

Abstract
The metabolic changes that occur in a cancer cell have been studied for a few decades, but our appreciation of the complexity and importance of those changes is now being realized. The metabolic switch from oxidative phosphorylation to aerobic glycolysis provides intermediates for cell growth and division and is regulated by both oncogenes and tumor suppressor genes. The p53 tumor suppressor gene has long been shown to play key roles in responding to DNA damage, hypoxia, and oncogenic activation. However, now p53 has added the ability to mediate metabolic changes in cells through the regulation of energy metabolism and oxidative stress to its repertoire of activities. It is therefore the focus of this review to discuss the metabolic pathways regulated by p53 and their cooperation in controlling cancer cell metabolism.
Keywords: p53, aerobic glycolysis, oxidative phosphorylation, oxidative stress
Introduction
The Warburg effect is defined as an increased dependence on glycolysis for ATP synthesis, even in the presence of abundant oxygen, instead of a cell using the more effective pathway of oxidative phosphorylation.1 This shift has even been termed a hallmark of cancer as it gives pathologists and clinicians a method to diagnosis cancer,2 and it helps to explain how cancerous processes prepare substrates for active cell growth and division. Because glycolysis produces ATP less efficiently than aerobic respiration, tumor cells compensate by taking up glucose at a higher rate than normal cells. This shift in metabolic activity is observed in many (but not all) cancer cells, and thus the increase in glucose usage provides an opportunity to image tumors using a glucose analog 2-(18F)-fluoro-2-deoxy-D-glucose (FDG) by positron emission tomography (PET). Although it is widely accepted that aerobic glycolysis is the metabolic pathway preferred in many cancer cells, its relationship to cancer progression is still unclear. In addition to aerobic glycolysis, metabolic transformation also includes a cell’s ability to undergo replicative division such that the cell can duplicate its genome, proteins, and lipids into 2 daughter cells. This means that the cell must take up the nutrients such as glucose and glutamine and drive them into the metabolic pathways that will produce the necessary biosynthetic precursors. Tumor cells do this through changing the levels of expression of key enzymes such as nutrient transporters, reducing agents, biosynthetic pathways, and energy production. Likewise, the way that metabolic pathways are controlled has revealed an important role for both oncogenes and tumor suppressor genes. Among them, p53 seems to play an important role in regulating several aspects of cellular metabolism that are now being explored not only in cancer but in other aspects of disease and development.3 This review will delve into some of the many ways that p53 functions to control various aspects of cellular metabolism.
Metabolism: Glycolysis in Cancer Development
Cellular metabolism of glucose to CO2 and H2O in normal adult cells is accomplished through the oxidative phosphorylation pathway that leads to the generation of ATP in the presence O2. However, under hypoxic conditions or when energy is needed rapidly, for instance, in cancer cells, glycolysis is the preferred method for energy production. Why glycolysis is the method of choice for cancer cells may seem surprising given that only 2 molecules of ATP per molecule of glucose are produced during glycolysis and 36 molecules of ATP are produced in oxidative phosphorylation.4 In the case of cancer cells, a switch in the metabolic pathways allows cancer cells that are sometimes reproducing at a rapid rate to be able to produce all the substrates for macromolecules to sustain growth and at the same time live under conditions of oxidative stress and hypoxia. Thus, tumors face three important obstacles that require them to change their cellular metabolism: 1) the need for high-energy production and lots of reducing agents (NADPH, glutathione) to “cool” the large turnover of glucose, 2) the requirement to support rapid cell growth and proliferation, and 3) the ability to survive periods of metabolic stress and low oxygen and prevent cell death.
Cells that are rapidly proliferating require favorable energetics, meaning high ATP/ADP and ATP/AMP ratios. Cancer cells accomplish this by using large amounts of glucose, which is a critical nutrient for cells, the primary substrate for ATP generation, and the carbon source for macromolecules. To generate energy, cancer cells use glucose whose entrance into the cells is differentially mediated by glucose transporters and then converted to pyruvate. Pyruvate is then further converted into lactate by lactate dehydrogenase (LDH-A) and most often secreted out of the cell. This step recovers the necessary NAD+ that is needed to maintain glycolysis, and when glucose is in excess, the flux through this pathway leads to high ATP amounts.
In addition to energy, glycolysis can produce the biomolecules that are important for tumor cell growth. Pyruvate kinase–M2 (PK-M2) is the embryonic splice variant of the rate-limiting enzyme for pyruvate generation that is preferentially expressed in tumor cells and regulates the flux of carbon into ribose-5-phosphate and nucleotide synthesis via phosphotyrosine signaling.5 Since PK-M2 can regulate the amounts of pyruvate that are produced by glycolysis, it can also regulate the flow of carbon into making nucleotides or fatty acids to promote further tumor growth that is being stimulated by high levels of certain growth factors. In addition, pyruvate can be shuttled into the mitochondria for use in the production of additional amino acids or fatty acid synthesis and used to maintain mitochondrial membrane potential. Thus, maintaining high rates of glycolysis allows cells to build up phosphorylated intermediates that meet the energetic and biosynthetic demands of anabolic synthesis and cell growth.
p53 and Energy Metabolism: Glycolysis and Oxidative Phosphorylation
To regulate energy metabolism, p53 interjects at many points in both glycolysis and oxidative phosphorylation. One of the ways that p53 functions is to slow down glycolysis and promote oxidative phosphorylation. Thus, by p53 balancing the use of glycolysis and oxidative phosphorylation, through transcriptional regulation of target genes, it provides a mechanism of blocking tumorigenesis and the Warburg effect that is mechanistically different from either apoptosis or senescence. Unfortunately, things are never that simple. p53 can both promote glycolysis and oxidative phosphorylation, as well as modulate down glycolysis. This complexity might stem from p53’s ability to regulate many different cellular pathways, and thus its loss can cause alterations in metabolism that can have effects on almost every aspect of cell behavior (i.e., proliferation, growth, and survival) under conditions of increasing and decreasing nutrient and oxygen levels. Furthermore, the difference between basal levels of p53 versus increased levels of p53 during severe or sustained stress might allow for the transcription of different sets of genes as well as tissue-specific contexts.6
p53 influences metabolic pathways by regulating the levels of a series of gene products that affect metabolic fates and metabolic products. For example, p53 increases the expression of synthesis of cytochrome c oxidase 2 (SCO2), glutaminase 2 (GLS2), the p53 modulator of apoptosis (PUMA), glucose transporter 1 and 4 (GLUT1, GLUT4), and TP53-induced glycolysis and apoptosis regulator (TIGAR), and p53 decreases the expression of phosphoglycerate mutase (PGM)7-16 (Figure 1). p53 influences glucose flux by regulating PGM, GLUT1, GLUT4, and TIGAR expression. The p53 protein induces the transcription of the TIGAR gene, which lowers the intracellular concentrations of fructose-2,6-bisphosphate and thus decreases glycolysis and overall levels of intracellular reactive oxygen species (ROS).8 The decrease in glycolysis allows for entry into the pentose phosphate pathway (PPP) and limits oxidative stress–induced cell death. Glycolysis is likewise decreased when p53 decreases PGM expression.12 Increases in PGM allow for enhanced glycolytic flux and indefinite proliferation, which might occur in some cancers that have a mutant p53. p53 also regulates the expression of GLUT1 and GLUT4 genes through a repression mechanism.7 Mutations in the DNA binding domain of p53 abolished the repressive effect on GLUT1 and GLUT4, allowing for increased glucose metabolism and cell energy supply.
Figure 1.
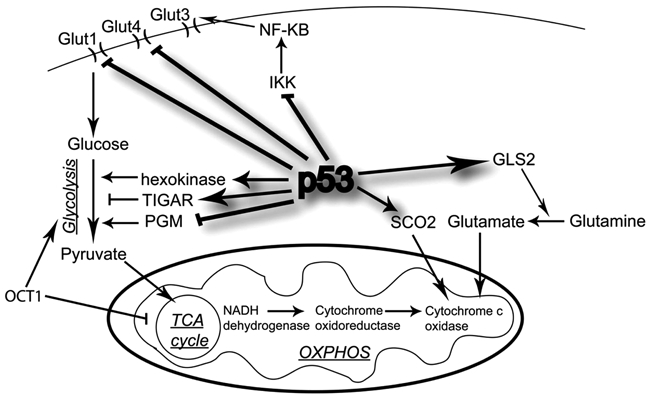
Regulation of metabolism by p53 in proliferating cells. p53 induces TP53-induced glycolysis and apoptosis regulator (TIGAR), hexokinase, inhibits phosphoglycerate mutase (PGM), and represses glucose transporter 1 and 4 (GLUT1 and GLUT4), resulting in inhibition of glycolysis and opposing the Warburg effect that is seen in many cancers. Oxidative phosphorylation is enhanced by p53 induction of synthesis of cytochrome c oxidase 2 (SCO2) and glutaminase 2 (GLS2). p53 can also regulate the glycolytic pathway through IKK/NF-κB signaling.
p53 also functions to promote the use of the oxidative phosphorylation pathway. It has been shown that ATP is produced at higher levels using oxidative phosphorylation in HCT116 cells expressing p53 versus similar cells lacking p53.17 One way p53 balances the usage of the respiratory and glycolytic pathways is through its downstream mediator, SCO2.13 SCO2 is critical for regulating a major site of oxygen utilization in the cell, the cytochrome c oxidase (COX) complex. Loss of p53 supports a switch from aerobic respiration to glycolysis through the disruption of COX function, and this decreases cellular dependence on oxygen. Another p53 target gene involved in energy production is glutaminase 2, which converts glutamine to glutamate and can be used to enhance the rate of the TCA cycle and oxidative phosphorylation.10,15 Upon p53 activation and subsequent glutaminase 2 activation, glutamate and α-ketoglutarate levels rise, as well as mitochondrial respiration rates and glutathione (GSH) levels. The regulation exhibited by p53 adds to its role in metabolism as well as antioxidant defense against tumorigenesis. Glycolysis is also affected through the increased levels of hexokinase 2 (HK2), which is highly expressed in tumors and facilitates high rates of glucose catabolism, which in turn promotes their rapid proliferation.18 Mutant p53 has been found to activate expression of HK2 by promoter binding in liver tumors, thus maintaining a high glycolytic rate and their link to loss of cell cycle control.
A transcription factor that potentially could mediate the balance between oxidative and glycolytic metabolism through p53 cooperatively is OCT1 (POU2F1).19 OCT1 might initiate this through supporting resistance to oxidative stress, which may work with the loss of p53 during transformation. OCT1 has also been shown to interact with glycolytic enzymes in the OCA-S complex, which may act as a metabolic sensor.20
There is also an interesting link between p53, glycolysis, and apoptosis where p53-mediated PUMA expression is suppressed by high glucose after growth factor withdrawal.16 Inhibition of p53 activation and PUMA induction is initiated by glucose metabolism that then promotes an antiapoptotic balance supporting cell survival. Similarly, inhibition of PUMA induction is mediated through activated Akt dependence on glycolysis.21 In contrast to the above target gene regulation, p53 can form a complex with the antiapoptotic proteins BCL-XL and PUMA, resulting in cytochrome c release from mitochondria and promotion of mitochondrial outer membrane permeabilization.14
p53’s role in the regulation of cellular metabolism is complex given the many genes that are under its transcriptional control. p53 is also mutated in many tumors and thus can influence aspects of both glycolysis and oxidative phosphorylation, and thus is significantly important in contributing to the Warburg effect. p53 loss may also allow for metabolic alterations that are vulnerable to therapeutic intervention.
p53 and Fatty Acid Metabolism
Fatty acids are important metabolic intermediates that are used for lipid synthesis and protein modification as well as can be broken down through mitochondrial beta-oxidation to generate ATP via oxidative phosphorylation. Cancer cells can alter their metabolism by increasing de novo fatty acid (FA) synthesis irrespectively of the levels of extracellular lipids.22 Also, de novo FA synthesis in cancer cells produces lipids (i.e., phosphatidylinositol, phosphatidyl serine, or phophatidyl choline) that can regulate various oncogenic pathways such as PI3K/AKT, Ras, or Wnt pathways.23
In response to genotoxic stress and glucose deprivation, p53 modulates creatine biosynthesis and fatty acid oxidation (FAO) by increasing guanidinoacetate N-methyltransferase (GAMT)11 levels (Figure 2). GAMT, in turn, converts the glycine metabolite guanidoacetate to creatine for ADP/ATP energy metabolism and promotes apoptosis. This suggests that p53 regulation of GAMT may allow for ATP to remain at a level that is sufficient for survival or apoptosis when energy generation by glycolysis is impaired. Similarly, increased FAO occurs in the livers of wild-type p53 animals, indicating that p53 might play a role in energy maintenance of the FAO pathway.24 In addition, fatty acid synthase, an enzyme capable of de novo fatty acid synthesis of long-chain fatty acids from acetyl-CoA, malonyl-CoA, and NADPH, is a conserved p53 family target gene.25,26 p53 has also been linked to β-oxidation, which is the breakdown of fatty acids to generate acetyl-CoA, which then enters the TCA cycle. Treatment of p53 wild-type cells with metformin, a drug proposed to function as an indirect activator of AMPK, has enhanced fatty acid β-oxidation and rate of glycolysis in a p53-dependent manner.27 Metabolic stress also stimulates β-oxidation through the carnitine palmitoyltransferase (Cpt1) enzyme, and PI3K and Akt modulate its expression to suppress β-oxidation during anabolic growth.28 In addition, AMPK inhibits fatty acid and cholesterol synthesis by phosphorylating the metabolic enzymes acetyl-CoA carboxylase 1 (ACC1), which carboxylates acetyl-CoA to malonyl-CoA, and HMG-CoA reductase (HMGCR).29
Figure 2.
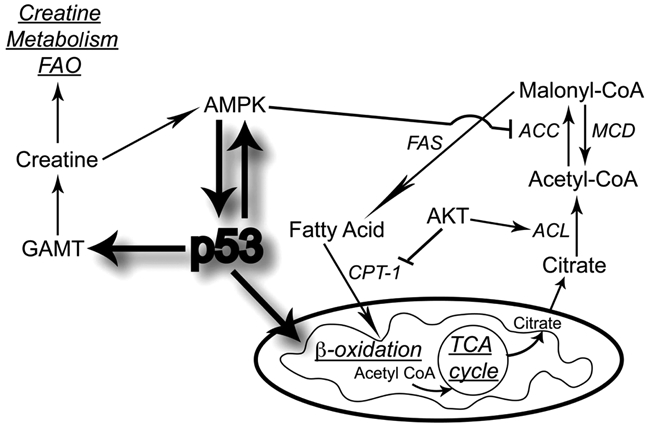
p53 modulates fatty acid metabolism. p53 can regulate fatty acid metabolism through guanidinoacetate N-methyltransferase (GAMT), AMP-activated protein kinase (AMPK), carnitine palmitoyltransferase (CPT-1), and β-oxidation. ACC, acetyl-CoA carboxylase; ACL, ATP citrate lyase; FAS, fatty acid synthase; FAO, fatty acid oxidation; MCD, malonyl-CoA decarboxylase.
Thus, it might be that p53-creatine and p53-GAMT function in either tumor suppressor mechanisms or keep the balance between the glycolytic and respiratory pathways and oppose the metabolic shift in tumorigenesis.24 Also, the fatty acid synthesis pathway might therefore be an important pathway for the use in diagnosis, treatment, and prevention of cancer.
p53’s Metabolic Response to Limited Nutrient Availability and Cell Growth
p53 senses many stress signals and acts to alleviate them, whether they are DNA damage or oncogene activation, which can then lead to cell death or senescence. One of the metabolic stresses that activate p53 is a limited supply of nutrients to a cell or deregulated nutrient-sensing pathways. The pathways that p53 modulates down in response to this type of stress are the IGF-1/AKT/mTOR pathways. This then limits the error frequency during cell growth and division. Under normal cellular conditions, the IGF-1/AKT/mTOR pathways signal for cell growth and division in response to high levels of glucose and amino acids to support growth. They allow for cells to undergo metabolic transformation by increasing the expression of nutrient transporters along its surface, thus increasing the uptake of glucose and amino acids, as well as enhancing the biosynthesis of macromolecules, AKT-dependent activation of hexokinase and phosphofructokinase, enhanced transcription of genes involved in glycolysis, and enhanced protein translation through AKT activation of mTOR.30,31 However, under reduced nutrient or energy levels, the AKT/mTOR pathways and AMPK fail to be activated, which can then induce p53. The way that p53 negatively regulates these pathways is to modulate the expression levels of IGF-BP3, PTEN, TSC2, AMPK β1, sestrins 1 and 2, and REDD132-36 (Figure 3).
Figure 3.
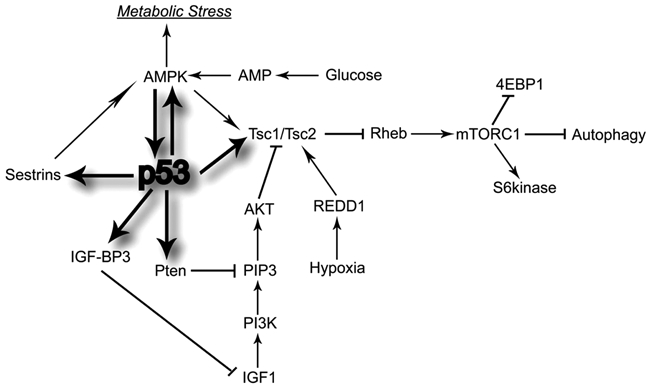
p53 regulates PI3kinase, Akt, and mTOR pathways to mediate a cell’s adaptation to stress. To do this, p53 regulates the transcription of 4 genes—PTEN, IGF-BP3, TSC2, and AMPK β—which then all negatively regulate Akt kinase and mTOR, leading to a decrease in cell growth. 4EBP1, 4E-binding protein 1; AMPK, AMP-activated protein kinase; IGF-BP3, insulin-like growth factor binding protein 3; IGF1, insulin-like growth factor 1; mTORC, mammalian target of rapamycin complex; PI3K, phosphatidylinositol-3 kinase; PIP3, phosphatidylinositol 3,4,5-trisphosphate; Pten, phosphatase and tensin homologue; Rheb, Ras homolog enriched in brain; S6 kinase, ribosomal protein S6 kinase; Tsc, tuberosclerosis complex.
IGF-1/AKT signaling is shut down through the binding of IGF-BP3 to IGF-1, thus subsequently inhibiting IGF-1 from binding to its receptor. Similarly, AKT can be inactivated through PTEN, a 403–amino acid polypeptide originally described as a dual-specificity protein phosphatase, which can decrease the function of PIP3 in the activation of PDK-1 and mTORC2. Also, the loss of the AKT activity increases the TSC1-TSC2 activity, which in turn decreases the mTORC1 activity. In addition, a reduction in AKT can lead to an increase in p53, which can activate PTEN expression and amplify this loop.36 In cancer cells, AKT activation is also sufficient to drive glycolysis and lactate production and to suppress macromolecular degradation.31 AMPK is a heterotrimeric protein consisting of 3 subunits—α, β, and γ—and drives the catabolic, energy-producing responses (i.e., fatty acid oxidation) that are needed under conditions of metabolic stress. AMPK β1 is activated by p53, leading to an increase in AMPK and TSC1-TSC2 activity and to deactivation of the mTOR pathway.34 p53 also directly up-regulates TSC2 levels, which also have a negative impact on mTOR activity.34,35 Furthermore, p53 up-regulates the expression of 2 target genes, sestrin 1 and sestrin 2, both of which activate AMPK, allowing AMPK to phosphorylate TSC2, which stimulates its GTPase activity to negatively regulate Rheb, the protein that activates mTOR complex 1 (mTORC1).32 mTORC1 can also directly or indirectly dephosphorylate p53 on Ser12 as a consequence of nutrient deprivation. REDD1 encodes a p53 and p63 transcriptional target that links ROS in a p53-dependent DNA damage response, and disruption of REDD1 shuts down hypoxia-induced inhibition of mTOR. REDD1 overexpression can also down-regulate S6K phosphorylation in a TSC1/TSC2-dependent manner.33,37 Thus, p53 shuts down IGF-1/AKT and mTOR pathways through the negative regulation of IGF-BP3, PTEN, TSC2, AMPK β1, and sestrins 1 and 2 and thus plays a critical role in cell growth and division, nutritional sensing, and metabolic regulation in response to stress (Figure 3).
The activities between p53 and the IGF-1/AKT/mTOR pathways show the importance of the coordination of many proteins to regulate proliferation and growth of cells in times of stress from nutrient deficiency. Also, many regulatory loops exist to keep these pathways in check and balance a response to cellular stress or a decision to grow and divide.38
p53’s Response to Oxidative Stress
Oxidative damage results from an imbalance between the production of free radicals (i.e., ROS) and their elimination by antioxidants (i.e., superoxide dismutase [SOD], glutathione peroxidase [GPx], and GSH) through protective mechanisms.39 Even though ROS products are part of normal cellular metabolism produced mainly through the mitochondrial respiratory chain and play roles in cell proliferation and survival signaling pathways, their imbalance can cause damage to biomolecules, cell structure, and function and induce somatic mutations and neoplastic transformation. Also an advantage for cancer cells is to switch their metabolic signaling and use glycolysis, which can help to lower ROS levels to allow for the promotion of further tumor growth and thus protection against senescence and apoptosis. One way that the cells can lower ROS levels is to use GSH. GSH accepts an electron from ROS and is then converted to its oxidized form, glutathione disulfide (GSSG). GSSG is then reduced back to GSH by glutathione reductase using NADPH. Another way is through p53, whose role in ROS regulation is more complex.
p53 has both pro-oxidant and antioxidant functions that both can contribute to tumor suppression, albeit through different methods and outcomes. The antioxidant functions of p53 involve the activation of several genes—namely, sestrin, NRF2, glutathione peroxidase, aldehyde dehydrogenase, GLS2, TIGAR, and tumor protein p53-inducible nuclear protein 1 (TP53INP1).10,40-44 For instance, GLS2, a mitochondrial glutaminase, catalyzes the hydrolysis of glutamine to glutamate, which can then be used to generate reduced glutathione. Induction of GLS2 by p53 leads to an increased ratio between glutathione and reduced glutathione that then protects the cell from oxidative stress–induced apoptosis. Similarly, TIGAR can lower the levels of fructose-2,6-bisphosphate, thereby slowing glycolysis and directing the glucose to the pentose phosphate pathway, which is a source of NADPH and is needed by reduced glutathione. Through this pathway, TIGAR allows for the decrease in ROS levels and lowered sensitivity to ROS-associated apoptosis. Furthermore, the transcription factor Nrf2 is a regulator of an intracellular antioxidant response through transcriptional activation of many genes that protect cells from toxic and carcinogenic chemicals. NRF2 binds to KEAP1, which targets NRF2 for degradation. However, under oxidative stress, p53 stimulates expression of p21, which prevents the binding of KEAP1 to NRF2 and allows for NRF2-mediated antioxidant response.44 The pro-oxidant functions of p53 involve activation of p53-induced gene 3 (PIG3), proline oxidase, BAX, PUMA, and P66SHC or the inhibition/alteration of expression of antioxidant genes superoxide dismutase 2 (SOD2), aldehyde dehydrogenase 4 (ALDH4), and glutathione peroxidase 1 (GPX1).43,45-51 These pro-oxidant genes result in an increase in ROS levels and promotion of p53-dependent cell death. For instance, SOD2, which plays a major role in a cell’s defense against oxidative damage, is repressed at the promoter by p53, and in contrast, overexpression of MnSOD (product of the SOD2 gene) results in a decrease in p53-mediated apoptosis and the potential to increase oxidative stress.45
So what determines if p53 will take the antioxidant or pro-oxidant path might depend on the strength or duration of the stress.3 The pro-oxidant functions of p53 might go into effect when the stress is mild and not persistent and thus allow the cell to repair any damage, allowing the cell to survive. The antioxidant functions, however, would be activated under extreme or prolonged stress to eliminate the cell either through cell death or senescence. In addition, cell type might also matter in p53’s decision to be pro- or antioxidant. In the brain, p53 is pro-oxidant but both in other cells.3 Last, ROS can also regulate p53, resulting in a feedback or feed-forward loop depending on whether p53 increases or decreases ROS levels.52
p53 Regulates NF-κB Signaling via Glycolysis
p53 and NF-κB have several differences in dealing with stress responses.53 In particular, NF-κB initiates a metabolic pathway that uses glucose at a high rate through aerobic glycolysis and the phosphate pentose shunt. p53, on the other hand, regulates genes that favor the use of mitochondrial oxidative phosphorylation, which generates more ATP per mole of glucose than glycolysis. Stress activates p53, which turns off mTOR and IGF-1/AKT pathways, stopping growth and preventing NF-κB from activating a response to allow for a metabolic state for cell division.
Kawauchi et al.54 showed that p53 can regulate the glycolytic pathway through IKK/NF-κB signaling (Figure 1). Specifically, p53 that is either basally expressed or activated via genotoxic stress inhibits the kinase activities of IKKα and IKKβ and the transcriptional activity of NF-κB. However, loss of p53 enhances NF-κB-dependent glucose metabolism, which is at least partly dependent on GLUT3. Therefore, the data suggest that a positive feedback loop exists so that glycolysis can drive activation, and when p53 is lost, the activation of this loop is important for oncogene-induced cell transformation. However, when both p53 and NF-κB are simultaneously activated under specific conditions in a cell, p53 appears to be dominant, repressing the synthesis of both Glut1 and Glut3 transporters, modulating down the high use of glucose and preventing the transformation of cells by NF-κB and other oncogenes.
Uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), which is used as a substrate for N- and O-linked glycosylation, is a modification similar to phosphorylation but achieves opposite effects in terms of function.55 Up to 5% of the glucose flux through the cells goes through the hexosamine biosynthetic pathway toward producing UDP-GlcNAc, by the enzyme O-GlcNAc transferase.56,57 Addition of O-linked N-acetylglucosamine (O-GlcNAc) to serine 149 of p53 results in an increase in stability, high life, and concentration in the cell.58 Furthermore, it was shown that IKKβ was constitutively modified at Ser 733 with O-linked β-N-acetylglucosamine (O-GlcNAc) in p53-deficient mouse embryonic fibroblasts (MEFs), which was induced by accelerated aerobic glycolysis by induction with high glucose.59 This novel mechanism allows for the increase of NF-κB activity through loss of p53, which then allows for positive feedback regulation from enhanced glucose metabolism via IKK. Similarly, it was shown that O-GlcNAcylation of the NF-κB p65 subunit at amino acid Thr-352 induced NF-κB translocation into the nucleus through inhibition of NF-κB and IκBα interaction and thus transcriptional activation of NF-κB under hyperglycemic conditions. These observations suggest that O-GlcNAc modifications that occur in the glycolytic pathway are powerful enhancers of NF-κB signaling. Therefore, glucose metabolism is a key component in the regulation of p53 and NF-κB signaling and, with O-GlcNAcylation modifications, can prove to be important not only for cellular homeostasis but also for carcinogenesis.
Conclusion
It is becoming evident that tumor suppressive and oncogenic pathways are altering cellular metabolism to further promote a tumor’s ability to grow and survive. These alterations allow for rapid ATP generation, increased biosynthesis of macromolecules, and maintenance of appropriate redox balance in a cell. It is becoming clearer that the p53 tumor suppressor is an important part of this metabolic shift as it can influence several aspects of metabolism through different mechanisms.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Preparation of this article was supported by funding from the New Jersey Commission on Cancer Research.
References
- 1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269-70 [PubMed] [Google Scholar]
- 2. Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis: the seventh hallmark of cancer. Cell Mol Life Sci. 2008;65(24):3981-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9(10):691-700 [DOI] [PubMed] [Google Scholar]
- 4. Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292(5516):504-7 [DOI] [PubMed] [Google Scholar]
- 5. Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230-3 [DOI] [PubMed] [Google Scholar]
- 6. Bensaad K, Vousden KH. p53: new roles in metabolism. Trends Cell Biol. 2007;17(6):286-91 [DOI] [PubMed] [Google Scholar]
- 7. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64(7):2627-33 [DOI] [PubMed] [Google Scholar]
- 8. Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107-20 [DOI] [PubMed] [Google Scholar]
- 9. Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309(5741):1732-5 [DOI] [PubMed] [Google Scholar]
- 10. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107(16):7455-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ide T, Brown-Endres L, Chu K, et al. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Mol Cell. 2009;36(3):379-92 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Kondoh H, Lleonart ME, Gil J, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65(1):177-85 [PubMed] [Google Scholar]
- 13. Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650-3 [DOI] [PubMed] [Google Scholar]
- 14. Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11(3):577-90 [DOI] [PubMed] [Google Scholar]
- 15. Suzuki S, Tanaka T, Poyurovsky MV, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107(16):7461-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao Y, Coloff JL, Ferguson EC, Jacobs SR, Cui K, Rathmell JC. Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J Biol Chem. 2008;283(52):36344-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma W, Sung HJ, Park JY, Matoba S, Hwang PM. A pivotal role for p53: balancing aerobic respiration and glycolysis. J Bioenerg Biomembr. 2007;39(3):243-6 [DOI] [PubMed] [Google Scholar]
- 18. Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells: the type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem. 1997;272(36):22776-80 [DOI] [PubMed] [Google Scholar]
- 19. Shakya A, Cooksey R, Cox JE, Wang V, McClain DA, Tantin D. Oct1 loss of function induces a coordinate metabolic shift that opposes tumorigenicity. Nat Cell Biol. 2009;11(3):320-7 [DOI] [PubMed] [Google Scholar]
- 20. Zheng L, Roeder RG, Luo Y. S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell. 2003;114(2):255-66 [DOI] [PubMed] [Google Scholar]
- 21. Coloff JL, Mason EF, Altman BJ, et al. AKT requires glucose metabolism to suppress puma expression and prevent apoptosis of leukemic T cells. J Biol Chem. 2011;286(7):5921-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue: IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953;13(1):27-9 [PubMed] [Google Scholar]
- 23. Fritz V, Fajas L. Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene. 2010;29(31):4369-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ide T, Chu K, Aaronson SA, Lee SW. GAMT joins the p53 network: branching into metabolism. Cell Cycle. 2010;9(9):1706-10 [DOI] [PubMed] [Google Scholar]
- 25. Wakil SJ. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry. 1989;28(11):4523-30 [DOI] [PubMed] [Google Scholar]
- 26. D’Erchia AM, Tullo A, Lefkimmiatis K, Saccone C, Sbisa E. The fatty acid synthase gene is a conserved p53 family target from worm to human. Cell Cycle. 2006;5(7):750-8 [DOI] [PubMed] [Google Scholar]
- 27. Buzzai M, Jones RG, Amaravadi RK, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67(14):6745-52 [DOI] [PubMed] [Google Scholar]
- 28. Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. 2006;281(49):37372-80 [DOI] [PubMed] [Google Scholar]
- 29. Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223(2):217-22 [DOI] [PubMed] [Google Scholar]
- 30. Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23(5):537-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11-20 [DOI] [PubMed] [Google Scholar]
- 32. Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134(3):451-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ellisen LW, Ramsayer KD, Johannessen CM, et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell. 2002;10(5):995-1005 [DOI] [PubMed] [Google Scholar]
- 34. Feng Z, Hu W, de Stanchina E, et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67(7):3043-53 [DOI] [PubMed] [Google Scholar]
- 35. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102(23):8204-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stambolic V, MacPherson D, Sas D, et al. Regulation of PTEN transcription by p53. Mol Cell. 2001;8(2):317-25 [DOI] [PubMed] [Google Scholar]
- 37. Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18(23):2893-904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010;20(7):427-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49(11):1603-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304(5670):596-600 [DOI] [PubMed] [Google Scholar]
- 41. Cano CE, Gommeaux J, Pietri S, et al. Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer Res. 2009;69(1):219-26 [DOI] [PubMed] [Google Scholar]
- 42. Tan M, Li S, Swaroop M, Guan K, Oberley LW, Sun Y. Transcriptional activation of the human glutathione peroxidase promoter by p53. J Biol Chem. 1999;274(17):12061-6 [DOI] [PubMed] [Google Scholar]
- 43. Yoon KA, Nakamura Y, Arakawa H. Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. J Hum Genet. 2004;49(3):134-40 [DOI] [PubMed] [Google Scholar]
- 44. Chen W, Sun Z, Wang XJ, et al. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34(6):663-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Drane P, Bravard A, Bouvard V, May E. Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis. Oncogene. 2001;20(4):430-9 [DOI] [PubMed] [Google Scholar]
- 46. Faraonio R, Vergara P, Di Marzo D, et al. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J Biol Chem. 2006;281(52):39776-84 [DOI] [PubMed] [Google Scholar]
- 47. Hussain SP, Amstad P, He P, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64(7):2350-6 [DOI] [PubMed] [Google Scholar]
- 48. Liu Z, Lu H, Shi H, et al. PUMA overexpression induces reactive oxygen species generation and proteasome-mediated stathmin degradation in colorectal cancer cells. Cancer Res. 2005;65(5):1647-54 [DOI] [PubMed] [Google Scholar]
- 49. Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389(6648):300-5 [DOI] [PubMed] [Google Scholar]
- 50. Rivera A, Maxwell SA. The p53-induced gene-6 (proline oxidase) mediates apoptosis through a calcineurin-dependent pathway. J Biol Chem. 2005;280(32):29346-54 [DOI] [PubMed] [Google Scholar]
- 51. Trinei M, Giorgio M, Cicalese A, et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress–induced apoptosis. Oncogene. 2002;21(24):3872-8 [DOI] [PubMed] [Google Scholar]
- 52. Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008;44(8):1529-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ak P, Levine AJ. p53 and NF-kappaB: different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010;24(10):3643-52 [DOI] [PubMed] [Google Scholar]
- 54. Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10(5):611-8 [DOI] [PubMed] [Google Scholar]
- 55. Wells L, Kreppel LK, Comer FI, Wadzinski BE, Hart GW. O-GlcNAc transferase is in a functional complex with protein phosphatase 1 catalytic subunits. J Biol Chem. 2004;279(37):38466-70 [DOI] [PubMed] [Google Scholar]
- 56. Haltiwanger RS, Holt GD, Hart GW. Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins: identification of a uridine diphospho-N-acetylglucosamine:peptide beta-N-acetylglucosaminyltransferase. J Biol Chem. 1990;265(5):2563-8 [PubMed] [Google Scholar]
- 57. Kornfeld S, Kornfeld R, Neufeld EF, O’Brien PJ. The feedback control of sugar nucleotide biosynthesis in liver. Proc Natl Acad Sci U S A. 1964;52:371-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang WH, Kim JE, Nam HW, et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol. 2006;8(10):1074-83 [DOI] [PubMed] [Google Scholar]
- 59. Kawauchi K, Araki K, Tobiume K, Tanaka N. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc Natl Acad Sci U S A. 2009;106(9):3431-6 [DOI] [PMC free article] [PubMed] [Google Scholar]