45RA+ effector memory (TEMRA) (CCR7− CD45RO−) (61). These subsets are differentially infected due in part to variation in coreceptor expression (50), cellular activation (26), and tissue localization (27). TEM and TEMRA cells are found predominantly in effector sites, including the rectal and cervicovaginal mucosae, while naïve and TCM cells are most abundant in the lymph nodes. TEM cells, the most abundant subset in mucosal effector sites, are preferentially infected and massively depleted during acute infection (reviewed in reference 42). Since potential target cells in the mucosae may be limiting during transmission, we hypothesized that T/F Envs may infect TEM and TEMRA cells preferentially relative to the matched chronic controls.
Peripheral blood CD4+ T cells from three normal uninfected donors were purified by negative selection and stimulated with anti-CD3/anti-CD28 and IL-2 for 3 days prior to infection with HIV-1 pseudotypes expressing a GFP reporter. At 3 days postinfection, the viability and expression of CD3, CD4, CCR7, CD45RO, and GFP were assessed by FACS analysis. Productively infected cells were defined as CD3+ GFP+, since CD4 was downregulated in the majority of infected cells (16). The gating strategy is shown in Fig. 3A. In all three donors, infected cells were predominantly TEM (∼65%), followed by TCM (∼30%), TEMRA (∼3%), and naïve (∼2%) cells (Fig. 3B). As X4-tropic viruses have been previously reported to readily infect naïve CD4+ T cells compared to R5-tropic viruses (21, 46, 48), this assay contains an important internal validation: the five viruses that could utilize CXCR4 for entry (one X4-tropic Env shown in cyan and four R5X4-tropic Envs shown in red in Fig. 3B) preferentially infected naïve cells. With the exception of these five Envs, no other pseudotypes were reproducibly outliers in their ability to mediate entry into any of the subsets, and there were no statistically significant differences or trends between the T/F and chronic Envs for any of the four cell subsets in any of the three donors examined (Fig. 3B). In addition, there was no statistically significant difference in overall infectivity between the T/F and chronic Env pseudotypes in any of the three donors examined, suggesting comparable Env fitness in peripheral CD4+ T cells (Fig. 3C). Together, these results suggest that transmission and early expansion are not due to differential infection of CD4+ T cells or their subsets between T/F and chronic Envs.
Fig. 3.

CD4+ T cell subset tropism. To assess human CD4+ T subset tropism of the T/F and chronic Envs, cells were infected with Env pseudotypes expressing GFP and then stained and analyzed by flow cytometry. (A) Cells were gated as shown. Infected cells (GFP+) were then back gated on the memory markers CCR7 and CD45RO to evaluate differential subset infection. Naïve, CCR7+ CD45RO−; central memory (TCM), CCR7+ CD45RO+; effector memory (TEM), CCR7− CD45RO+; effector memory RA (TEMRA), CCR7− CD45RO−. (B) T/F and chronic Envs infected all four CD4+ T cell subsets comparably. TEM and TCM cells were infected most readily, followed by naïve and TEMRA cells. As expected, Envs that could utilize CXCR4 preferentially infected naïve cells compared to Envs that used exclusively CCR5. (C) T/F and chronic Env pseudotypes have comparable overall CD4+ T cell infection frequencies in each of the three donors examined. R5X4-tropic Envs are shown in red, and the one X4-tropic Env is shown in cyan. Tropism was assessed in cells obtained from three different, uninfected normal donors as indicated (ND218, ND335, and ND337). The horizontal lines indicate the mean value for each group of Envs.
DC-mediated trans-infection.
DCs can enhance HIV-1 infection in trans by efficiently capturing virus particles and presenting them to CD4+ T cells. In vitro, coculture of monocyte-derived DCs with CD4+ T cells results in enhanced virus infection, particularly at low virus inocula (reviewed in reference 51). To assess whether DCs preferentially bind and transfer T/F compared to chronic Env pseudoviruses, we performed DC/CD4+ T cell coculture experiments. Viral pseudotype stocks were normalized for infectivity on NP2 cells to control for differences in viral titer. A relatively limiting amount of virus (80 RLUs on NP2 cells) was bound to DCs, which were then washed to remove cell-free virus and cocultured with CD4+ T cells. All Envs were assessed in at least three independent experiments, each time using DCs and CD4+ T cells from different normal donors. Adding this amount of virus to 3 × 104 CD4+ T cells and then washing as with the DCs resulted in infection at background levels. However, adding virus associated with DCs markedly increased infection. Nonetheless, the magnitude of DC/CD4+ T cell trans-infection was not different between T/F and chronic Envs (Fig. 4) (P = 0.44 by Mann-Whitney test). In addition, there was no difference in CD4+ T cell infectivity in the absence of DCs, and there was no detectable infection of DC control cultures in the absence of CD4+ T cells (data not shown). The absence of any difference between T/F and chronic pseudoviruses in this trans-infection assay suggests that, at least when presented with an equal amount of infectious pseudovirus, DCs bind and transfer T/F and chronic Env pseudotypes similarly.
Fig. 4.
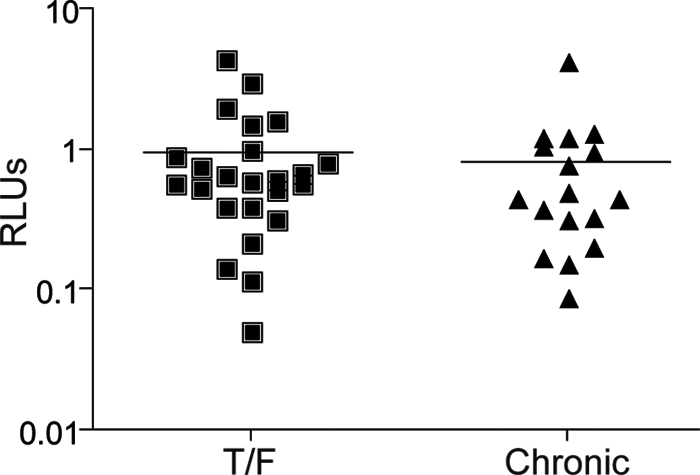
Dendritic cell (DC) trans-infection. To assess differential DC binding and CD4+ T cell trans-infection of T/F and chronic Envs, we pulsed DCs with luciferase-expressing Env pseudotypes and then washed off unbound virus and added CD4+ T cells. Relative light units (RLUs) were then measured as a surrogate for infection. DC trans-infection efficiency was comparable between the T/F and chronic Envs (P = 0.44). Viral input was normalized based upon infectivity on NP2 cell lines. The data shown are from one of at least three independent experiments with cells from different donors, each done in at least triplicate. Data were analyzed by a Mann-Whitney test.
Entry kinetics and enfuvirtide sensitivity.
Productive entry of HIV-1 into cells may occur following internalization and delivery to endosomes, albeit in a pH-independent manner (43). If so, then the rate at which a virus is internalized, fuses, and enters cells could affect viral tropism. In addition, the rate at which a virus fuses is a measure of how well it productively engages CD4 and coreceptor. Hypothesizing that faster-fusing viruses may preferentially overcome mucosal barriers to transmission, we indirectly assessed the entry kinetics of the T/F and chronic pseudoviruses using a time-of-addition experiment with the fusion inhibitor enfuvirtide. As enfuvirtide is not membrane permeative, the time to enfuvirtide escape may reflect the rate of viral endocytosis, fusion, or some combination thereof. HIV-1 pseudotypes were added to NP2 cells on ice. Cells were spinoculated at 4°C to facilitate HIV-1 binding, and then cold medium was removed and replaced immediately with prewarmed medium. Saturating concentrations of enfuvirtide were then added at 0, 5, 10, 20, 40, 80, and 160 min postwarming, and then infectivity was normalized to that of a no-drug control. To control for experimental variation, the prototypic R5-tropic virus JRFL was included in all experiments. There was no significant difference or trend in the rates at which T/F and chronic Env pseudotypes productively entered NP2 cells, thus becoming resistant to enfuvirtide addition. The median times to half-maximal resistance (t1/2 max) postwarming were 32.5 min for the T/F Envs and 31.4 min for the chronic Envs (P = 0.55 by Mann-Whitney test). Interestingly, JRFL became resistant to enfuvirtide significantly faster (t1/2 max = 15.9 min) than all 41 T/F and chronic Envs (Fig. 5A). In addition, we assessed enfuvirtide potency, a measure of pre-hairpin bundle exposure that also reflects the kinetics of CD4/coreceptor engagement and endocytosis (44). There was no difference between the sensitivities of T/F and chronic Envs to enfuvirtide (mean IC50 of 0.10 versus 0.13 μg/ml; P = 0.53) (Fig. 5B). Together, these results suggest that the kinetics of viral entry/endocytosis are comparable for T/F and chronic Envs.
Fig. 5.

Entry kinetics and enfuvirtide sensitivity. (A) To examine differences in T/F and chronic Env endocytosis/fusion kinetics, we employed an indirect assay in which viral pseudotypes were bound to NP2 cells in the cold prior to the addition of prewarmed medium. A saturating concentration of enfuvirtide was added at various times postwarming. The time to half-maximal resistance to enfuvirtide (t1/2 max) was then calculated. The T/F and chronic Envs became resistant to enfuvirtide at equal rates (P = 0.55), with all of the Envs acquiring resistance to enfuvirtide more slowly than a prototypic R5-tropic HIV-1 control, JRFL. (B) Enfuvirtide potency, a compound measure of fusion kinetics and affinity, was assessed for all T/F and chronic Envs. There was no difference in enfuvirtide IC50 between the T/F and chronic Envs (P = 0.53), further suggesting that there is no difference in endocytosis/fusion rates between T/F and chronic Envs. Each infection condition was done in triplicate (A) or duplicate (B) for each Env in each of at least three independent experiments. Data were analyzed by a Mann-Whitney test.
Sensitivity to broadly neutralizing antibodies and HIV Ig.
It has previously been reported that Envs derived from acutely infected individuals may exhibit enhanced sensitivity to antibody-mediated neutralization because of changes in glycosylation and/or variable-loop length (19). This finding raised the possibility that such Envs might be able to bind to CD4 and coreceptor more efficiently. To examine this, we measured the sensitivities of the T/F and chronic Envs to four broadly neutralizing MAbs. MAbs b12 (12) and VRC01 (78) neutralize Env by engaging the CD4 binding site (CD4bs), while the epitopes for PG9 and PG16 (68), distinct germ line variants from the same individual, are glycosylation dependent and include parts of the V1/2 and V3 loops (20). To assess neutralization sensitivity, pseudoviruses were preincubated with a single concentration (10 μg/ml) of each MAb for 60 min prior to infection of NP2 cells. The maximal percent inhibition was then determined by normalizing to a control without antibody. Interestingly, T/F Envs were more sensitive than chronic Envs to both b12 (mean MPI of 66% versus 17%; P = 0.0003) (Fig. 6A) and VRC01 (mean MPI of 89% versus 50%; P = 0.0077) (Fig. 6B) (compare T/F to Chronic 1). There was also a trend toward enhanced sensitivity to neutralization by PG9 (Fig. 6C) and PG16 (Fig. 6D). To confirm these differences, the neutralization sensitivities of T/F Envs were independently examined using a different backbone (SG3) and HIV-1 reporter cell line (TZMbl), with both MPI and IC50s being determined. The results confirmed the NP2 cell data in that the T/F Envs were more sensitive to neutralization by b12 (Fig. 6E) and VRC01 (Fig. 6F). In addition, the T/F Envs exhibited a trend toward increased neutralization sensitivity to both PG9 (Fig. 6G) and PG16 (Fig. 6H). While this did not reach statistical significance, it is consistent with a more neutralization-sensitive phenotype of T/F than of chronic Envs.
Fig. 6.

Neutralization sensitivity. (A to H) The sensitivity to monoclonal antibodies b12, VRC01, PG9, and PG16 was assessed on both NP2 cells (A to D) and TZMbl cells (E to H). Neutralization sensitivity on NP2 cells was assessed by determining the maximal percent inhibition (MPI) to 10 μg/ml of the indicated MAb. IC50s were determined in the TZMbl assay. Clade B T/F Envs were more sensitive to b12 and VRC01 than the geographically matched panel of chronic Envs (Chronic 1). To confirm this finding, we assessed an independent panel of clade B chronic Envs from Washington state (Chronic 2). “All chronic” includes clade B chronic panels 1 and 2. (I) Clade B T/F Envs are also more sensitive to clade B HIV Ig on NP2 cells as measured by IC50. P values shown are from Mann-Whitney tests with the corresponding T/F Envs. NP2 and TZMbl experiments were performed in at least three and two independent experiments, respectively.
To assess whether the neutralization-sensitive phenotype of our T/F Envs depended on the particular panel of chronic Envs used, we examined the neutralization sensitivities of 14 clade B control Envs derived from six additional chronically infected individuals (Chronic 2 in Fig. 6A to D). Similarly to the initial test set of chronic Envs (Chronic 1 in Fig. 6A to D), this validation set exhibited increased resistance to b12 compared to T/F Envs (P = 0.0001 by Mann-Whitney test). However, unlike the initial chronic Env panel, the validation Envs were similar to the T/F Envs in their sensitivity to VRC01 (P = 0.14 by Mann-Whitney test). Finally, there were no differences in PG9 and PG16 sensitivity between the T/F and the validation Envs (Fig. 6C and D).
While broadly neutralizing MAbs are useful tools for examining neutralization sensitivity, they are rare in HIV-1-infected individuals and thus may give a biased view of HIV-1 neutralization. Thus, we examined the neutralization sensitivities of the T/F and chronic Envs to pooled sera from patients infected with clade B HIV-1 strains (clade B HIV Ig). The T/F Envs (median IC50, 741 μg/ml) were approximately 2-fold more neutralization sensitive than the chronic test panel (median IC50, 1,179 μg/ml; P = 0.062), the chronic validation panel (median IC50, 1,500 μg/ml; P = 0.0095), and the combined clade B chronic panel (median IC50, 1,324 μg/ml; P = 0.0078) (Fig. 6I).
To examine the basis for the enhanced b12 and VRC01 neutralization sensitivity of T/F Envs, we measured the binding of the two MAbs to both T/F and chronic Envs. Binding to the trimeric form of the Env expressed on the surface of cells was measured using a cell-based ELISA system (32). To obtain an accurate measure of antibody binding affinity, we corrected binding measurements for the levels of cell surface expression of the different Envs. For this purpose, the binding efficiency of b12 and VRC01 was expressed as a fraction of the binding of a CD4-Ig probe added at saturating concentrations. CD4-Ig is a fusion protein that consists of two copies of the two N-terminal domains of CD4 that are linked to the Fc region of human IgG1.
For the entire group of Envs (i.e., T/F and both chronic Envs groups combined), a very strong correlation was observed between the binding of the MAbs to the trimeric Envs and their sensitivity to inhibition. Spearman rank order correlation coefficients of 0.62 (P < 0.0001) and 0.77 (P < 0.0001) were obtained for b12 and VRC01, respectively (Fig. 7A and B). Comparison of MAb binding to the T/F and chronic Envs showed clear differences between the two groups for both b12 and VRC01. Binding of b12 to the T/F Envs was significantly increased relative to that to both groups of chronic Envs (Fig. 7C). Binding of VRC01 to the T/F Envs was increased relative to that to the original group of chronic Envs (P = 0.004) (Fig. 7D). The differential formation/exposure of these epitopes suggests the existence of at least modest structural differences within or near the CD4 binding sites of T/F and chronic Envs. No significant differences were observed between VRC01 binding to the T/F and the Chronic 2 Envs (P = 0.21).
Fig. 7.

Correlation between MAb binding and neutralization. (A and B) Env was expressed on the surface of cells, and then binding to b12 (A) and VRC01 (B) relative to a CD4 control was assessed by ELISA. There is a strong positive correlation between binding and Env pseudotype neutralization sensitivity for both b12 and VRC01 for the T/F Envs and both panels of chronic Envs serving to validate the assay. (C and D) To assess the mechanism of enhanced neutralization sensitivity, we compared b12 (C) and VRC01 (D) binding between T/F and chronic Envs. This suggests that differences in MAb binding explain neutralization differences between T/F and chronic Envs. The data shown are the means from two independent experiments.
DISCUSSION
The genetic bottleneck that occurs during mucosal transmission of HIV-1 results from the fact that most often only a single founder virus is successfully transmitted from among a diverse swarm of viruses present in the donor (36). It is evident that a significant degree of selection is manifest at this step, since transmission of R5-tropic virus strains is far more efficient than that of X4-tropic and even R5X4-tropic viruses (62, 79). Whether there is selection for additional viral phenotypes beyond coreceptor use or whether viral transmission is essentially a stochastic process, in which any reasonably fit R5-tropic HIV-1 strain can be transmitted, has not yet been determined. Addressing this question is of practical importance since properties associated with preferential viral transmission could potentially be exploited by vaccine or other antiviral approaches.
Genetic, immunologic, and phenotypic signatures associated with transmitted HIV-1 Envs have been sought in a number of previous studies, most entailing Envs obtained from early infections (acute Envs) (2, 13, 19, 35, 57, 72, 79) as opposed to true T/F Envs obtained by SGA analyses (36, 39, 60). Several studies concluded that T/F and acute Envs have on average shorter variable loops and fewer PNGs than Envs derived from chronically infected individuals (19, 72). While such differences have been noted for Envs from multiple HIV-1 clades, they are relatively subtle, variable in location, and far from predictive, with some being evident only when larger numbers of sequences are compared. The 24 T/F Envs examined here, for example, exhibited no consistent genetic differences from the chronic controls. Nonetheless, a much larger sequence comparison that included all but one of the Envs examined here identified a small number of sequence signatures associated with transmission, including specific sites in the signal sequence and gp41 cytoplasmic domain that could affect Env processing, localization, and incorporation into virus particles as well as changes in the receptor binding regions in gp120 and in N-linked glycosylation sites (Gnanakaran et al., submitted). Thus, existing evidence points to an array of genetic features that may be associated with enhanced HIV-1 transmission across mucosal surfaces by unknown mechanisms.
The identification of genetic motifs in env that are enriched in T/F viruses is consistent with the possibility that specific phenotypic properties that might provide a selective advantage to transmitted viruses can be identified. This is clearly the case at a global level, in that T/F Envs are almost invariably R5-tropic and replicate well in CD4+ T cells but poorly in monocyte-derived macrophages (with the exception of clade D viruses [G. M. Shaw and J. Baalwa, unpublished data]) (60). More-detailed phenotypic studies of recently transmitted viruses are generally lacking, although donor and recipient Envs from eight transmission pairs exhibited no differences in CD4 or CCR5 utilization, and a second study using some of these same Envs did not find consistent differences in primary cell infection or use of receptors other than CCR5 and CXCR4 (2, 35). As genetic signatures associated with transmission can be both variable and subtle, we employed a more-detailed series of functional assays to seek differences between viral pseudotypes bearing the T/F Envs and those expressing Envs from chronic controls. We found no phenotypic differences between the T/F and chronic Envs examined here in assays designed to probe the efficiency and rate of membrane fusion, the efficiency of coreceptor use, the ability to infect primary CD4+ T cell subsets from different donors, and the ability of virus to be captured by DCs and transferred to adjoining CD4+ T cells. One could ask whether the assays we employed are sufficiently sensitive to detect functional differences between viruses bearing different Env glycoproteins. We feel that they are, as we and others have used these and similar assays to identify significant functional differences between Envs at the level of primary CD4+ T cell tropism, membrane fusion kinetics, the efficiency of CD4 and coreceptor utilization, and attachment to C-type lectins such as DC-SIGN (25, 53, 54, 67). Even single amino changes in Env can affect these properties to extents that can be easily detected. The CD4+ T cell subset tropism assay that we have developed, which can determine the efficiency with which a given virus infects TCM, TEM, TEMRA, and naïve T cells, is a particularly sensitive measure of CD4 and coreceptor use, as these receptors are expressed differently on various CD4+ T cell subsets (7, 28, 38, 45, 50). The fact that that 24 T/F Envs here were functionally equivalent to the chronic Env controls in all of the assays employed argues that any phenotypic differences between these and chronic Env controls are apt to be slight in magnitude.
A second consideration regarding the presence or absence of phenotypic traits associated with enhanced virus transmission is whether the assays we employed effectively recapitulate the key events during the earliest stages of HIV-1 transmission (reviewed in reference 31). Following mucosal transmission of HIV-1, virus is not detected in the circulation for about 10 days, a period termed the eclipse phase (reviewed in reference 41). Detailed studies in the macaque model show that after vaginal exposure small clusters of infected cells are found in the endocervical region, which is lined by a single layer of epithelial cells (77). The recruitment of plasmacytoid DCs, T cells, and macrophages over several days transforms the initial focus of infection into a CD4+ T cell-rich environment. Similar studies have not yet been conducted to assess penile or rectal transmission in the rhesus model, the likely mode of transmission in the predominantly male cohort assessed in this study. Conceivably, Env properties that promote entry into resting and activated CD4+ T cells in the submucosa as well as transmission between cells could increase the possibility that an initial focus of infection will successfully propagate and eventually lead to dissemination to regional lymph nodes and a systemic infection. The CD4+ T cell subset tropism assay we employed, while more detailed and sensitive than bulk CD4+ T cell infection assays, may not produce CD4+ T cells with properties identical to those found in the rectal or cervicovaginal mucosa. In addition, the DC/CD4+ T cell transmission assay we used is but a surrogate for the likely more complex cell-cell interactions found in the initial foci of infection. It is important to keep in mind that since virus appears to replicate locally for a period of at least a few days to a week, even a relatively subtle change in an Env property that might enhance infection could result in a significant selective advantage over the course of multiple rounds of infection. The single-cycle assays we employed, while sensitive and well validated, cannot capture the impact of more subtle differences in Env fitness over time. Future studies employing T/F infectious molecular clones in both primary cell and tissue explant cultures might be better suited for the identification of early fitness differences associated with T/F viruses.
In addition to genetic signatures, differences at the level of sensitivity to antibody-mediated neutralization have been found in some studies of recently transmitted viruses (19, 36). We found that the panel of clade B T/F Envs was more sensitive to the CD4 binding site MAbs b12 and VRC01, as well as clade B HIV Ig, but not to the broadly neutralizing antibodies PG9 and PG16. These differences were approximately 2-fold in magnitude and were partially dependent upon the control group employed. Specifically, when a second panel of chronic Envs was used as a control, enhanced sensitivity to VRC01 was not observed, though MAb b12 and clade B HIV Ig continued to neutralize the T/F Envs more efficiently. The relatively modest differences that were observed, along with the fact that enhanced neutralization was not seen between all study groups, raises several important questions: do T/F Envs exhibit features that generally enhance their sensitivity to certain types of neutralizing antibodies, and if so, what is the basis for these differences and what are the implications for virus transmission?
One limitation of this study is the selection of chronic control Envs. Ideally, chronic control Envs would be selected from longitudinal samples or confirmed transmission pairs; however, such samples are difficult to find in sufficient numbers, especially since the great majority of acute clade B infections are treated with antiretroviral therapy. It would also be preferable to obtain chronic Envs from semen or genital secretions, the likely source of the viral inoculum, but again such samples are exceedingly scarce. In addition, the majority of Envs used in this study were from males who likely acquired HIV by penile or rectal transmission. Thus, further work is needed to characterize the transmission bottleneck that occurs during vaginal transmission. Our results emphasize the importance of selecting appropriate matched chronic controls, since the chronic test and validation sets differed in their neutralization profiles to VRC01 (though not to MAb b12 and clade B HIV Ig) despite no obvious differences in length of infection, transmission risk factor, patient demographics, or phylogenetic relationships to the T/F Envs. Of course, since we are unable to reliably predict neutralization sensitivity from sequence information alone, a control group could by chance differ immunologically from the T/F Envs despite being otherwise well matched. To mitigate this, selecting chronic Env controls from geographically matched individuals may be important. For example, we previously reported that clade B T/F Envs are more resistant than chronic Envs to b12 and the membrane-proximal external region (MPER) antibodies 2F5 and 4E10 (36), seemingly in contradiction with our current findings. However, reexamination of the data reported by Keele et al. (36) showed that this was due to the predominance of neutralization-sensitive Envs derived from chronically infected individuals in Trinidad. These Trinidad Envs form a subcluster within the other clade B Envs used in this study (Fig. 1), have a Thr deletion in the V3 loop compared to the clade B consensus, were overrepresented in the chronic controls, and were more sensitive to neutralization by MAbs b12, 2F5, and 4E10 (14). Thus, the previously reported 2F5 and 4E10 neutralization difference between T/F and chronic Envs was due to bias resulting from disproportionate representation of Envs from Trinidad in the chronic controls.
Several other studies that have assessed the neutralization sensitivity of clade B Envs did not use geographically matched chronic controls, raising the possibility that the results from these studies could be complicated by genotypic differences linked to geographic location (64, 68, 73). In addition to the location, it may also be important to match the time of sample collection when developing well-matched chronic control groups. For example, Bunnik et al. reported that HIV-1 has become more neutralization resistant over the course of the epidemic, and thus patient sampling times may bias comparisons between T/F and chronic Envs (10). Here, the chronic Envs were sampled four calendar years before the T/F Envs on average. However, this difference is significantly shorter than the 14- to 21-year time span between contemporary and historic Envs assessed by Bunnik et al. (10). In addition, we detected no correlation between sampling time and neutralization sensitivity, and thus this cannot account for the neutralization differences between the T/F Envs and the chronic controls. It is also of note that multiple chronic Envs from the same individual were treated as independent events in this study. Reanalyzing the data to include only one chronic Env value (mean of the multiple Envs) per individual did not change the magnitude of the neutralization difference, though it did decrease the P values above the level of significance for VRC01 and HIV Ig, but not b12, likely due to decreased sample size. In summary, more-detailed studies involving larger numbers of T/F Envs with appropriately matched control Envs, including Envs derived from the same individuals over time, and a greater number of broadly neutralizing MAbs and human sera will be needed to draw definitive conclusions about the neutralization sensitivities of transmitted virus strains.
When our data are considered along with other published studies on T/F and acute Envs, several conclusions can be drawn. First, we believe that HIV-1 transmission is in part stochastic, with any reasonably fit R5-tropic virus being capable of initiating an infection (36, 56, 63, 79). With a now relatively large number of T/F and acute Envs having been examined, it is evident that no single major genetic, phenotypic or immunologic signature is required for transmission beyond the use of CCR5. Second, an array of genetic traits, including but not limited to shorter variable loops and reduced numbers of N-linked glycosylation sites, are associated with enhanced virus transmission. The structural implications of these signatures are not well understood, and it is not yet clear if these or as-yet-unidentified other genetic traits are responsible for the modestly enhanced sensitivity to antibody-mediated neutralization that is characteristic of some T/F and acute Envs. Third, the presence of genetic signatures linked to transmission implies some impact on function that enhances transmission. If so, then the functional impact is apt to be modest given the variable nature of the genetic signatures and the fact that neither we nor others have observed clear differences between T/F and acute Envs with chronic controls. However, the possibility exists that relatively subtle alterations of Env function, perhaps in the context of full-length T/F viral genomes, could provide a sufficiently robust selective advantage during the eclipse phase of HIV-1 transmission to result in preferential transmission of viruses with specific properties. The growing application of SGA technology coupled with increasingly sophisticated cell-to-cell and ex vivo tissue systems will make it possible to more rigorously identify immunologic and phenotypic traits associated with HIV-1 transmission.
ACKNOWLEDGMENTS
We thank the University of Pennsylvania's Center for AIDS Research (CFAR) Immunology Core for human CD4+ T cells and monocytes, the University of Washington's CFAR HIV Specimen Repository for plasma, Dennis Burton and John Mascola for generously providing PG9/16 and VRC01, respectively, Gerald Learn for phylogenetic expertise, Peter Hraber for determining variable-loop lengths of T/F Envs, and Drew Weissman, Truman Grayson, Chuanxi Sun, James A. Hoxie, Yanji Yi, and Ronald G. Collman for technical expertise and helpful discussions.
This work was funded by the Center for HIV/AIDS Vaccine Immunology, National Institutes of Health grants (AI67854, AI27767, and AI40880), the Bill and Melinda Gates Foundation grants 37874 and 38619, and the International AIDS Vaccine Initiative. C.B.W. and N.F.P. were supported by grants T32 AI000632 and T32 GM008361, respectively.
Footnotes
Published ahead of print on 29 June 2011.
REFERENCES
- 1. Abrahams M. R., et al. 2009. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-Poisson distribution of transmitted variants. J. Virol. 83:3556–3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander M., et al. 2010. Donor and recipient envs from heterosexual human immunodeficiency virus subtype C transmission pairs require high receptor levels for entry. J. Virol. 84:4100–4104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balotta C., et al. 1997. Homozygous delta 32 deletion of the CCR-5 chemokine receptor gene in an HIV-1-infected patient. AIDS 11:F67–F71 [DOI] [PubMed] [Google Scholar]
- 4. Bar K. J., et al. 2010. Wide variation in the multiplicity of HIV-1 infection among injection drug users. J. Virol. 84:6241–6247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barbas C. F., III, et al. 1992. Recombinant human Fab fragments neutralize human type 1 immunodeficiency virus in vitro. Proc. Natl. Acad. Sci. U. S. A. 89:9339–9343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biswas P., Tambussi G., Lazzarin A. 2007. Access denied? The status of co-receptor inhibition to counter HIV entry. Expert Opin. Pharmacother. 8:923–933 [DOI] [PubMed] [Google Scholar]
- 7. Bleul C. C., Wu L., Hoxie J. A., Springer T. A., Mackay C. R. 1997. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 94:1925–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borrow P., et al. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3:205–211 [DOI] [PubMed] [Google Scholar]
- 9. Bridger G. J., et al. 1995. Synthesis and structure-activity relationships of phenylenebis(methylene)-linked bis-tetraazamacrocycles that inhibit HIV replication. Effects of macrocyclic ring size and substituents on the aromatic linker. J. Med. Chem. 38:366–378 [DOI] [PubMed] [Google Scholar]
- 10. Bunnik E. M., et al. 2010. Adaptation of HIV-1 envelope gp120 to humoral immunity at a population level. Nat. Med. 16:995–997 [DOI] [PubMed] [Google Scholar]
- 11. Burton D. R., et al. 1991. A large array of human monoclonal antibodies to type 1 human immunodeficiency virus from combinatorial libraries of asymptomatic seropositive individuals. Proc. Natl. Acad. Sci. U. S. A. 88:10134–10137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burton D. R., et al. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024–1027 [DOI] [PubMed] [Google Scholar]
- 13. Chohan B., et al. 2005. Selection for human immunodeficiency virus type 1 envelope glycosylation variants with shorter V1-V2 loop sequences occurs during transmission of certain genetic subtypes and may impact viral RNA levels. J. Virol. 79:6528–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cleghorn F. R., et al. 2000. A distinctive clade B HIV type 1 is heterosexually transmitted in Trinidad and Tobago. Proc. Natl. Acad. Sci. U. S. A. 97:10532–10537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Curlin M. E., et al. 2010. HIV-1 envelope subregion length variation during disease progression. PLoS Pathog. 6:e1001228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dalgleish A. G., et al. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763–767 [DOI] [PubMed] [Google Scholar]
- 17. Decker J. M., et al. 2005. Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J. Exp. Med. 201:1407–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Clercq E., et al. 1994. Highly potent and selective inhibition of human immunodeficiency virus by the bicyclam derivative JM3100. Antimicrob. Agents Chemother. 38:668–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Derdeyn C. A., et al. 2004. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303:2019–2022 [DOI] [PubMed] [Google Scholar]
- 20. Doores K. J., Burton D. R. 2010. Variable loop glycan dependency of the broad and potent HIV-1-neutralizing antibodies PG9 and PG16. J. Virol. 84:10510–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eckstein D. A., et al. 2001. HIV-1 actively replicates in naive CD4(+) T cells residing within human lymphoid tissues. Immunity 15:671–682 [DOI] [PubMed] [Google Scholar]
- 22. Emmelkamp J. M., Rockstroh J. K. 2007. CCR5 antagonists: comparison of efficacy, side effects, pharmacokinetics and interactions—review of the literature. Eur. J. Med. Res. 12:409–417 [PubMed] [Google Scholar]
- 23. Fiebig E. W., et al. 2003. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. AIDS 17:1871–1879 [DOI] [PubMed] [Google Scholar]
- 24. Frost S. D., et al. 2005. Characterization of human immunodeficiency virus type 1 (HIV-1) envelope variation and neutralizing antibody responses during transmission of HIV-1 subtype B. J. Virol. 79:6523–6527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geijtenbeek T. B., et al. 2000. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100:587–597 [DOI] [PubMed] [Google Scholar]
- 26. Giorgi J. V., et al. 1999. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J. Infect. Dis. 179:859–870 [DOI] [PubMed] [Google Scholar]
- 27. Gordon S. N., et al. 2010. Disruption of intestinal CD4+ T cell homeostasis is a key marker of systemic CD4+ T cell activation in HIV-infected individuals. J. Immunol. 185:5169–5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Groot F., van Capel T. M., Schuitemaker J., Berkhout B., de Jong E. C. 2006. Differential susceptibility of naive, central memory and effector memory T cells to dendritic cell-mediated HIV-1 transmission. Retrovirology 3:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guindon S., Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704 [DOI] [PubMed] [Google Scholar]
- 30. Haaland R. E., et al. 2009. Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLoS Pathog. 5:e1000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haase A. T. 2011. Early events in sexual transmission of HIV and SIV and opportunities for interventions. Annu. Rev. Med. 62:127–139 [DOI] [PubMed] [Google Scholar]
- 32. Haim H., et al. 2009. Soluble CD4 and CD4-mimetic compounds inhibit HIV-1 infection by induction of a short-lived activated state. PLoS Pathog. 5:e1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harrison J. E., et al. 2008. Baseline resistance of primary human immunodeficiency virus type 1 strains to the CXCR4 inhibitor AMD3100. J. Virol. 82:11695–11704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hendrix C. W., et al. 2000. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob. Agents Chemother. 44:1667–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Isaacman-Beck J., et al. 2009. Heterosexual transmission of human immunodeficiency virus type 1 subtype C: macrophage tropism, alternative coreceptor use, and the molecular anatomy of CCR5 utilization. J. Virol. 83:8208–8220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Keele B. F., et al. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105:7552–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Klatzmann D., et al. 1984. Selective tropism of lymphadenopathy associated virus (LAV) for helper-inducer T lymphocytes. Science 225:59–63 [DOI] [PubMed] [Google Scholar]
- 38. Lee B., et al. 1999. Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J. Biol. Chem. 274:9617–9626 [DOI] [PubMed] [Google Scholar]
- 39. Li H., et al. 2010. High multiplicity infection by HIV-1 in men who have sex with men. PLoS Pathog. 6:e1000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li M., et al. 2006. Genetic and neutralization properties of subtype C human immunodeficiency virus type 1 molecular env clones from acute and early heterosexually acquired infections in Southern Africa. J. Virol. 80:11776–11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McMichael A. J., Borrow P., Tomaras G. D., Goonetilleke N., Haynes B. F. 2010. The immune response during acute HIV-1 infection: clues for vaccine development. Nat. Rev. Immunol. 10:11–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mehandru S., Tenner-Racz K., Racz P., Markowitz M. 2005. The gastrointestinal tract is critical to the pathogenesis of acute HIV-1 infection. J. Allergy Clin. Immunol. 116:419–422 [DOI] [PubMed] [Google Scholar]
- 43. Miyauchi K., Kim Y., Latinovic O., Morozov V., Melikyan G. B. 2009. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 137:433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miyauchi K., Kozlov M. M., Melikyan G. B. 2009. Early steps of HIV-1 fusion define the sensitivity to inhibitory peptides that block 6-helix bundle formation. PLoS Pathog. 5:e1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mo H., et al. 1998. Expression patterns of the HIV type 1 coreceptors CCR5 and CXCR4 on CD4+ T cells and monocytes from cord and adult blood. AIDS Res. Hum. Retroviruses 14:607–617 [DOI] [PubMed] [Google Scholar]
- 46. Nishimura Y., et al. 2005. Resting naive CD4+ T cells are massively infected and eliminated by X4-tropic simian-human immunodeficiency viruses in macaques. Proc. Natl. Acad. Sci. U. S. A. 102:8000–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. O'Brien T. R., et al. 1997. HIV-1 infection in a man homozygous for CCR5 delta 32. Lancet 349:1219. [DOI] [PubMed] [Google Scholar]
- 48. Ostrowski M. A., et al. 1999. Both memory and CD45RA+/CD62L+ naive CD4(+) T cells are infected in human immunodeficiency virus type 1-infected individuals. J. Virol. 73:6430–6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pang S., et al. 1992. Rapid generation of sequence variation during primary HIV-1 infection. AIDS 6:453–460 [DOI] [PubMed] [Google Scholar]
- 50. Pfaff J. M., et al. 2010. HIV-1 resistance to CCR5 antagonists associated with highly efficient use of CCR5 and altered tropism on primary CD4+ T cells. J. Virol. 84:6505–6514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Piguet V., Steinman R. M. 2007. The interaction of HIV with dendritic cells: outcomes and pathways. Trends Immunol. 28:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Poss M., et al. 1995. Diversity in virus populations from genital secretions and peripheral blood from women recently infected with human immunodeficiency virus type 1. J. Virol. 69:8118–8122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Puffer B. A., Altamura L. A., Pierson T. C., Doms R. W. 2004. Determinants within gp120 and gp41 contribute to CD4 independence of SIV Envs. Virology 327:16–25 [DOI] [PubMed] [Google Scholar]
- 54. Reeves J. D., et al. 2002. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. U. S. A. 99:16249–16254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Roben P., et al. 1994. Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1. J. Virol. 68:4821–4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Roos M. T., et al. 1992. Viral phenotype and immune response in primary human immunodeficiency virus type 1 infection. J. Infect. Dis. 165:427–432 [DOI] [PubMed] [Google Scholar]
- 57. Rusert P., et al. 2005. Virus isolates during acute and chronic human immunodeficiency virus type 1 infection show distinct patterns of sensitivity to entry inhibitors. J. Virol. 79:8454–8469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sagar M., et al. 2009. Selection of HIV variants with signature genotypic characteristics during heterosexual transmission. J. Infect. Dis. 199:580–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Salazar-Gonzalez J. F., et al. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 82:3952–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Salazar-Gonzalez J. F., et al. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 206:1273–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sallusto F., Lenig D., Forster R., Lipp M., Lanzavecchia A. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712 [DOI] [PubMed] [Google Scholar]
- 62. Scarlatti G., et al. 1997. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat. Med. 3:1259–1265 [DOI] [PubMed] [Google Scholar]
- 63. Schuitemaker H., et al. 1992. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 66:1354–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Seaman M. S., et al. 2010. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J. Virol. 84:1439–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Stephenson J. 2007. Researchers buoyed by novel HIV drugs: will expand drug arsenal against resistant virus. JAMA 297:1535–1536 [DOI] [PubMed] [Google Scholar]
- 66. Theodorou I., Meyer L., Magierowska M., Katlama C., Rouzioux C. 1997. HIV-1 infection in an individual homozygous for CCR5 delta 32. Seroco Study Group. Lancet 349:1219–1220 [PubMed] [Google Scholar]
- 67. Tilton J. C., et al. 2010. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J. Virol. 84:10863–10876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Walker L. M., et al. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wei X., et al. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307–312 [DOI] [PubMed] [Google Scholar]
- 70. Westby M., et al. 2007. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 81:2359–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wolfs T. F., Zwart G., Bakker M., Goudsmit J. 1992. HIV-1 genomic RNA diversification following sexual and parenteral virus transmission. Virology 189:103–110 [DOI] [PubMed] [Google Scholar]
- 72. Wolinsky S. M., et al. 1992. Selective transmission of human immunodeficiency virus type-1 variants from mothers to infants. Science 255:1134–1137 [DOI] [PubMed] [Google Scholar]
- 73. Wu X., et al. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang H., et al. 2004. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J. Virol. 78:1718–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang L. Q., et al. 1993. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J. Virol. 67:3345–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang M., et al. 2004. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology 14:1229–1246 [DOI] [PubMed] [Google Scholar]
- 77. Zhang Z., et al. 1999. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science 286:1353–1357 [DOI] [PubMed] [Google Scholar]
- 78. Zhou T., et al. 2010. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329:811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhu T., et al. 1993. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 261:1179–1181 [DOI] [PubMed] [Google Scholar]