

Abstract
The ability of heat shock proteins to (1) chaperone peptides, including antigenic peptides; (2) interact with antigen-presenting cells through a receptor; (3) stimulate antigen-presenting cells to secrete inflammatory cytokines; and (4) mediate maturation of dendritic cells, makes them a unique starting point for generation of immune responses. These properties also permit the use of heat shock proteins for development of a new generation of prophylactic and therapeutic vaccines against cancers and infectious diseases.
INTRODUCTION
Quae medicamenta non sanat, ferrum sanat.
Quae ferrum non sanat, ignis sanat.
Quae vero ignis non sanat,
insanabilia reportari oportet.
(That which drugs fail to cure, the scalpel can cure. That which the scalpel fails to cure, heat can cure. That which heat cannot cure, must be determined to be incurable.)—Hippocrates
THE THIN EDGE OF THE WEDGE: HEAT SHOCK PROTEINS ELICIT IMMUNITY TO CANCERS
Inbred mice immunized with irradiated syngeneic cancer cells were observed, as early as 1943, to be resistant to subsequent challenges with the cancer cells used for immunization (Srivastava and Old 1988). These experiments generated 2 tenets. First, cancers are immunogenic in syngeneic hosts. This was confirmed for cancers of a wide variety, including those induced by chemical carcinogens and UV radiation and spontaneous cancers as well. Second, immunity to cancer was specific for each individual cancer. Mice were protected from challenge only with the specific cancer that was used to immunize. Thus, the cancer antigens were not cancer specific or cancer type specific (eg, hepatoma specific) but were individually specific. Cross-reactive protection was observed in some studies, but, with notable exceptions (Donawho and Kripke 1990; Frey and Cestari 1995), such immunity was substantially weaker than the individually specific immunity (Coggin and Anderson 1974).
The 2 tenets led to a search for individually cancer-specific antigens, the philosopher's stones of cancer immunology. In early approaches, cancers and their normal counterparts were analyzed by high-resolution electrophoretic methods to identify cancer-specific bands or spots. Extensive studies with antibodies, first polyclonal and then monoclonal, scanned the surfaces of human and mouse cancer cells to define cancer-specific antigens (Old 1981). Although these studies were immensely productive in definition of the composition of the mammalian cell surface and helped identify a number of important molecules, no truly cancer-specific molecules were identified and characterized as a result. Identification of cancer-specific molecules recognized by T lymphocytes did not come into vogue until recently (Monach et al 1995; Boon and van der Bruggen 1996), because methods for doing so were previously unavailable. With few exceptions (Coulie et al 1995; Monach et al 1995; Robbins et al 1996; Ikeda et al 1997), these studies too have generally identified molecules shared between cancers and normal tissues.
Another approach to the problem was to look for the cancer-specific antigens by their ability to elicit protective immunity to cancer challenges, ie, by the very assay that pointed to their existence. This approach typically involved fractionation of cancer homogenates into various protein components by conventional chromatographic methods (see Srivastava et al 1998 for review). The fractions thus obtained were used to immunize animals, which were then challenged with live cancer cells. The fractions that elicited protection against the cancer were then refractionated and the cancer rejection assay repeated until apparently homogeneous preparations were obtained. This approach, with variations, led to identification of cancer-rejection molecules from cancers of diverse histological origins, induced in mice and rats of different haplotypes by chemicals or UV radiation, or of spontaneous origin. The cancers ranged in immunogenicity from the nonimmunogenic (eg, the Lewis lung carcinoma) to the highly immunogenic regressor cancers induced by UV radiation. Surprisingly, all the well-characterized molecules identified by this method turned out to be heat shock proteins (Hsps) of the Hsp90 or the Hsp70 family, including gp96, calreticulin (Basu and Srivastava 1999), Hsp70, and Hsp90. The first of the 2 tenets of cancer immunity, that cancers elicit protective immunity in syngeneic hosts, had thus achieved a molecular definition. Consistent with tenet 2, the Hsps purified from a given cancer were observed to elicit protective immunity specific to that particular cancer. Hsps derived from normal tissues did not elicit protective immunity to any cancers tested (Udono and Srivastava 1994). The observed specificity of immunogenicity of cancer-derived Hsps would suggest Hsps to display somatic polymorphism, such that Hsps would differ between cancers and normal tissues and from one cancer to another. However, extensive sequencing studies of Hsp complementary DNAs of cancers and normal tissues did not support this idea (Srivastava 1993). What is the origin of the specificity of immunogenicity?
SEARCH FOR SPECIFICITY: DISCOVERY OF Hsp-ASSOCIATED PEPTIDES
Because the Hsp preparations used to immunize were homogeneous by all criteria tested, the possibility was envisaged that low-molecular-weight substances, not detectable by polyacrylamide gel electrophoresis, are associated with Hsps and are responsible for the specificity of immunogenicity of Hsp preparations (Srivastava and Heike 1991; Srivastava and Maki 1991). This idea gained some credence when a seemingly large collection of peptides was shown to be associated with apparently homogeneous gp96 preparations (Li and Srivastava 1993). Formal proof for the idea came when depletion of peptides from tumor-protective Hsp70 preparations rendered them ineffective in immunizing against cancer cells (Udono and Srivastava 1993).
Recent studies from a number of laboratories have begun to provide structural support for peptide-binding activity of certain Hsps. Recently, Zhu et al (1996) have shown the presence of a definite peptide-binding pocket in a bacterial Hsp70 molecule. The peptide-binding activity of gp96 has also been demonstrated independently (Wearsch and Nicchitta 1997), and although the mechanism and the site of peptide binding by gp96 are yet unknown, studies with fluorescent probes point to the presence of a possible hydrophobic peptide-binding pocket (Wearsch et al 1998; Sastry and Linderoth 1999; Linderoth et al 2000). The cytosolic homologue of gp96, Hsp90, is also being studied extensively in this regard. Studies by Scheibel et al (1998) suggest the existence of 2 substrate-binding sites in Hsp90, whereas ongoing crystal structure studies with Hsp90 are in the process of clarifying the identity and structure of the peptide-binding pocket of Hsp90 (Prodromou et al 1997a; Prodromou et al 1997b; Stebbins et al 1997).
The idea of Hsps as chaperones of antigenic peptides has also been strengthened by a series of independent immunological observations. Arnold et al (1995, 1997) showed that immunization with gp96 preparations isolated from cells transfected with the gene encoding β-galactosidase elicited cytotoxic T lymphocytes (CTLs) specific for an Ld-restricted epitope of β-galactosidase; similarly, immunization with gp96 preparations purified from cells expressing certain minor histocompatibility complex (MHC) antigens elicited CTL responses against the particular minor antigens. Nieland et al (1996) first identified a known viral epitope found associated with gp96 purified from virus-infected cells; such peptides could not be detected in gp96 preparations from uninfected cells. Blachere et al (1997) could reconstitute gp96-peptide and Hsp70-peptide complexes in vitro and demonstrate with a large panel of peptides that, although Hsps and peptides were nonimmunogenic by themselves, peptides chaperoned by Hsps elicited antigen-specific CD8+ CTLs. The quantity of antigenic peptides associated with an immunogenic dose of gp96-peptide complexes was found to be extremely small (∼1–2 ng). Significantly, immunogenicity did not result when peptides were complexed with another peptide-binding protein, mouse serum albumin, suggesting that the ability to elicit CTLs is unique to Hsp-chaperoned peptides. Roman and Moreno (1996) showed that peptides complexed with a xenogeneic (mycobacterial) Hsp70 could also elicit peptide-specific T cells in mice. Recently, Houghton and colleagues (Moroi et al 2000) have used a variation of the approach where Hsp70 is complexed with peptides designed to have a high affinity for Hsp70, and such complexes are shown to immunize and elicit peptide-specific CD8+ T-cell responses.
Hsp-PEPTIDE COMPLEXES FOR IMMUNOTHERAPY OF CANCERS: THEORETICAL CONSIDERATIONS AND RESULTS OF THE FIRST FEW HUMAN CLINICAL TRIALS
The observation that the immunogenicity of Hsp preparations from a large variety of cancers actually originated from bound peptides returned the spotlight to the identity of the cancer-specific antigens. Which of the Hsp-bound peptides are unique to cancer and therefore immunogenic? The chromatographic profiles of peptides eluted from Hsp preparations (as well as MHC class I preparations) of cancers and normal tissues are very much alike, and it is impossible to identify the cancer-specific peaks in an overwhelming background of common peptides. The problem is compounded by the individual antigenic specificity of cancers, which would indicate that the antigen(s) identified for a given cancer must be different from those for another cancer. This question, viewed in the context of the limited size of the genome, is reminiscent of the early debates regarding the origin of diversity of immunoglobulins and may have a fundamentally similar solution (Srivastava 1993). In this view, (1) the immunogenicity of cancers results not from one or a few cancer-specific peptides but from a large and complex array of them; (2) cancer-specific antigens are generated by random mutations that accumulate in cancers because of continuous cell division and the genomic instability of cancers (Cahill et al 1998); the mutated peptides become antigenic by virtue of their presentation by the MHC alleles of the tumor; (3) the randomness of the mutational process leads to an individually specific “antigenic fingerprint” for each cancer, accounting for the individually specific immunogenicity of nearly all cancers tested; and finally, (4) the mutational repertoire that becomes immunogenic is incidental to the transformation process; thus, the transforming mutations such as those in ras, p53, and other such molecules do not contribute significantly to the immunogenicity of cancers, which results from other random, unrelated mutations.
Although this hypothesis explains the individually distinct antigenicity of cancers and of the Hsp-peptide complexes derived from them, it is apparently inconsistent with recent studies in which a number of CTL epitopes on human melanomas have been identified and found to be common to melanomas and normal tissues (Boon and van der Bruggen 1996). A close look reveals that the inconsistency results not from species-specific differences but from the assays used to identify the antigens: observations on murine cancers are based on cancer rejection assays in vivo, whereas studies with human melanomas are, of necessity, based on CTL assays in vitro, which do not distinguish between an active response in vivo and an inactive response in vivo that is expandable in vitro because of lack of regulatory constraints (see Srivastava 1996 for full discussion). Indeed, when murine cancer antigens are examined by CTL assays in vitro, common CTL epitopes are readily detected (Rohrer et al 1994) as in human melanomas (Boon and van der Bruggen 1996). However, there is little evidence thus far that the known unmutated CTL epitopes of human cancers are immunoprotective. Studies with unmutated CTL epitopes of murine cancers show that although such epitopes elicit CD8+ CTL responses as measured in vitro, immunization with their peptides does not lead to protection against a tumor challenge in vivo (Ramarathinan et al 1994; Huang et al 1996).
Mutated CTL epitopes have also been identified on human cancers (see Boon and van der Bruggen 1966 for review; Coulie et al 1995; Wolfe et al 1995; Robbins et al 1996). Based on experience with murine cancers (Monach et al 1995; Ikeda et al 1997), the mutated human cancer epitopes would be expected to elicit protective immunity to human cancers. However, such epitopes are expressed specifically on single individual cancers or a small number of cancers and are, therefore, inappropriate for general use, and identification of mutated epitopes on individual human cancers is daunting and impractical. The use of cancer-derived Hsp-peptide complexes, which, in principle, elicit immunity to the entire array of mutated epitopes of the cancers from which they are isolated, circumvents this dilemma, because such complexes can be readily isolated from cancers without need of prior identification of the epitopes from individual cancers and have been shown to effectively cure preexisting micrometastatic disease (Tamura et al 1997; Nicchitta 1998). Clearly, appropriately controlled clinical trials with defined unmutated CTL epitopes common to cancers and with Hsp-chaperoned, naturally processed epitopes, specific for individual cancers, will resolve this issue in the near future. Although no such trials have been initiated thus far, a number of preliminary clinical trials that begin to test the use of Hsps derived from autologous human cancers in immunotherapy of human cancers have now begun and some have been completed. Of these, 4 trials are relatively more mature and are discussed herein.
Janetzki et al (2000) prepared individual gp96 vaccines derived from the tumors of 16 patients with advanced malignancies. Each patient received a 25-μg subcutaneous injection of tumor-derived gp96, once weekly for 4 weeks. Blood samples recovered from patients revealed a CD8+-restricted response against autologous tumor in 6 of 12 patients where such responses could be tested. Postvaccination stabilization of disease for 3 to 7 months was observed in 4 patients. One patient with hepatocellular carcinoma was observed to have necrosis of more than 50% of her tumor, coincident with vaccination. Interestingly, this patient had synchronous liver metastases of a different primary tumor that did not respond to the vaccine prepared from her primary liver tumor. No evidence of autoimmune reactions or of other significant toxic effects associated with Hsp vaccine administration was observed in any patient in the study.
Amato et al (1999) have treated patients with renal cell carcinoma with gp96 vaccines. Patients received 1 of 3 doses of autologous tumor gp96 vaccines: 2.5, 25, or 100 μg. The vaccine was administered once a week for 4 weeks, with a follow-up dose at 12 weeks or 20 weeks if the patient's tumor showed stabilization or regression, respectively. Among the 16 patients who received the 25-μg dose, 1 patient had a complete response, 3 patients had partial responses, and 3 additional patients showed prolonged stabilization of disease (52 or more weeks). In a follow-up trial with a substantially larger number of renal carcinoma patients, similar results were obtained (Amato et al 2000).
Eton et al (2000) have used gp96 vaccines in metastatic melanoma using the same dosing regimen as described above for renal cancers. Among the patients with stage IV disease who were rendered apparently disease free by surgery and who were vaccinated with autologous tumor-derived gp96, more than 75% are alive and disease free 17 months after surgery. The median survival in historical controls of a similar patient population is approximately 12 months. However, reliance on historical controls is fraught with obvious risks, and randomized clinical trials are needed to test the efficacy of gp96 vaccination.
All of the clinical trials described above are, of necessity, nonrandomized, and the clinical data generated thus far can only be suggestive if that. To the extent that one may draw any inferences from them, the results thus far are consistent with the murine data that showed that the autologous gp96 vaccine was highly effective in the adjuvant setting, where most treated animals remained disease free for their entire lives, and less effective in animals with progressive tumors, where only disease stabilization was achieved (Tamura et al 1997; Srivastava 2000). A randomized multicenter phase III trial in patients with renal cancer in the adjuvant setting has now been initiated, and other randomized phase III trials are being planned.
Hsp-PEPTIDE COMPLEXES FOR THERAPY OF INFECTIOUS DISEASES: Hsps AS AGENTS OF CROSS-PRIMING AND AS ADJUVANTS
It was suggested in 1991 (Srivastava and Maki 1991) that if Hsps chaperone a substantial proportion of the antigenic repertoire, immunization with Hsp-peptide complexes derived from virus-transformed or infected cells should elicit viral immunity. Subsequently, gp96 preparations isolated from SV40-transformed cells were shown to elicit specific, MHC I–restricted CTL response (Blachere et al 1993). Similar results were obtained in the influenza system by Heikema et al (1997), with gp96 purified from influenza-infected cells. Recently, immunization of mice with gp96 or Hsp70 molecules reconstituted with specific CTL epitopes derived from SV40, influenza virus, vesicular stomatitis virus (VSV), and lymphocytic choriomeningitis virus (LCMV) has been shown to elicit cognate virus-specific CTL responses (Suto and Srivastava 1995; Blachere et al 1997; Ciupitu et al 1998). In the case of LCMV, Ciupitu et al (1998) showed that mice immunized with Hsp70 molecules mixed with a known MHC class I binding epitope were also protected against subsequent LCMV infection.
Hsps are nonpolymorphic; that is, individual hsp genes do not have more than one allele. It was, therefore, reasoned that Hsp preparations isolated from cells infected with a given virus should contain the same repertoire of peptides regardless of the MHC haplotype of the cells. Hence, Hsp preparations isolated from virus-infected cells of one haplotype should be able to prime antiviral response in mice of another haplotype (Srivastava et al 1994), a phenomenon known as cross-priming (Bevan 1976). This was shown in the case of VSV, since gp96 preparations from VSV-infected cells of the b or the d haplotype were equally effective in eliciting b haplotype–specific, antigen-specific anti-VSV CTLs (Suto and Srivastava 1995). At the same time, Arnold et al (1995) demonstrated the ability of gp96 preparations to cross-prime CTL responses in the case of minor histocompatibility complex antigens. The ability of Hsp-peptide complexes to cross-prime is significant from the vantage point of their potential use as vaccines against intracellular infections, where CD8+ responses have a protective value.
In addition to the studies with syngeneic (ie, mouse) Hsps, a number of interesting observations have been made with the mycobacterial Hsp-peptide complexes. Lussow et al (1991) and Barrios et al (1992) first observed that covalent complexes of mycobacterial Hsp65 or Hsp70 and peptides could be used to elicit potent and specific antipeptide antibodies, without the use of additional adjuvants. The responses were shown to be T-cell dependent. Vaccination with noncovalent mycobacterial Hsp70-peptide complexes has also been shown to elicit antipeptide CD4+ T cells (Roman and Moreno 1996); CD8+ responses were not tested in this study. In an interesting variation on this general theme, CTL responses and T-cell–dependent protective immunity have been elicited recently by immunization of mice with a mycobacterial Hsp70-ovalbumin fusion protein (Suzue et al 1997). Several new facts, as well as questions, emerge from these studies. Clearly, these studies show that immunization with Hsp-peptide complexes can elicit not only CTL but also antibody responses. Whether this is a unique property of covalent Hsp-peptide complexes is unclear, since the ability of noncovalent Hsp-peptide complexes to elicit such antipeptide antibodies has not been tested. It further remains to be determined if covalent complexes of syngeneic Hsps with peptides will be effective in eliciting antipeptide antibodies in mice or if the xenogeneicity of the mycobacterial Hsps makes them particularly able to do so. The fact that fusion proteins incorporating mycobacterial Hsps can elicit CD8+ T-cell responses in mice suggests that in addition to syngeneic Hsps, xenogeneic Hsps can also elicit a CD8+ immune response to the associated peptides.
The immunogenicity of Hsp-peptide complexes has a number of significant implications for vaccination against intracellular infections and treatment of preexisting infections, including viruses, mycobacteria, and certain parasites. Such vaccination does not require identification of immunogenic epitopes or attenuation of the agents normally required to create vaccines. Because immunization with Hsp-peptide complexes derived from infected cells is directed against the entire antigenic repertoire, it drastically reduces if not eliminates the possibility of development of escape variants. Should vaccination against a new variant be necessary, as in the case of highly mutable infectious agents, creation of a vaccine against the new variant depends only on its isolation and does not require its characterization. Furthermore, since cancer-derived Hsp-peptide complexes have been used to elicit regression, even cure of preexisting cancers, a reasonable case can be made that Hsp-peptide complexes from infected cells may be used not only for prophylaxis against infections but also for therapy of chronic infections.
The adjuvanticity of mammalian Hsps is unique in many respects (Blachere et al 1997). They are the first and thus far the only adjuvants of mammalian origin. Being self-antigens, they do not elicit an immune response to themselves. They are nonlive, and yet they elicit CD8+ T-cell responses, in addition to antibody responses, and finally, their apparent promiscuity in peptide binding makes them applicable to a wide variety of antigens. These properties are particularly interesting in light of the paucity of safe and effective adjuvants.
MECHANISMS OF IMMUNOGENICITY OF Hsp-PEPTIDE COMPLEXES: Hsps AS THE “SWISS ARMY KNIFE OF THE IMMUNE SYSTEM”
The first clue into the mechanism through which immunization with Hsp-peptide complexes elicits antigen-specific CD8+ T cells came from depletion studies that showed that priming of immune response by Hsp-peptide complexes was exquisitely sensitive to abrogation of function of phagocytic cells but did not require CD4+ T cells (Udono et al 1994). This observation, coupled with the finding that that extremely small quantities of Hsp-peptide complexes were effective in eliciting specific immunity, led to the suggestion that professional antigen-presenting cells (APCs) possess Hsp receptors, which take up Hsp-peptide complexes with specificity (Srivastava et al 1994). Subsequently, it was shown that macrophage, but not B cells, fibroblasts, or other nonprofessional APCs, take up gp96-peptide complexes and re-present the gp96-chaperoned peptides on the MHC I molecules of the macrophages; re-presentation does not occur by transfer of peptides from the gp96 molecules to MHC I on the cell surface (Suto and Srivastava 1995; Fig 1, top panel). The re-presentation involves trafficking of the peptides through a nonacidic compartment and is sensitive to brefeldin A. The evidence for an Hsp receptor has come from several independent sources (Binder et al 1998; Arnold-Schild et al 1999; Wassenberg et al 1999; Binder et al 2000a) and a gp96 receptor, CD91, has recently been identified in our laboratory (Binder et al 2000b). Further details of the mechanism, including the identity of the macrophage compartment that takes up the Hsp-peptide complexes, the mechanism by which the peptides arrive at the endoplasmic reticulum, the proteasome dependence and transporter associated with antigen dependence of processing and transport, and the fate of the gp96 itself, are still unclear. The mechanism of immunogenicity of other Hsp preparations, the Hsp90- and Hsp70-peptide complexes, also remain to be clarified, although interesting facets of these mechanisms, which are essentially consistent with the observations on the immunogenicity of gp96-peptide complexes, are gradually emerging (Castellino et al 2000; Singh-Jasuja et al 2000). The role of Hsps in mediating antigen-specific T-cell immune responses is depicted in Figure 1 (top panel).
Fig 1.
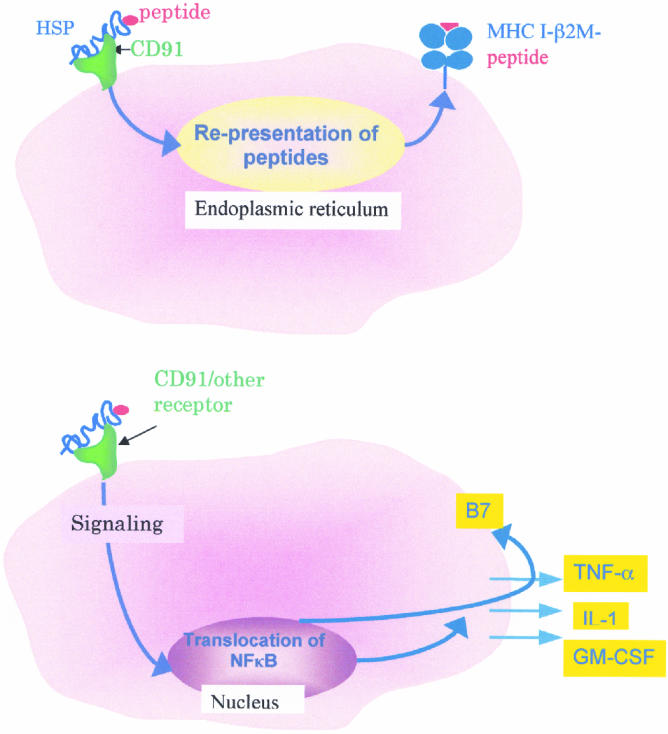
Two consequences of Hsp-APC interaction. The top panel shows the uptake of Hsp-peptide complex through the CD91 gp96 receptor, followed by re-presentation of Hsp-chaperoned peptides by the MHC I molecules of the APC. The MHC I–peptide complexes stimulate the cognate CD8+ T lymphocytes. This scheme is supported by a number of studies (Suto and Srivastava 1995; Binder et al 2000b). The bottom panel shows that interaction of Hsps with APCs (macrophages or dendritic cells) leads to translocation of NFκB into the nucleus, followed by secretion of a number of inflammatory cytokines and expression of antigen-presenting and costimulatory molecules. This scheme of the role of Hsps in innate immune response is supported by a number of recent studies (Basu et al 2000; Asea et al 2000)
During the analysis of re-presentation of Hsp-peptide complexes by APCs, another facet of the Hsp-APC interaction came to light (Fig 1, bottom panel). Exposure of APCs to preparations of gp96, Hsp90, or Hsp70 was observed to stimulate macrophages and dendritic cells to secrete cytokines (Basu et al 1998) and to express antigen-presenting and costimulatory molecules, regardless of the peptides associated with them (Basu et al 2000; Fig 1, bottom panel). Interestingly, the interaction of gp96 with the APCs was found to be associated with translocation of NFκB into the nucleus of the APC, one of the most highly conserved signal transduction pathways of the immune system. Asea et al (2000) have also recently observed a subset of these properties with respect to Hsp70 molecules. Macrophages and dendritic cells are key components of innate and adaptive immune responses. The identity of endogenous signals that activate APCs has been a crucial question in immunology. The observation that Hsps, the most abundant and conserved mammalian molecules, constitute such an internal signal places Hsps at a crucial intersection of adaptive and innate immune responses. The role of Hsps in innate immunity is also consistent with our previous observation that immunization with cancer-derived Hsp-peptide complexes elicits, in addition to the CD8+ and CD4+ T-cell response, an NK response that is crucial for eliciting protective immunity (Tamura et al 1997). Presumably, the interleukin 12 secreted by the Hsp-APC interaction is responsible for the expansion of the NK population, as also seen in a human clinical trial where patients received gp96 (Janetzki et al 2000).
The ability of Hsps to elicit such a diverse array of immune responses lead Rammensee and Schild to label Hsps the Swiss Army knife of the immune system (Rammensee and Schild, 2000); the title of this section is derived from that nomination.
Hsps, NECROTIC CELL DEATH, Hsp RECEPTORS, AND ACTIVATION OF APCs: THE PRIMORDIAL IMMUNOLOGICAL PROGRAM?
Contemplation of these ideas leads to some interesting insights. Hsps are among the most primitive proteins in living systems, and macrophage-like cells of one kind or another are present in the earliest multicellular living systems (Beck and Habicht 1996). It is conceivable that in a less polymorphic era, when adaptive immune response was but a distant gleam in the evolutionary eye, the interaction of Hsps with macrophage-like cells, leading to stimulation of the macrophage-like cells to secrete interleukin 1 and other messengers, was the primary ‘innate’ defense mechanism. Because Hsps are the most abundant soluble molecules in cells, they can be reasonably expected to be the most reliable messengers of cell death or, at any rate, cell lysis, which would result if the organism were in stress or danger, eg, being consumed by a predator. This chain of events would also explain the otherwise-strange presence of a surface receptor for Hsps, which are normally intracellular. In this view, the Hsps would have been transformed from being protectors of cells from cellular stress in the single-celled organisms to being messengers of stress and protectors against such stress in the first multicellular organisms or at least among those in which a differentiated macrophage-like cell had evolved. By all accounts, this happened quite early, since phagocytes of echinoderms, mollusks, annelids, and tunicates had already acquired the ability to secrete interleukin 1–like molecules (Ottaviani and Franceschi 1997). From this initial point, it is relatively straightforward to see how the Hsp-APC interaction was incorporated into future versions of the developing defense mechanisms, until, with the arrival of specific immunity and its paraphernalia of specificity of peptide-binding and T-cell receptors, the primal peptide-binding proteins, ie, the Hsps, would become accessories in antigen presentation by MHC molecules. The evolutionary implications of a convergence of the peptide-binding functions of the Hsps and the MHC molecules has been discussed in detail elsewhere (Srivastava and Heike 1991).
In the proposed role of Hsps as harbingers of cell death and, therefore, of danger to an organism (Matzinger 1994), a kind of death that results in an encounter of Hsps with the immune system will be more productive, ie, communicative of danger, and protective from it than a ‘silent death,’ which does not lead to Hsp-APC interaction. We have recently reported that necrotic but not apoptotic cell death leads to release of Hsps (Basu et al, 2000) and that necrotic but not apoptotic cells activate translocation of NFκB into the nucleus of APCs. Because Hsps are intracellular, abundant, and soluble, their presence in the extracellular milieu and the consequent activation of APCs constitute an excellent mechanism for response to cell death. Because Hsps are conserved from bacteria to mammals, the ability of Hsps to activate APCs provides a unified mechanism for response to internal and external stimuli. It is our belief that immunogenicity of cells, infectious agents, and cancers is defined, to a significant degree, by the extent to which cell death leads to induction of Hsps and consequent Hsp-APC interaction. It is safe to suggest that the recent fascination of immunologists with death has atavistic origins in the evolutionary past when the presence of extracellular Hsps signaled physical disintegration.
REFERENCES
- Amato R, Murray L, Wood L, Savary C, Tomasovic S, Reitsma D. Active specific immunotherapy in patients with renal cell carcinoma (RCC) using autologous tumor derived heat shock protein-peptide complex-96 (HSPP-96) vaccine [abstract] ASCO Meeting. 20002000 [Google Scholar]
- Amato RJ, Murray L, Wood LA, Savary C, Tomasovic S, Srivastava PK, Reitsma D. Active specific immunotherapy in patients with renal cell carcinoma (RCC) using autologous tumor derived heat shock protein-peptide complex-96 (HSPPC-96) vaccine [abstract] ASCO Meeting. 19991999 [Google Scholar]
- Arnold D, Faath S, Rammensee H, Schild H. Cross-priming of minor histocompatibility antigen-specific cytotoxic T cells upon immunization with the heat shock protein gp96. J Exp Med. 1995;182:885–889. doi: 10.1084/jem.182.3.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold D, Wahl C, Faath S, Rammensee HG, Schild H. Influences of transporter associated with antigen processing (TAP) on the repertoire of peptides associated with the endoplasmic reticulum-resident stress protein gp96. J Exp Med. 1997;186:461–466. doi: 10.1084/jem.186.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold-Schild D, Hanau D, Spehner D, Schmid C, Rammensee HG, de la Salle H, Schild H. Cutting edge: receptor-mediated endocytosis of heat shock proteins by professional antigen-presenting cells. J Immunol. 1999;162:3757–3760. [PubMed] [Google Scholar]
- Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Calderwood SK. Hsp70 stimulates cytokine production through a CD14-dependent pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–442. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- Barrios C, Lussow AR, van Embden J, et al. Mycobacterial heat shock proteins as carrier molecules II: the use of the 70 kda mycobacterial heat shock protein as carrier for conjugated vaccines can circumvent the need for adjuvants and BCG priming. Eur J Immunol. 1992;22:1365. doi: 10.1002/eji.1830220606. [DOI] [PubMed] [Google Scholar]
- Basu S, Binder RJ, Suto R, Anderson K, and Srivastava PK 2000 Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NKκB pathway. Intl Immunol. in press. [DOI] [PubMed] [Google Scholar]
- Basu S, Srivastava PK. Calreticulin, a peptide-binding chaperone of the endoplasmic reticulum, elicits tumor- and peptide-specific immunity. J Exp Med. 1999;189:797–802. doi: 10.1084/jem.189.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Suto R, Binder RJ, Srivastava PK. Heat shock proteins as novel mediators of cytokine secretion by macrophages. Cell Stress Chaperones. 1998;3:11. [Google Scholar]
- Beck G, Habicht GS. Immunity and Invertebrates. Sci Am. 1996;275:60–63. doi: 10.1038/scientificamerican1196-60. [DOI] [PubMed] [Google Scholar]
- Bevan M. Minor H antigens introduced on H-2 different stimulating cells cross react at the cytotoxic T cell level during in vivo priming. J Immunol. 1976;117:2233–2238. [PubMed] [Google Scholar]
- Binder RJ, Harris ML, Ménoret A, Srivastava PK. Saturation, competition and specificity in interaction of heat shock proteins (hsp) gp96, hsp90 and hsp70 with CD11b+ cells. J Immunol. 2000a;165:2582–2587. doi: 10.4049/jimmunol.165.5.2582. [DOI] [PubMed] [Google Scholar]
- Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nat Immunol. 2000 doi: 10.1038/77835. [DOI] [PubMed] [Google Scholar]
- Binder RJ, Ménoret A, Srivastava PK. Events involved in re-presentation of heat shock protein-chaperoned peptides. Cell Stress Chaperones. 1998;3:2. [Google Scholar]
- Blachere NE, Li Z, Chandawarkar RY, Suto R, Jaikaria NS, Basu S, Udono H, Srivastava PK. Heat shock protein-peptide complexes, reconstituted in vitro, elicit peptide-specific cytotoxic T lymphocyte response and tumor immunity. J Exp Med. 1997;186:1315–1322. doi: 10.1084/jem.186.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blachere NE, Udono H, Janetzki S, Li Z, Heike M, Srivastava PK. Heat shock protein vaccines against cancer. J Immunother. 1993;14:352–356. doi: 10.1097/00002371-199311000-00016. [DOI] [PubMed] [Google Scholar]
- Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183:725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill DP, Lengauer C, Yu J, Riggins GJ, Wilson KKV, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- Castellino F, Boucher PE, Eichelberg K, Mayhew M, Rothman JE, Houghton AN, Germain RN. Receptor-mediated uptake of antigen/heat shock protein complexes results in major histocompatibility complex class I antigen presentation via two distinct processing pathways. J Exp Med. 2000;191:1957–1964. doi: 10.1084/jem.191.11.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciupitu AMT, Petersson M, O'Donnell CL, Williams K, Jindal S, Kiessling R, Welsh RM. Immunization with a lymphocytic choriomeningitis virus peptide mixed with heat shock protein 70 results in protective antiviral immunity and specific cytotoxic T lymphocytes. J Exp Med. 1998;187:685–691. doi: 10.1084/jem.187.5.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggin JH Jr,, Anderson NG. Cancer, differentiation and embryonic antigens: some central problems. Adv Cancer Res. 1974;19:105–165. doi: 10.1016/s0065-230x(08)60053-6. [DOI] [PubMed] [Google Scholar]
- Coulie PG, Lehmann F, Lethe B, Herman J, Lurquin C, Andrawiss M, Boon T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc Natl Acad Sci USA. 1995;92:7976–7980. doi: 10.1073/pnas.92.17.7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donawho C, Kripke ML. Immunogenicity and cross-reactivity of syngeneic murine melanomas. Cancer Commun. 1990;2:101–107. doi: 10.3727/095535490820874597. [DOI] [PubMed] [Google Scholar]
- Eton O, East MJ, Ross M, Savary C, Tomasovic S, Reitsma D, Hawkins E, Srivastava PK. Autologous tumor-derived heat-shock protein peptide complex-96 (HSPPC-96) in patients (PTS) with metastatic melanoma. Proc Am Assoc Cancer Res. 2000;41:543. [Google Scholar]
- Frey AB, Cestari S. Expression of activated H-rasval 12 in nontumorigenic and non-cross-reactive syngeneic cells induces tumor antigens cross-reactive with rat mammary adenocarcinoma 13762. J Immunol. 1995;155:4783–4789. [PubMed] [Google Scholar]
- Heikema A, Agsteribbe E, Wiscjut J, Huckriede A. Generation of heat shock protein-based vaccines by intracellular loading of gp96 with antigenic peptides. Immunol Lett. 1997;57:69–74. doi: 10.1016/s0165-2478(97)00048-5. [DOI] [PubMed] [Google Scholar]
- Huang AY, Gulden PH, Woods AS, et al. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci USA. 1996;93:9730–9735. doi: 10.1073/pnas.93.18.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Ohta N, Furukawa K, Miyazaki H, Wang L, Kuribayashi K, Old LJ, Shiku H. Mutated mitogen-activated protein kinase: a tumor rejection antigen of mouse sarcoma. Proc Natl Acad Sci USA. 1997;94:6375–6379. doi: 10.1073/pnas.94.12.6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetzki S, Polla D, Rosenhauer, Lochs H, Lewis J, Srivastava PK. Immunization of cancer patients with autologous cancer-derived heat shock protein gp96: a pilot study. Intl J Cancer. 2000;88:232–238. doi: 10.1002/1097-0215(20001015)88:2<232::aid-ijc14>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Li Z, Srivastava PK. Tumor rejection antigen gp96/grp94 is an ATPase: implications for protein folding and antigen presentation. EMBO J. 1993;12:3143–3151. doi: 10.1002/j.1460-2075.1993.tb05983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linderoth NA, Popowicz A, Sastry S. Identification of the peptide-binding site in the heat shock chaperone/tumor rejection antigen gp96 (Grp94) J Biol Chem. 2000;275:5472–5477. doi: 10.1074/jbc.275.8.5472. [DOI] [PubMed] [Google Scholar]
- Lussow AR, Barrios C, van Embden J, et al. Mycobacterial heat shock proteins as carrier molecules. Eur J Immunol. 1991;22:1365–1372. doi: 10.1002/eji.1830211002. [DOI] [PubMed] [Google Scholar]
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- Monach PA, Meredith SC, Siegel CT, Schreiber H. A unique tumor antigen produced by a single amino acid substitution. Immunity. 1995;2:45–59. doi: 10.1016/1074-7613(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Moroi Y, Mayhew M, Trcka J, Hoe MH, Takechi Y, Hartl FU, Rothman JE, Houghton AN. Induction of cellular immunity by immunization with novel hybrid peptides complexed to heat shock protein 70. Proc Natl Acad Sci USA. 2000;97:3485–3490. doi: 10.1073/pnas.070550797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicchitta C. Biochemical, cell biological and immunological issues surrounding the endoplasmic reticulum chaperone GRP94/gp96. Curr Opin Immunol. 1998;10:103–109. doi: 10.1016/s0952-7915(98)80039-3. [DOI] [PubMed] [Google Scholar]
- Nieland TJ, Tan MC, Monne-van Muijen M, Koning F, Kruisbeek AM, van Bleek GM. Isolation of an immunodominant viral peptide that is endogenously bound to the stress protein GP96/GRP94. Proc Natl Acad Sci USA. 1996;93:6135–6139. doi: 10.1073/pnas.93.12.6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Old LJ. Cancer immunology: the search for specificity–G. H. A. Clowes Memorial lecture. Cancer Res. 1981;41:361–375. [PubMed] [Google Scholar]
- Ottaviani E, Franceschi C. The invertebrate phagocytic immunocyte: clues to a common evolution of immune and neuroendocrine systems. Immunol Today. 1997;18:169–174. doi: 10.1016/s0167-5699(97)84663-4. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the AYP/ADP-binding site in the hsp90 molecular chaperone. Cell. 1997a;90:65–70. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, Piper PW, Pearl LH. A molecular clamp in the crystal structure of the N-terminal domain of the yeast hsp90-chaperone. Nat Struct Biol. 1997b;4:477–482. doi: 10.1038/nsb0697-477. [DOI] [PubMed] [Google Scholar]
- Ramarathinam L, Castle M, Wu Y, Liu Y. T cell costimulation by B7/BB1 induces CD8 T cell-dependent tumor rejection: an important role of B7/BB1 in the induction, recruitment, and effector function of antitumor T cells. J Exp Med. 1994;179:1205–1214. doi: 10.1084/jem.179.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammensee HG, Schild H-J. gp96 is the Swiss Army Knife of the immune system. Nat Immunol. 2000 doi: 10.1038/77770. [DOI] [PubMed] [Google Scholar]
- Robbins PF, El-Gamil M, Li YF, Kawakami Y, Loftus D, Appella E, Rosenberg SA. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med. 1996;183:1185–1192. doi: 10.1084/jem.183.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JW, Rohrer SD, Barsoum A, Coggin JH Jr. Differential recognition of murine tumor-associated oncofetal transplantation antigen and individually specific tumor transplantation antigens by syngeneic cloned BALB/c and RFM mouse T cells. Immunology. 1994;152:754–764. [PubMed] [Google Scholar]
- Roman E, Moreno C. Synthetic peptides non-covalently bound to bacterial hsp 70 elicit peptide-specific T-cell responses in vivo. Immunology. 1996;88:487–492. doi: 10.1046/j.1365-2567.1996.d01-697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry S, Linderoth N. Molecular mechanisms of peptide loading by the tumor rejection antigen/heat shock chaperone gp96 (GRP94) J Biol Chem. 1999;274:12023–35. doi: 10.1074/jbc.274.17.12023. [DOI] [PubMed] [Google Scholar]
- Scheibel T, Weikl T, Buchner J. Two chaperone sites in Hsp90 differing in substrate specificity and ATP dependence. Proc Natl Acad Sci USA. 1998;95:1495–1499. doi: 10.1073/pnas.95.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Jasuja H, Toes RE, Spee P, et al. Cross-presentation of glycoprotein 96-associated antigens on major histocompatibility complex class I molecules requires receptor-mediated endocytosis. J Exp Med. 2000;191:1965–1974. doi: 10.1084/jem.191.11.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava PK. Peptide-binding heat shock proteins in the endoplasmic reticulum: role in immune response to cancer and in antigen presentation. Adv Cancer Res. 1993;62:153–177. doi: 10.1016/s0065-230x(08)60318-8. [DOI] [PubMed] [Google Scholar]
- Srivastava PK. Do human cancers express shared protective antigens? or the necessity of remembrance of things past. Semin Immunol. 1996;8:295–302. doi: 10.1006/smim.1996.0038. [DOI] [PubMed] [Google Scholar]
- Srivastava PK. Immunotherapy of human cancer: lessons from mice. Nature Immunol. 2000;1:363–366. doi: 10.1038/808795. [DOI] [PubMed] [Google Scholar]
- Srivastava PK, Heike M. Tumor-specific immunogenicity of stress-induced proteins: convergence of two evolutionary pathways of antigen presentation? Semin Immunol. 1991;3:57–64. [PubMed] [Google Scholar]
- Srivastava PK, Maki RG. Stress-induced proteins in immune response to cancer. Curr Top Microbiol Immunol. 1991;167:109–123. doi: 10.1007/978-3-642-75875-1_7. [DOI] [PubMed] [Google Scholar]
- Srivastava PK, Ménoret A, Basu S, Binder R, McQuade K. Heat shock proteins come of age: primitive functions acquire new roles in an adaptive world. Immunity. 1998;8:657–665. doi: 10.1016/s1074-7613(00)80570-1. [DOI] [PubMed] [Google Scholar]
- Srivastava PK, Old LJ. Individually distinct transplantation antigens of chemically induced mouse tumors. Immunol Today. 1988;9:78–83. doi: 10.1016/0167-5699(88)91269-8. [DOI] [PubMed] [Google Scholar]
- Srivastava PK, Udono H, Blachere NE, Li Z. Heat shock proteins transfer peptides during antigen processing and CTL priming. Immunogenetics. 1994;39:93–98. doi: 10.1007/BF00188611. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Russo AA, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an anti-tumor agent. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science. 1995;269:1585–1588. doi: 10.1126/science.7545313. [DOI] [PubMed] [Google Scholar]
- Suzue K, Zhou X, Eisen HN, Young RA. Heat shock fusion proteins as vehicles for antigen delivery into the major histocompatibility complex class I presentation pathway. Proc Natl Acad Sci USA. 1997;94:13146–13151. doi: 10.1073/pnas.94.24.13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Peng P, Kang L, Daou M, Srivastava PK. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science. 1997;278:117–120. doi: 10.1126/science.278.5335.117. [DOI] [PubMed] [Google Scholar]
- Udono H, Levey DL, Srivastava PK. Cellular requirements for tumor-specific immunity elicited by heat shock proteins: tumor rejection antigen gp96 primes CD8+ T cells in vivo. Proc Natl Acad Sci USA. 1994;91:3077–3081. doi: 10.1073/pnas.91.8.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udono H, Srivastava PK. Heat shock protein 70-associated peptides elicit specific cancer immunity. J Exp Med. 1993;178:1391–1396. doi: 10.1084/jem.178.4.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udono H, Srivastava PK. Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90, and hsp70. J Immunol. 1994;152:5398–5403. [PubMed] [Google Scholar]
- Wassenberg JJ, Dezfulian C, Nicchitta CV. Receptor mediated and fluid phase pathways for internalization of the ER Hsp90 chaperone GRP94 in murine macrophages. J Cell Sci. 1999;12(0):2167–2175. doi: 10.1242/jcs.112.13.2167. [DOI] [PubMed] [Google Scholar]
- Wearsch PA, Nicchitta CV. Interaction of endoplasmic reticulum chaperone GRP94 with peptide substrates is adenine nucleotide-independent. J Biol Chem. 1997;272:5152–5156. doi: 10.1074/jbc.272.8.5152. [DOI] [PubMed] [Google Scholar]
- Wearsch PA, Voglino L, Nicchitta CV. Structural transitions accompanying the activation of peptide binding to the endoplasmic reticulum hsp90 chaperone GRP94. Biochemistry. 1998;37:5709–5719. doi: 10.1021/bi9801006. [DOI] [PubMed] [Google Scholar]
- Wolfe T, Hauer M, Schneider J, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–1284. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]