

Abstract
The autism spectrum disorder tuberous sclerosis complex (TSC) is caused by mutations in the Tsc1 or Tsc2 genes, whose protein products form a heterodimeric complex that negatively regulates mammalian target of rapamycin-dependent protein translation. Although several forms of synaptic plasticity, including metabotropic glutamate receptor (mGluR)-dependent long-term depression (LTD), depend on protein translation at the time of induction, it is unknown whether these forms of plasticity require signaling through the Tsc1/2 complex. To examine this possibility, we postnatally deleted Tsc1 in vivo in a subset of hippocampal CA1 neurons using viral delivery of Cre recombinase in mice. We found that hippocampal mGluR-LTD was abolished by loss of Tsc1, whereas a protein synthesis-independent form of NMDA receptor-dependent LTD was preserved. Additionally, AMPA and NMDA receptor-mediated EPSCs and miniature spontaneous EPSC frequency were enhanced in Tsc1 KO neurons. These changes in synaptic function occurred in the absence of alterations in spine density, morphology, or presynaptic release probability. Our findings indicate that signaling through Tsc1/2 is required for the expression of specific forms of hippocampal synaptic plasticity as well as the maintenance of normal excitatory synaptic strength. Furthermore, these data suggest that perturbations of synaptic signaling may contribute to the pathogenesis of TSC.
Introduction
Mutations in the tuberous sclerosis complex 1 or 2 genes (Tsc1 or Tsc2), which encode the tumor suppressor proteins hamartin and tuberin, respectively, cause the disease tuberous sclerosis complex (TSC). TSC is associated with benign tumors in multiple organs as well as neurological deficits, including varying degrees of cognitive dysfunction, epilepsy, and autism (Joinson et al., 2003; DiMario, 2004; Wiznitzer, 2004; Goorden et al., 2007). In many cell types, loss of Tsc1 or Tsc2 disrupts the Tsc1/2 complex and results in aberrant activation of the mammalian target of rapamycin complex 1 (mTORC1), upregulation of protein translation, and increased cell growth and proliferation (Tee et al., 2002; Chong-Kopera et al., 2006; Huang and Manning, 2008).
While the functions of Tsc1/2 in neurons are not well understood, they have been shown to regulate hippocampal-dependent learning and synapse function (Tavazoie et al., 2005; Goorden et al., 2007; Ehninger et al., 2008). In addition, alterations in synaptic plasticity, experience-dependent processes thought to be essential for learning and memory, may contribute to the neurological manifestations of TSC (von der Brelie et al., 2006; Ehninger et al., 2008). Several forms of synaptic plasticity require new protein synthesis and are prevented by the mTORC1 inhibitor rapamycin (Tang et al., 2002; Hou and Klann, 2004; Ronesi and Huber, 2008a; Sharma et al., 2010). Selective perturbation of these types of plasticity due to deregulation of translation has been proposed as a common pathogenic mechanism among autism spectrum disorders (ASDs) (Kelleher and Bear, 2008).
Long-term depression (LTD) induced by activation of group 1 metabotropic glutamate receptors (mGluRs) in the CA1 region of the hippocampus (or mGluR-LTD) requires protein translation in the postsynaptic cell and is mediated by internalization of glutamate receptors (Oliet et al., 1997; Huber et al., 2000, 2001; Hou and Klann, 2004; Ronesi and Huber, 2008a; Waung et al., 2008). This form of synaptic plasticity is deregulated in mouse models of fragile X syndrome (FXS), an X-linked form of mental retardation associated with a high prevalence of autism and caused by loss of the translational repressor fragile X mental retardation protein (FMRP, the protein product of the Fmr1 gene) (Jin and Warren, 2003). In Fmr1 knock-out mice, mGluR-LTD is enhanced and, interestingly, insensitive to blockade of protein synthesis (Huber et al., 2002; Nosyreva and Huber, 2006; Sharma et al., 2010). Since the Tsc1/2 complex also negative regulates protein translation, mGluR-LTD may be similarly enhanced in models of TSC (Kelleher and Bear, 2008).
To test this hypothesis, we deleted Tsc1 in a sparse subset of hippocampal CA1 neurons in vivo and examined the basal properties of glutamatergic synapses as well as two forms of synaptic depression. Relative to neighboring neurons in acute brain slices, cells lacking Tsc1 exhibited enhanced AMPA receptor (AMPAR)- and NMDA receptor (NMDAR)-mediated EPSCs without changes in dendritic spines or presynaptic function. Contrary to observations in Fmr1 KO mice, loss of Tsc1 abolished mGluR-LTD without affecting NMDA receptor-mediated LTD. Thus, Tsc1 is required for the expression of specific forms of protein translation-dependent synaptic plasticity, and the perturbations in synaptic function following loss of Tsc1 are distinct from those observed after loss of FMRP.
Materials and Methods
All animal handling was performed in accordance with guidelines approved by the Harvard Institutional Animal Care and Use Committee and federal guidelines.
Dissociated cultures and Western blotting.
Primary hippocampal cultures were prepared from P0–P1 Tsc1fl/fl mice of either sex using standard protocols. A total of 2 × 105 cells were plated onto 24 well plates precoated with poly-d-lysine. On day 2 in culture, lentivirus expressing either synapsin-driven GFP (6 × 108 IU/ml, control) or GFP-IRES-Cre (2.1 × 108 IU/ml, Tsc1 KO) was added at 2–4 IU/cell. At 12–14 DIV, cells were harvested in lysis buffer containing 2 mm EDTA, 2 mm EGTA, 1% Triton-X, 0.5% SDS in PBS with Halt phosphatase inhibitor (Thermo Scientific) and Complete mini EDTA-free protease inhibitor (Roche). Total protein was determined by BCA assay (Pierce), and 12.5 μg of protein were loaded onto Tris-HCl gels (Bio-Rad). Proteins were transferred to PVDF membranes, blocked in 5% milk in TBS-Tween for 1 h at room temperature (RT), and incubated with primary antibodies against Tsc1, Tsc2 (Bethyl Laboratories), β-Actin (Sigma), p-S6 (S240/244), total S6, p-p70S6K (T389), and total p70S6K (all from Cell Signaling Technology) overnight at 4°C. Blots were incubated with HRP-conjugated secondary antibodies (Bio-Rad) for 1 h at RT, washed, incubated with chemiluminescence substrate (PerkinElmer), and developed on Kodak Bio-Max film. Bands were quantified by densitometry using ImageJ software. Tsc1 and Tsc2 levels were normalized to a β-actin loading control. p-p70S6K and p-S6 were normalized to total p70S6K and S6 levels, respectively.
Immunohistochemistry.
Mice were perfused transcardially with 1× PBS and 4% paraformaldehyde. Brains were postfixed in 4% paraformaldehyde overnight and sectioned at 40 μm. Sections were blocked for 1 h at RT in 2% normal serum and incubated overnight at 4°C with an antibody against phosphorylated (Ser240/244) S6 ribosomal protein (Cell Signaling Technology). The following day, sections were washed and incubated for 1 h at RT with an Alexa Fluor 594-conjugated secondary antibody (Invitrogen). Sections were mounted onto slides using ProLong Gold antifade reagent with DAPI (Invitrogen). Images were taken on an Olympus IX71 inverted microscope with an Andor camera using the same exposure and acquisition settings for each section.
Stereotaxic virus injection.
P14–P16 Tsc1fl/fl mice of either sex were anesthetized with isofluorane and mounted on a stereotaxic frame (Stoetling) equipped with ear cups (Kopf). Unilateral injections into the CA1 region of the hippocampus were made anteroposterior −3.0 mm, medioventral −3.0 mm, and dorsoventral 2.2 mm relative to bregma. One microliter of an adenoassociated virus (AAV) encoding CreEGFP (1.2 × 1013 genome copy/ml, diluted 1:5 in PBS) was injected at a rate of 100 nl/min. The viral construct was provided by Matthew During, Ohio State University, Columbus, OH (Lu et al., 2009), and the virus was prepared by the Harvard Gene Therapy Initiative using standard techniques. Unless otherwise specified, mice were used for experiments 10–14 d following the virus injection.
Two-photon laser scanning microscopy and image analysis.
Acute slices were prepared from virus-injected Tsc1fl/fl mice as for electrophysiology (see below). CreEGFP-expressing (Tsc1 KO) and CreEGFP-negative (control) CA1 neurons were loaded with 40 μm Alexa Fluor 594 via a patch pipette, and two to five images per neuron were taken from secondary apical dendrites (512 × 512 pixels; 36 × 36 μm) using a custom-built 2-photon microscope and a Chameleon Ti-Sapphire laser tuned to 840 nm. Multiple slices at a separation of 1.0 μm were acquired to image the three-dimensional extent of the dendritic field. Six to 12 neurons were imaged per genotype. Images were analyzed by an observer who was blind to genotype using custom software written in Matlab (Mathworks) (Tavazoie et al., 2005). Spine lengths were measured from the junction with the dendritic shaft to the tip. Soma cross-sectional area was measured in the maximum intensity projection of a low-power image stack by quantifying the number of pixels within an outline drawn around the soma.
Electrophysiology.
Hippocampal slices from P24–P31 virus-injected Tsc1fl/fl or Tsc1w/w mice were cut (300 μm) in ice-cold external solution containing the following (in mm): 110 choline, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 7 MgCl2, 0.5 CaCl2, 25 glucose, 11.6 sodium ascorbate, and 3.1 sodium pyruvate. Slices were transferred to ACSF containing the following (in mm): 127 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 1 MgCl2, 2 CaCl2, and 25 glucose.
For miniature EPSC (mEPSC) recordings, the external solution was comprised of ASCF with (in μm) 1 TTX, 10 CPP, 50 picrotoxin, 10 mibefradil, 1 ω-conotoxin MVIIC, and 10 nimodipine to isolate AMPA receptor-mediated EPSCs and to improve the quality of voltage-clamp and space-clamp. Recording pipettes (∼3 MΩ) were filled with cesium-based internal solution containing the following (in mm): 120 CsMeSO3, 15 CsCl, 8 NaCl, 10 TEA-Cl, 10 HEPES, 2 QX 314 chloride, 4 Mg-ATP, 0.3 NaGTP, and 0.5 EGTA. Starting 3 min after break-in mEPSCs were recorded in voltage-clamp at −70 mV for 10 min with the amplifier Bessel filter set at 3 kHz. Traces were analyzed in Igor Pro using custom software to identify miniature events and accumulate distributions of mEPSC amplitudes and interevent intervals. Recordings with series resistance >20 MΩ or holding current above −200 pA were rejected.
To measure evoked responses, paired voltage-clamp recordings were obtained from neighboring CreEGFP-positive and CreEGFP-negative CA1 neurons in ACSF containing 50 μm picrotoxin. Schaffer collaterals were stimulated at 0.33 Hz to evoke primarily AMPAR-mediated (holding potential at −70 mV) or NMDA-mediated (holding potential at +40 mV) EPSCs. For this and all plasticity experiments, a cut was made between CA3 and CA1 to prevent recurrent excitation. Paired-pulse (PP) ratio was determined in voltage-clamp at −70 mV by delivering two stimuli at various interstimulus intervals (ISIs) (10–500 ms) and measuring the ratio of the peak amplitude of the second EPSC to the first EPSC, after subtracting the first EPSC from the second to remove any residual current.
For mGluR-LTD experiments, slices were allowed to recover 3–5 h before LTD induction. For the (RS)-3,5-dihydroxyphenylglycine (DHPG)-LTD experiment, voltage-clamp recordings at −70 mV in cesium-based internal solution were performed at RT in ACSF with 50 μm picrotoxin and 40 nm 2-chloroadenosine to reduce polysynaptic responses. LTD was induced by wash-in of 100 μm DHPG (Tocris Bioscience). Reconstituted DHPG solutions (50 mm) were kept at −80°C and used within 1 week of preparation. The PP-low-frequency synaptic stimulation (LFS)-LTD experiments were performed at 30°C in ACSF with 50 μm picrotoxin and 50 μm d-APV (Tocris Bioscience). Neurons were voltage-clamped at −60 mV in a potassium-based internal solution containing the following (in mm): 135 KMeSO4, 10 HEPES, 4 MgCl2, 4 NaATP, 0.4 NaGTP, 10 creatine phosphate, 125 EGTA, and 0.2 EDTA. LTD was induced by 900 paired-pulse (50 ms ISI) stimuli delivered at low frequency (1 Hz) (Huber et al., 2001).
For NMDA receptor (NMDAR)-dependent LTD, neurons were voltage-clamped at −70 mV in cesium-based internal solution and recordings were performed at 30°C in ACSF with 50 μm picrotoxin. NMDAR-dependent LTD was induced by 5 Hz stimulation for 3 min while cells were held at −40 mV (Morishita et al., 2005). For all plasticity experiments, series resistance was 10–30 MΩ, remained stable during the course of the experiment, and was not compensated. The peak EPSC amplitude was calculated using Igor Pro, and values were normalized to the 10 min baseline period for each recording. The percentage of LTD was calculated from the average of the responses 35–45 min (DHPG-LTD and PP-LFS-LTD) or 25–35 min (NMDAR-LTD) after induction of LTD and was expressed as a percentage of baseline.
Statistical analysis.
Two-tailed, paired or unpaired t tests were used for comparisons between two groups. In cases where the variance was found to be significantly different between two groups, a Welch's t test was used. To determine whether the late phase of plasticity was significantly different than baseline responses within an experiment, a one-sample t test was used.
Results
Sparse deletion of Tsc1 in CA1 neurons
We confirmed that conditional loss of Tsc1 resulted in upregulation of the mTOR pathway in hippocampal neurons. To do this, we generated dissociated hippocampal cultures from Tsc1fl/fl (Kwiatkowski et al., 2002) conditional knock-out mice and at 2 d in vitro added a high-efficiency lentivirus expressing Cre recombinase (GFP-IRES-Cre) under the synapsin promoter to delete Tsc1. A similar lentivirus encoding GFP alone was used to infect control cultures. After 12–14 d in vitro, we observed near complete loss of Tsc1 protein measured by Western blotting and a significant reduction in its binding partner Tsc2 (Chong-Kopera et al., 2006) (Tsc1, 5.2 ± 1.7% of control; Tsc2, 29.2 ± 2.4% of control; n = 12, p < 0.001) (Fig. 1A,B). Upregulation of mTOR signaling was evidenced by increased phosphorylation of the mTORC1 substrate p70S6 kinase (Thr389, 290 ± 60.6% of control; n = 10, p < 0.01) (Fig. 1A,B) and of its target ribosomal protein S6 (Ser240/244, 163.7 ± 12.8% of control, n = 16, p < 0.001) (Fig. 1A,B).
Figure 1.
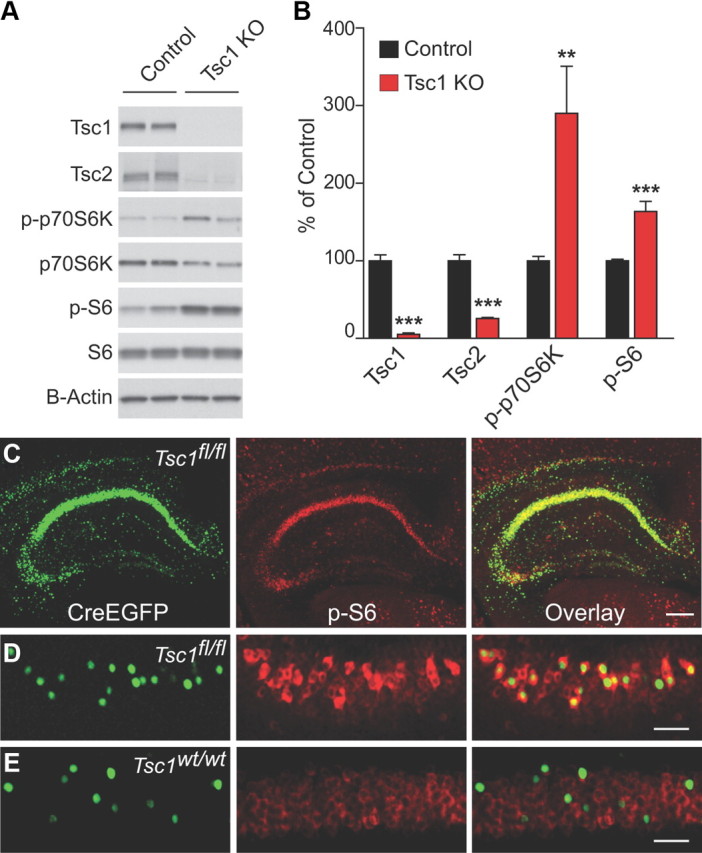
Conditional deletion of Tsc1 in hippocampal neurons. A, Representative Western blots from Tsc1fl/fl-dissociated hippocampal cultures. Control, treated with GFP lentivirus; Tsc1 KO, treated with GFP-IRES-Cre lentivirus. B, Quantification of Western blots for the indicated proteins expressed as a percentage of control. Bars represent mean ± SEM (n = 8–17; **p < 0.01; ***p < 0.001). C, Section from a Tsc1fl/fl mouse stereotaxically injected with AAV-CreEGFP demonstrating robust hippocampal expression. Green, Nuclear CreEGFP fluorescence; Red, immunohistochemistry staining for phosphorylated S6 (p-S6, Ser240/244). Right, Overlaid CreEGFP and anti-pS6 fluorescence. Scale bar, 250 μm. D, CreEGFP-expressing CA1 neurons from a sparsely infected Tsc1fl/fl mouse show upregulation of p-S6. Scale bar, 100 μm. E, CreEGFP-expressing CA1 neurons from a Tsc1w/w mouse do not show upregulation of p-S6.
To conditionally delete Tsc1 in vivo in a sparse subset of hippocampal neurons, we stereotaxically injected an AAV encoding a CreEGFP fusion protein (Lu et al., 2009) into the CA1 region of the hippocampus of P14–P16 Tsc1fl/fl mice and performed experiments 10–14 d later. Immunohistochemical analysis of hippocampal sections from virus-injected Tsc1fl/fl mice revealed upregulation of p-S6 (Ser240/244) in CreEGFP-expressing neurons (Tsc1 KO), consistent with functional upregulation of mTORC1 activity (Fig. 1C,D). CreEGFP-expressing neurons from a wild-type mouse did not show upregulation of p-S6 compared with neighboring cells, confirming the specificity of the genetic manipulation (Fig. 1E). In subsequent experiments, slices exhibiting sparse-to-moderate expression of CreEGFP in the CA1 region (Fig. 1D,E) (∼1–50% of CA1 cells) were used to investigate the cell-autonomous effects of loss of Tsc1 in an otherwise normal circuit.
Neuronal and spine morphology in Tsc1 KO neurons
To determine whether acute deletion of Tsc1 in vivo affected neuronal morphology, CA1 neurons from acute slices were filled through a whole-cell recording pipette with the red fluorescent dye Alexa Fluor 594 and imaged using 2-photon laser scanning microscopy (2PLSM). CreEGFP-positive Tsc1 KO neurons generally exhibited normal neuronal structure and dendrite branching (Fig. 2A); however, soma cross-sectional area was significantly increased compared with CreEGFP-negative control cells (control, 103.8 ± 5.1 μm2, n = 19; Tsc1 KO, 146.4 ± 10.4 μm2, n = 17; p < 0.01) (Fig. 2B,C). This finding is consistent with the somatic hypertrophy observed in many cell types due to loss of Tsc1/2 and may reflect overactivation of mTOR-regulated cell growth processes (Hay and Sonenberg, 2004; Tavazoie et al., 2005; Meikle et al., 2007; Huang and Manning, 2008).
Figure 2.
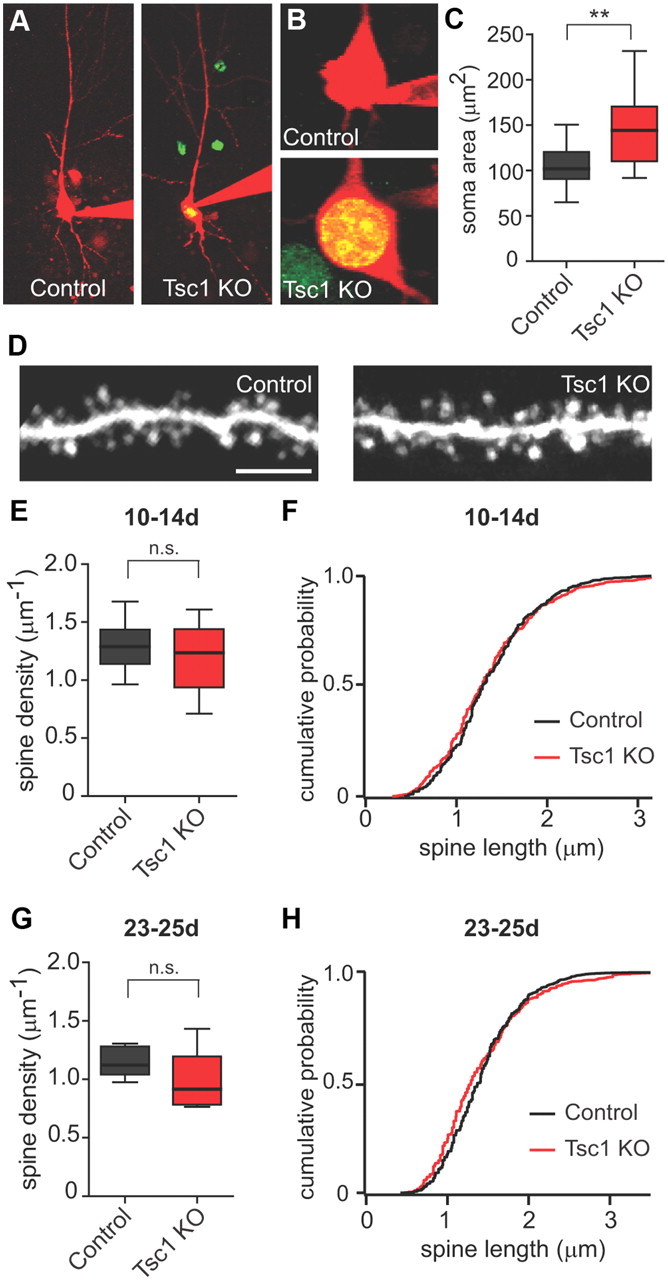
Neuronal and spine morphology of Tsc1 KO neurons in vivo. A, 2-photon images of Control (CreEGFP negative) and Tsc1 KO (CreEGFP positive) CA1 neurons 10–14 d following AAV-CreEGFP injection into a Tsc1fl/fl mouse. B, 2-photon images of somas from Control (top) and Tsc1 KO (bottom) neurons. C, Box-plot summary of soma area (in μm2) (n = 17–19; **p < 0.01). D, 2-photon images of apical dendritic spines from control and Tsc1 KO neurons. Scale bar, 5 μm. E, G, Box-plot summaries of spine density (spines per μm of dendrite) 10–14 d (E) or 23–25 d (G) post-injection (n = 11–12 neurons. n.s., Not significant. F, H, Cumulative distributions of spine length (in micrometers) from control (black traces) and Tsc1 KO (red traces) neurons 10–14 d (F) or 23–25 d (H) post-injection.
We previously observed changes in spine density and morphology in neurons from organotypic hippocampal cultures following prolonged loss of Tsc1 or Tsc2 (Tavazoie et al., 2005). To determine whether deletion of Tsc1 in vivo similarly affected spine density or structure, we filled control and Tsc1 KO cells with Alexa Fluor 594 and imaged spines on secondary apical dendrite branches using 2PLSM (Fig. 2D). We observed no significant changes in spine density (control, 1.29 ± 0.06 μm−1, n = 12 cells; Tsc1 KO, 1.18 ± 0.08 μm−1, n = 11 cells) (Fig. 2E) or spine length (control, 1.15 ± 0.03 μm, n = 12 cells; Tsc1 KO, 1.18 ± 0.03 μm, n = 11 cells) (Fig. 2F) between control and Tsc1 KO neurons 10–14 d after virus injection. We also assessed spine density and length 23–25 d post-virus injection, a time point at which significant changes in spine structure were observed in organotypic cultures, but did not detect any significant changes in spine number or morphology (control density 1.15 ± 0.05 μm−1, control length 1.47 ± 0.04 μm, n = 7 cells; Tsc1 KO density 0.989 ± 0.10 μm−1, Tsc1 KO length 1.41 ± 0.04 μm, n = 6 cells) (Fig. 2G,H).
Alterations in excitatory synaptic currents in Tsc1 KO neurons
To determine whether excitatory synapse number and strength were functionally altered by loss of Tsc1, we performed whole-cell voltage-clamp recordings of AMPA receptor-mediated mEPSCs. Viral delivery of CreEGFP did not by itself affect synapse function because mEPSC amplitude and frequency were similar between CreEGFP-expressing (WT-Cre+) and neighboring nonexpressing (WT-Cre−) CA1 neurons in wild-type mice [WT-Cre− amp 10.40 ± 0.47 pA; WT-Cre+ amp 9.7 ± 0.49 pA; WT-Cre− inter-event interval (IEI) 863 ± 139 ms; WT-Cre+ IEI 963 ± 111 ms; n = 6] (Fig. 3A–C). In paired recordings from CA1 neurons in virus-injected Tsc1fl/fl mice, mEPSC amplitude was not different between Tsc1 KO and neighboring control neurons (control 10.91 ± 0.69 pA; Tsc1 KO 11.09 ± 1.0 pA; n = 9) (Fig. 3D,E); however, there was a significant decrease in the IEI, indicative of increased mEPSC frequency in Tsc1 KO neurons (control 1173 ± 229 ms; Tsc1 KO 719 ± 145 ms; n = 9, p < 0.05) (Fig. 3D,F). In addition, Tsc1 KO neurons exhibited increased membrane capacitance (control 96.2 ± 7.7 pF, n = 14; Tsc1 KO 160.72 ± 17.1 pF, n = 11; p < 0.01; WT-Cre+ 84.2 ± 7.9 pF, n = 8) (Fig. 3H) and decreased input resistance (control 268.2 ± 27.35 MΩ, n = 14; Tsc1 KO 191.0 ± 14.57 MΩ, n = 11; p < 0.05; WT-Cre+ 307.4 ± 28.8 MΩ, n = 8) (Fig. 3I) compared with control neurons or to CreEGFP-positive neurons from WT mice.
Figure 3.
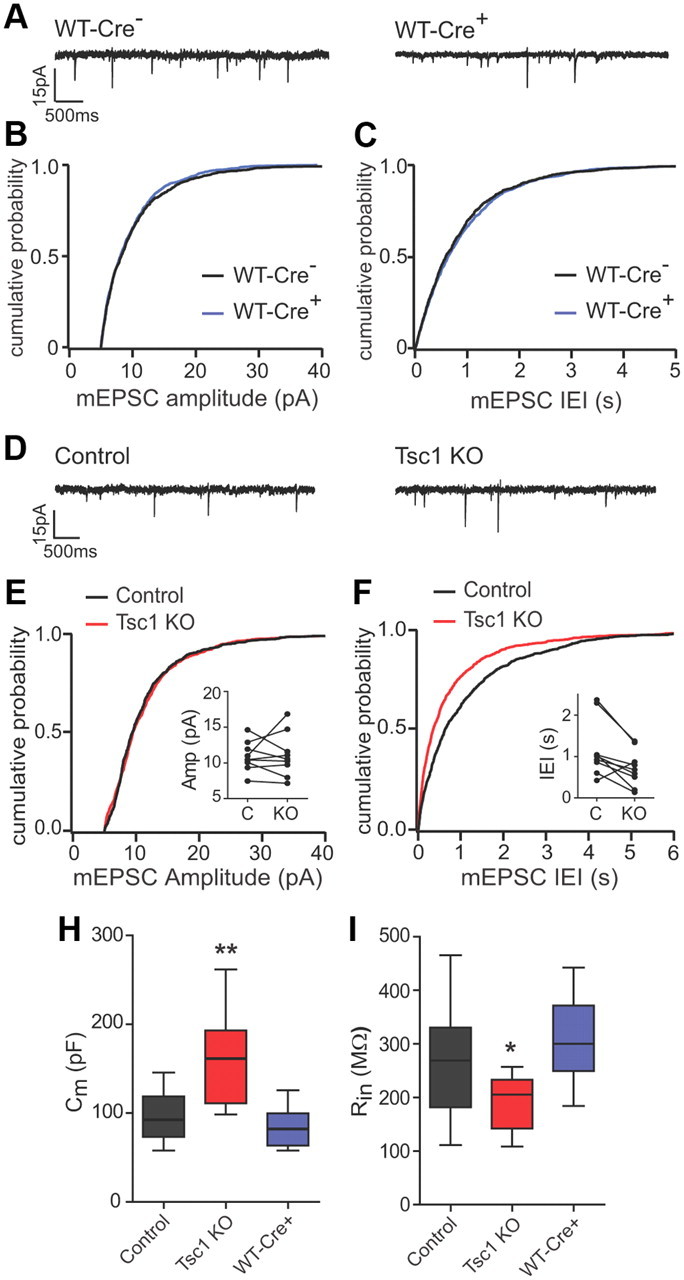
Basal synaptic function and intrinsic membrane properties in Tsc1 KO neurons. A, D, Representative traces of mEPSCs recorded from CreEGFP-negative (left) and CreEGFP-positive (right) CA1 neurons from virus-injected C57BL/6 (WT, A) or Tsc1fl/fl mice (D). B, C, Cumulative distributions of mEPSC amplitudes (pA) (B) and inter-event intervals (in seconds) (C) from CreEGFP-negative (black traces) and CreEGFP-positive (blue traces) neurons from C57BL/6 mice. E, F, Cumulative distributions of mEPSC amplitudes (pA) (E) and interevent intervals (in seconds) (F) from control (black traces) and Tsc1 KO (red traces) neurons from Tsc1fl/fl mice. Insets show pairwise comparisons of the averages for each cell. H, I, Box-plot summaries of membrane capacitance (H) and resting membrane resistance (I) from CA1 neurons of the indicated genotypes measured 10–14 d after AAV-CreEGFP injection. (n = 8–14; *p < 0.05; **p < 0.01). Control, CreEGFP-negative neurons from a Tsc1fl/fl mouse; Tsc1 KO, CreEGFP-positive neurons from a Tsc1fl/fl mouse, WT-Cre+; CreEGFP-positive neurons from a C57BL/6 mouse.
To investigate whether loss of Tsc1 altered evoked excitatory synaptic responses, we performed simultaneous paired recordings of neighboring CreEGFP-negative (control) and CreEGFP-positive (Tsc1 KO) CA1 neurons while stimulating Schaffer collaterals to elicit AMPAR and NMDAR EPSCs (Fig. 4A). We observed a significant increase in AMPAR EPSC amplitude in Tsc1 KO neurons recorded at −70 mV (control 173.0 ± 13.5 pA; Tsc1 KO 296.7 ± 33.6 pA, n = 16, p < 0.01) (Fig. 4B). Similarly, NMDAR-mediated currents in compound EPSCs measured at +40 mV were also strongly increased in Tsc1 KO cells (control: 114.3 ± 13.1 pA; Tsc1 KO: 174.3 ± 20.6 pA, n = 12, p < 0.01) (Fig. 4C). The ratio of AMPAR to NMDAR responses within each cell, however, was similar between control and Tsc1 KO neurons (control 1.55 ± 0.15; Tsc1 KO 1.60 ± 0.14, n = 12) (Fig. 4D), indicating a parallel increase in both AMPAR- and NMDAR-mediated synaptic currents.
Figure 4.

Evoked AMPA and NMDA receptor synaptic currents are increased in Tsc1 KO neurons. A, Differential interference contrast image overlaid with EGFP fluorescence. Example of a paired recording between neighboring CreEGFP-negative (control, left) and CreEGFP-positive (Tsc1 KO, right) neurons. EPSCs were induced by Schaffer collateral stimulation. B, C, Scatter plots of EPSC amplitude from pairs of control and Tsc1 KO CA1 neurons recorded at −70 mV (B, primarily AMPAR currents) and at +40 mV (C, primarily NMDAR currents). Insets show representative traces from the same pair of neurons. Control, Black traces; Tsc1 KO, red traces. D, Mean AMPA/NMDA ratio ± SEM from control and Tsc1 KO neurons (n = 13). E, Mean paired-pulse ratio ± SEM from control and Tsc1 KO neurons measured at different ISIs (n = 10–11). PPR >1 indicates facilitation. Representative overlaid traces from individual neurons showing pairs of EPSCs recorded at −70 mV for each ISI.
To determine whether the observed changes in excitatory synaptic transmission were due to alterations in presynaptic function, we examined paired-pulse facilitation, a form of short-term presynaptic plasticity measured by the ratio (PPR) of two stimuli delivered in rapid succession. PPRs measured at several interstimulus intervals were similar between control and Tsc1 KO neurons (10 ms: control 1.60 ± 0.07, Tsc1 KO 1.51 ± 0.06; 20 ms: control 1.64 ± 0.06, Tsc1 KO 1.60 ± 0.07; 50 ms: control 1.56 ± 0.06, Tsc1 KO 1.44 ± 0.06; 100 ms: control 1.33 ± 0.05, Tsc1 KO 1.28 ± 0.03; 200 ms: control 1.11 ± 0.03, Tsc1 KO 1.09 ± 0.03; 500 ms: control 1.03 ± 0.03, Tsc1 KO 1.02 ± 0.01; n = 9–11) (Fig. 4E), demonstrating normal basal presynaptic release probability. Together, these data indicate that loss of Tsc1 enhances postsynaptic glutamatergic function. Furthermore, since loss of Tsc1 had no effect on mEPSC amplitude but increased mEPSC frequency, it is likely that loss of Tsc1 increases the number of synapses formed onto the postsynaptic neuron.
mGluR-LTD is abolished in Tsc1 knock-out neurons
Based on the reported deregulation of the protein translation-dependent phase of mGluR-LTD observed in the ASD-related disorder FXS, we examined whether postsynaptic loss of Tsc1 affected the expression of mGluR-LTD. In response to a 5 min application of the group 1 mGluR agonist DHPG, both Tsc1 KO and control neurons exhibited a decrease in the magnitude of EPSCs evoked by Schaffer collateral stimulation (Fig. 5A). However, whereas control neurons displayed a long-lasting reduction in EPSC amplitude measured 35–45 min after DHPG (66.7 ± 2.7% of baseline; n = 9, p < 0.001) (Fig. 5A,E), responses from Tsc1 KO neurons returned to baseline levels within 20 min of DHPG application, indicating an absence of LTD (103.8 ± 8.2% of baseline; n = 7) (Fig. 5A,E). Series resistance (Rs) and membrane holding current (Im) remained stable over the course of the experiment (time period 1 vs time period 2) (Fig. 5A) for each genotype [Rs: control (1) 20.9 ± 1.8 MΩ, (2) 22.6 ± 2.2 MΩ; Tsc1 KO (1) 18.1 ± 1.3 MΩ, (2) 20.2 ± 2.8 MΩ; Im: control (1) −74.6 ± 13.70 pA, (2) −78.9 ± 21.4 pA; Tsc1 KO (1) −170.5 ± 33.7 pA, (2) −131.4 ± 17.1 pA]. Importantly, DHPG-induced LTD was normal in CreEGFP-expressing neurons from wild-type mice (WT-Cre+), confirming that loss of Tsc1 was responsible for this effect (59.6 ± 6.0% of baseline, n = 6, p < 0.01) (Fig. 5B,E).
Figure 5.
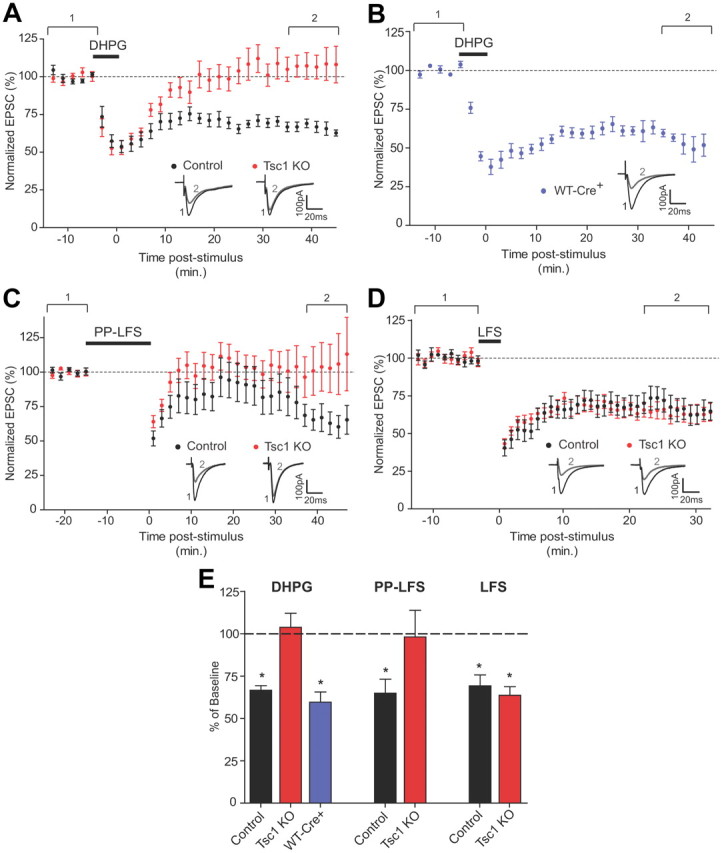
mGluR-LTD is selectively disrupted in Tsc1 KO neurons. A–D, Mean EPSC amplitude ± SEM expressed as a percentage of baseline plotted versus time in minutes. Dashed lines indicate the average baseline response. Insets show representative EPSCs. Black traces represent the average response from the 10 min baseline period (1). Gray traces represent the average response during the last 10 min (2). A, B, mGluR-LTD was induced by a 5 min wash-in of 100 μm DHPG. A, Tsc1fl/fl mice: control, CreEGFP-negative neurons (n = 9 cells); Tsc1 KO, CreEGFP-positive neurons (n = 8 cells). B, WT-Cre+, CreEGFP-positive neurons from C57BL/6 mice (n = 8). C, mGluR-LTD was induced by PP-LFS, consisting of 900 pairs of stimuli (50 ms ISI) delivered at 1 Hz in the presence of d-APV (n = 10). D, NMDA-dependent LTD was induced by LFS consisting of 5 Hz stimulation for 3 min at −40 mV (n = 8–9). E, Summary data showing the mean ± SEM response measured during the last 10 min of each recording expressed as a percentage of the 10 min baseline period for the indicated genotypes. Dashed line at 100% indicates the baseline. Left bars (DHPG) represent data from A and B, middle bars (PP-LFS) are from C, right bars (LFS) are from D. *Significant difference from 100%, p < 0.01.
In addition to acute application of DHPG, mGluR-dependent LTD can be elicited in extracellular field recordings by PP-LFS in the presence of an NMDAR antagonist (Huber et al., 2000, 2001). We determined whether PP-LFS could induce LTD in whole-cell recordings and found that the magnitude of depression elicited by this protocol in patch-clamp configuration was quite variable between cells. However, on average, control cells exhibited significant LTD 35–45 min poststimulus (64.9 ± 8.2% of baseline, n = 7, p < 0.01). In contrast, Tsc1 KO neurons did not exhibit LTD over this time period (98.1 ± 15.7% of baseline, n = 9) (Fig. 5C,E) further confirming the lack of mGluR-LTD in these cells. Passive cell properties remained stable over the course of the experiment for both genotypes [Rs: control (1) 16.6 ± 1.3 MΩ, (2) 17.3 ± 2.4 MΩ; Tsc1 KO (1) 15.5 ± 1.8 MΩ, (2) 13.4 ± 1.7 MΩ; Im: control (1) −52.7 ± 30.1 pA, (2) −38.4 ± 29.6 pA; Tsc1 KO (1) 7.9 ± 5.3 pA, (2) 11.7 ± 16.0 pA).
Tsc1 knock-out neurons exhibit normal NMDAR-LTD
Whereas mGluR-LTD requires new protein synthesis (Waung et al., 2008), hippocampal LTD dependent upon the activation of NMDA receptors (NMDAR-LTD) is independent of protein translation (Huber et al., 2000). NMDAR-LTD is mechanistically distinct from mGluR-LTD (Collingridge et al., 2010) and can be elicited by LFS, leading to low levels of NMDAR-mediated calcium influx (Oliet et al., 1997; Morishita et al., 2005). We examined NMDAR-LTD in Tsc1 KO neurons to test whether the defect in mGluR-LTD observed following loss of Tsc1 was generalizable to multiple forms of long-term depression. We found that both control and Tsc1 KO neurons showed stable and robust depression of EPSCs in response to LFS (control 69.2 ± 6.4% of baseline, n = 9, p < 0.01; Tsc1 KO 63.7 ± 5.0% of baseline, n = 9, p < 0.001) (Fig. 5D,E), and the magnitude of LTD measured 25–35 min postinduction was indistinguishable between control and Tsc1 KO neurons (p = 0.89). These results indicate that loss of Tsc1 specifically impairs mGluR-dependent forms of LTD in the hippocampus.
Discussion
In this study, we investigated how in vivo loss of the mTORC1 regulator Tsc1 in hippocampal CA1 neurons affected the structure and function of synapses. We found that glutamatergic synaptic function was enhanced in Tsc1 KO neurons, evidenced by larger evoked AMPAR and NMDAR currents and increased frequency of mEPSCs. Additionally, we found that the protein translation-dependent form of synaptic plasticity, mGluR-LTD, was absent in Tsc1 KO neurons, whereas NMDAR-LTD was unperturbed. These effects occurred in the absence of changes in dendritic spine number, morphology, or presynaptic release probability. Together, our results suggest that following loss of Tsc1 and deregulation of mTORC1 signaling, hippocampal neurons are unable to activate the relevant signaling pathways necessary for the stabilization of protein translation-dependent LTD. This may lead to enhanced excitatory drive that could ultimately have consequences for circuit information processing and network excitability.
Sparse deletion of Tsc1 in vivo
To investigate the effects of loss of Tsc1 in vivo, we used a single-cell genetic approach to selectively delete Tsc1 postsynaptically in CA1 neurons by stereotaxic injection of a virus expressing a CreEGFP fusion protein (Lu et al., 2009). Using this approach, we could precisely control the timing and location of Cre expression and investigate the cell-autonomous effects of deletion of Tsc1 in an otherwise unperturbed circuit. This system has advantages for the study of synapse function over other mouse models where Tsc1 is deleted in all excitatory forebrain neurons. These mice display severe macroencephaly and die shortly following loss of Tsc1 from seizures, complicating the analysis of synaptic function (Ehninger et al., 2008). Additionally, by deleting Tsc1 selectively in a subset of CA1 neurons, we could identify alterations in postsynaptic function resulting directly from perturbation of excitatory neurons, rather than secondary changes due to macroscopic disorganization or alterations in glia (Uhlmann et al., 2002; Way et al., 2009). Based on estimated time delays for viral infection, Cre expression, gene deletion, and protein loss (Nagy, 2000), we expect that Tsc1 was deleted for a few days to a week at the time of analysis. Therefore, with this model we could investigate the consequences of relatively acute, postsynaptic loss of Tsc1.
A previous study from our laboratory reported that sparse disruption of Tsc1 or Tsc2 in organotypic hippocampal slice cultures led to changes in CA1 pyramidal neuron soma size, spine morphology, spine density, and intrinsic membrane properties (Tavazoie et al., 2005). Similar to these results from organotypic cultures, we observed enlarged cell somas, increased cell capacitance, and decreased resting membrane resistance following loss of Tsc1 in vivo. However, Tsc1 KO neurons in acute slices did not exhibit significant changes in spine density or morphology. These differences are not likely due to the relatively short loss of Tsc1 in our model since even prolonged loss of Tsc1 did not result in significant changes in spine structure in vivo. It is likely, therefore, that the differing hippocampal activity levels in vivo and in slice cultures influences the appearance of these phenotypes. Indeed, CA1 neurons in organotypic cultures show more complex dendritic branching and significantly increased frequency of mEPSCs compared with acute slices without a parallel change in inhibition (De Simoni et al., 2003). Organotypic slices also display high glutamatergic connectivity contributing to increased network synchronization and activity, which is sufficiently high to basally trigger the expression of activity-dependent forms of synaptic plasticity (Zhu et al., 2000; Mohajerani and Cherubini, 2005). Therefore, it is possible that challenging virus-injected Tsc1fl/fl mice with an enriched environment or other activity-promoting stimulus in vivo may be required to uncover changes in spine morphology.
Increased glutamatergic function in Tsc1 KO neurons
We found a robust increase in the amplitude of both AMPAR- and NMDAR-mediated currents in Tsc1 KO neurons compared with paired neighboring control neurons, indicative of enhanced glutamatergic transmission. Deletion of Tsc1 was limited to postsynaptic CA1 neurons, and retrograde alterations in presynaptic release probability did not occur. We also observed a significant increase in the frequency of AMPAR-mediated miniature EPSC events in Tsc1 KO neurons; therefore, an increase in excitatory synaptic connections between CA3 and CA1 neurons may explain the enhanced evoked responses. This enhanced glutamatergic transmission could be a direct consequence of the loss of mGluR-dependent LTD discussed below or may arise through an independent pathway. Regardless, a change in excitatory synaptic transmission is likely to have important consequences for circuit function and network excitability. Patients with TSC exhibit a high prevalence of epilepsy, and increased excitatory circuit activity may contribute to the development of this pathological state (Tsai and Sahin, 2011). Interestingly, hyperexcitability of cortical circuits has also been reported in mouse models of the autism- and epilepsy-related disorder FXS (Gibson et al., 2008).
Disruption of mGluR-LTD following deletion of Tsc1
In the Fmr1 KO mouse, hippocampal mGluR-LTD is enhanced and does not require new protein synthesis (Huber et al., 2002; Nosyreva and Huber, 2006). A proposed explanation for these findings is that, in the absence of FMRP, protein translation is basally upregulated and a critical factor for the expression of mGluR-LTD is constitutively translated (Ronesi and Huber, 2008b). Based on the fact that, like FMRP, the Tsc1/2 complex negatively regulates protein translation, it has been proposed that loss of Tsc1/2 may also result in an enhancement of mGluR-LTD (Kelleher and Bear, 2008). Contrary to this hypothesis, hippocampal mGluR-LTD induced either with the mGluR agonist DHPG or synaptic stimulation was abolished in CA1 neurons following loss of Tsc1. Our results are similar to the disruption in mGluR-LTD observed with the mTORC1 inhibitor rapamycin (Hou and Klann, 2004), indicating that constitutive upregulation, as seen in Tsc1 KO neurons, or blockade of mTORC1 signaling are both disruptive to this process. Therefore, temporally precise activation of mTORC1 via the Tsc1/2 complex is necessary for mGluR-LTD. Furthermore, our results suggest that the recently reported steady-state upregulation of mTOR signaling in Fmr1 KO mice is not likely to account for the enhancement in mGluR-LTD in observed models of FXS (Sharma et al., 2010).
One possible explanation for the observed differences is that FMRP and Tsc1/2-mTORC1 may regulate the translation of distinct subsets of transcripts involved in mGluR-LTD. Indeed, mTOR regulates the synthesis of the general protein translation machinery itself (Costa-Mattioli et al., 2009), while FMRP is thought to regulate translation of a subset of dendritic mRNAs (De Rubeis and Bagni, 2010). Alternatively, FMRP can be phosphorylated by the mTORC1 target, p70S6 kinase (Narayanan et al., 2008), a modification associated with translational repression (Ceman et al., 2003; Narayanan et al., 2007). Since loss of Tsc1 results in overactivation of p70S6K, it is possible that hyperphosphorylation of FMRP in Tsc1 KO neurons could prevent mGluR-stimulated translation of effector proteins (Narayanan et al., 2008). It is also possible that loss of Tsc1 causes mTOR-independent alterations in neurons that could affect the actin cytoskeleton or receptor trafficking, resulting in disruption of mGluR-LTD (Lamb et al., 2000; Tavazoie et al., 2005; Huang et al., 2008).
Interestingly, we found that the deficit in mGluR-LTD did not extend to a form of hippocampal LTD that is independent of protein synthesis at the time scale investigated here (Oliet et al., 1997; Huber et al., 2000). This type of LTD requires activation of NMDA receptors that initiate protein phosphatase and kinase cascades that ultimately destabilize synaptic glutamate receptors leading to internalization and synaptic depression (Collingridge et al., 2010). We found that NMDAR-LTD was unaltered in Tsc1 KO neurons, indicating that the signaling pathways underlying this form of plasticity as well as the basic mechanisms involved in AMPA receptor endocytosis are unperturbed following loss of Tsc1.
Relevance for autism spectrum disorders
One motivation for this study was to relate abnormalities uncovered here to what has been reported for other models of ASDs to potentially identify synaptic phenotypes that could underlie autism. We find that loss of Tsc1 in the hippocampus results in a constellation of synaptic phenotypes that are largely distinct from those reported in models of FXS (Pfeiffer and Huber, 2009). Thus, our findings support the idea that synapses may be the locus for abnormalities underlying autism (Bourgeron, 2009), and perturbations in protein translation-dependent forms of synaptic plasticity may indeed be a common dysfunction among these diseases (Kelleher and Bear, 2008). However, disease-specific mechanisms likely determine how synapse structure and function are affected in each ASD.
Footnotes
This work was supported by NINDS Grant NS052707 to B.L.S. H.S.B. is supported by a Nancy Lurie Marks Postdoctoral Fellowship. We thank the members of the Sabatini laboratory for helpful discussions and constructive comments, Caroline Johnson for technical support, and Jessica Hauser for pilot experiments. The CreEGFP viral construct was kindly provided by Matthew During.
References
- Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. 2009;19:231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Ceman S, O'Donnell WT, Reed M, Patton S, Pohl J, Warren ST. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet. 2003;12:3295–3305. doi: 10.1093/hmg/ddg350. [DOI] [PubMed] [Google Scholar]
- Chong-Kopera H, Inoki K, Li Y, Zhu T, Garcia-Gonzalo FR, Rosa JL, Guan KL. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J Biol Chem. 2006;281:8313–8316. doi: 10.1074/jbc.C500451200. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, Bagni C. Fragile X mental retardation protein control of neuronal mRNA metabolism: insights into mRNA stability. Mol Cell Neurosci. 2010;43:43–50. doi: 10.1016/j.mcn.2009.09.013. [DOI] [PubMed] [Google Scholar]
- De Simoni A, Griesinger CB, Edwards FA. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J Physiol. 2003;550:135–147. doi: 10.1113/jphysiol.2003.039099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMario FJ., Jr Brain abnormalities in tuberous sclerosis complex. J Child Neurol. 2004;19:650–657. doi: 10.1177/08830738040190090401. [DOI] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2(+/-) mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. 2008;100:2615–2626. doi: 10.1152/jn.90752.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/- mice in the absence of cerebral lesions and seizures. Ann Neurol. 2007;62:648–655. doi: 10.1002/ana.21317. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28:4104–4115. doi: 10.1128/MCB.00289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Warren ST. New insights into fragile X syndrome: from molecules to neurobehaviors. Trends Biochem Sci. 2003;28:152–158. doi: 10.1016/S0968-0004(03)00033-1. [DOI] [PubMed] [Google Scholar]
- Joinson C, O'Callaghan FJ, Osborne JP, Martyn C, Harris T, Bolton PF. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med. 2003;33:335–344. doi: 10.1017/s0033291702007092. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, Onda H. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11:525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- Lamb RF, Roy C, Diefenbach TJ, Vinters HV, Johnson MW, Jay DG, Hall A. The TSC1 tumour suppressor hamartin regulates cell adhesion through ERM proteins and the GTPase Rho. Nat Cell Biol. 2000;2:281–287. doi: 10.1038/35010550. [DOI] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohajerani MH, Cherubini E. Spontaneous recurrent network activity in organotypic rat hippocampal slices. Eur J Neurosci. 2005;22:107–118. doi: 10.1111/j.1460-9568.2005.04198.x. [DOI] [PubMed] [Google Scholar]
- Morishita W, Marie H, Malenka RC. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci. 2005;8:1043–1050. doi: 10.1038/nn1506. [DOI] [PubMed] [Google Scholar]
- Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- Narayanan U, Nalavadi V, Nakamoto M, Pallas DC, Ceman S, Bassell GJ, Warren ST. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J Neurosci. 2007;27:14349–14357. doi: 10.1523/JNEUROSCI.2969-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U, Nalavadi V, Nakamoto M, Thomas G, Ceman S, Bassell GJ, Warren ST. S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J Biol Chem. 2008;283:18478–18482. doi: 10.1074/jbc.C800055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. The state of synapses in fragile X syndrome. Neuroscientist. 2009;15:549–567. doi: 10.1177/1073858409333075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J Neurosci. 2008a;28:543–547. doi: 10.1523/JNEUROSCI.5019-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Metabotropic glutamate receptors and fragile x mental retardation protein: partners in translational regulation at the synapse. Sci Signal. 2008b;1:pe6. doi: 10.1126/stke.15pe6. [DOI] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai P, Sahin M. Mechanisms of neurocognitive dysfunction and therapeutic considerations in tuberous sclerosis complex. Curr Opin Neurol. 2011;24:106–113. doi: 10.1097/WCO.0b013e32834451c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, Yamada K, Gutmann DH. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- von der Brelie C, Waltereit R, Zhang L, Beck H, Kirschstein T. Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur J Neurosci. 2006;23:686–692. doi: 10.1111/j.1460-9568.2006.04594.x. [DOI] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way SW, McKenna J, 3rd, Mietzsch U, Reith RM, Wu HC, Gambello MJ. Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum Mol Genet. 2009;18:1252–1265. doi: 10.1093/hmg/ddp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiznitzer M. Autism and tuberous sclerosis. J Child Neurol. 2004;19:675–679. doi: 10.1177/08830738040190090701. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]