

Abstract
The polycomb group protein BMI1 has been linked to proliferation, senescence, cancer progression and stem cell phenotype. At present, very little is known about its regulation. Here, we report that BMI1 contains a functional recognition motif for the F box protein βTrCP, which regulates ubiquitination and proteasome-mediated degradation of various proteins. We show that overexpression of wild-type βTrCP but not the ΔF mutant of it promotes BMI1 ubiquitination and degradation, and knockdown of βTrCP results in increased expression of BMI1. Furthermore, a mutant of BMI1 with an altered βTrCP recognition motif is much more stable than wild-type BMI1. We also show that wild-type BMI1 but not the mutant BMI1 interacts with βTrCP. Accordingly, compared to wild-type BMI1, mutant protein exhibited increased pro-oncogenic activity. In summary, our findings suggest that βTrCP regulates turnover of BMI1 and its function relevant to oncogenesis, cellular senescence and aging.
Key words: BMI1, βTrCP, polycomb group proteins, senescence, breast cancer
Introduction
Polycomb group (PcG) proteins are important regulators of cell fate decisions.1 These proteins promote cell proliferation, and negatively regulate cellular senescence and differentiation in mammalian cells. Many of the PcG proteins are aberrantly expressed in human cancers. For example, BMI1 is overexpressed in a number of cancers including breast and prostate cancers.2–4 In addition to its role in cancer, BMI1 is known to be required for self-renewal of neural, hematopoietic, intestinal and mammary stem cells.5–7 It has been proposed that BMI1 overexpression promotes stem-ness in tumor cells, which promotes therapy resistance in cancer patients.2 Recent mouse xenograft studies using BMI1 and H-RAS co-overexpressing human mammary epithelial cells (HMECs) strongly support the oncogenic roles of BMI1 in breast cancer development and metastasis of breast cancer cells to various organs including brain.8,9
In response to telomere shortening or dysfunction, and certain stress signals, normal human cells undergo senescence, which constitutes a powerful barrier to oncogenesis.10 Overexpression of BMI1 results in bypass of this barrier and extension of replicative life span in human cells.11,12 The senescence bypass by BMI1 in human fibroblasts most probably results due to downregulation of p16INK4a by BMI1,12 and possibly repression of other differentiation-related genes and tumor suppressor targets of BMI1. BMI1 also upregulates AKT, ERK and telomerase activities via p16INK4a-independent pathways.3,8,11
Despite its well-documented role in cellular senescence, cancer and stem cell phenotype, at present very little is known about the regulation of BMI1. With respect to transcriptional regulation, we and others have shown that, BMI1 is positively regulated by c-Myc,13,14 and the E2F family of transcription factors.15 In particular, very little is known about the posttranslational regulation of BMI1. Very recently, we reported that BMI1 is a short-lived protein, which undergoes rapid turnover.14,16 However, the molecular basis of BMI1 turnover is not known.
Posttranslational regulation of proteins, in particular ubiquitin-proteasome mediated proteolysis plays a major role in cellular homeostasis and its deregulation contributes to pathological conditions.17 The two major E3-ubiquitin ligases, SCF (SKP1-cullin-F-box) and APC (anaphase-promoting complex) are the core components of the ubiquitin-proteasome system.18,19 The beta-transducin repeat containing protein (βTrCP) is an F box component of the SCF type E3 ubiquitin ligase complex, and is involved in substrate recognition.20 The substrates of βTrCP1 and βTrCP2, which are alternatively spliced forms and encode biochemically similar proteins (collectively referred to as βTrCP) regulate signaling, growth regulatory and circadian clock proteins such as IκB, β-catenin, CDC25A, Claspin, Gli, Mcl-1 and YAP.20,21
The conserved site known as degron recognized by βTRCP is DSG(X)2 + n (S).20 The DSG motif is usually followed by two but in some cases several non conserved residues before the highly conserved S residue. Our recent data suggest that the c-terminal region of BMI1, described as PS (proline-serine rich) domain may regulate the stability of BMI1.16 Here, we report that the PS region of BMI1 contains a functional βTrCP recognition motif (DSGsdkanS). We show that βTrCP binds to its putative recognition motif of BMI1, and that it regulates BMI1 stability via ubiquitination and 26S proteasome-mediated degradation.
Results
BMI1 undergoes proteasome-mediated degradation.
To examine the mechanism of posttranslational regulation of BMI1, we determined half life of BMI1 in MCF10A-BMI1 by inhibiting the de novo protein synthesis using cycloheximide (CHX) treatment for different time points (0–60 min). The BMI1 protein remaining after each time point was examined using western blot analysis and quantified by densitometry. Our data indicated that BMI1 is rapidly turned over with a half life of ∼40 min (Fig. 1A). On longer exposure of the western blot, we noticed several slow mobility bands of BMI1, which increased in intensity upon CHX treatment (Fig. 1A). We reasoned that these bands may represent phosphorylated forms of BMI1. Since, the multiple bands of BMI1 could complicate the half life analysis, we also determined half life of BMI1 after treating the extracts with calf-intestinal phosphatase (CIP) (Fig. 1A, lower part). The total cell extracts from each time point were treated with CIP in vitro, run on a gel, analyzed by western blot analysis and the half life of BMI1 was determined as above. The data indicated that CIP treatment results in disappereance of slow mobility bands and appearance of a faster mobility band, which represent the dephosphorylated form of the BMI1. Our data also indicated that the half life of CIP treated BMI1 is also ∼40 min (Fig. 1A and lower part).
Figure 1.
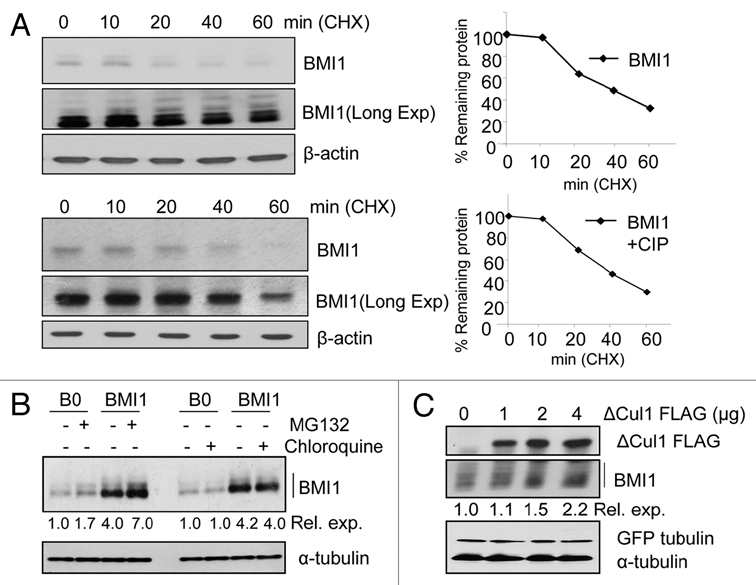
BMI1 is postranslationally regulated by ubiquitin proteasome system. (A) MCF10A-BMI1 cells were treated with CHX for the indicated time points and the immunoblot (IB) of BMI1 and β-actin (loading control) was performed (left part). The densitometric quantification of BMI1 normalized to β-actin was plotted against various time points to determine its half life (right parts). For densitometric analysis the light exposure of the blot was chosen in each case. The half life of BMI1 was also determined after further treating CHX-treated extracts with CIP, and performing western blot analysis and densitometry of BMI1 signal present at each time point (bottom part). (B) 26S-Proteasomal machinery orchestrates BMI1 proteolysis. Asynchronously growing MCF10A-B0 and MCF10A-BMI1 cells were treated with MG132 (10 µM), and lysosomal inhibitor Chloroquine (25 µM) as indicated. BMI1 was detected by western blot analysis. The relative expression (Rel. exp.) of BMI1 was determined by densitometric quantification of BMI1 bands normalized to α-tubulin levels. (C) SCF complex targets BMI1 for degradation. U2OS cells transiently transfected with ΔCul 1 FLAG plasmid as well as GFP-tubulin as transfection control. Cell lysates were analyzed after 48 hr. Densitometric values (relative expression) were obtained after BMI1 levels were normalized with the GFP-tubulin levels.
The turnover of most cellular proteins involves either lysosomal or proteasomal degradation pathway. To determine whether BMI1 turnover involves lysosomal and/or proteasomal pathway, we used pharmacological inhibitors of these pathways. Our data indicated that treatment of cells with the 26S proteasomal inhibitor MG132 but not the lysosomal inhibitor chloroquine accumulates BMI1 in both MCF10A-BMI1 overexpressed as well as MCF10A-B0 cells. Thus exogenously expressed BMI1 (MCF10A-BMI1 cells) as well as endogenous BMI1 (MCF10A-B0 cells) are degraded via 26S-proteasome pathway (Fig. 1B).
As the proteasome degradation pathway is often mediated by the APC or the SCF ubiquitin ligase complexes,18,19 and we did not observe any consensus APC degron motif in BMI1, we investigated the possibility of SCF-type E3 ligase complex regulating BMI1 turnover. To examine whether BMI1 undergoes degradation with the help of SCF type E3 ubiquitin ligase complex, we used a dominant negative (DN) mutant of the scaffolding protein Cullin 1 (ΔCul1), which has been shown to stabilize proteins known to be degraded by the SCF machinery. Our data indicated that the transient overexpression of ΔCul1-Flag accumulated BMI1 dose dependently in U2OS cells (Fig. 1C). These data indicate that the BMI1 is degraded via 26S-proteasome pathway involving the SCF complex.
βTrCP, the F-box component of SCF regulates BMI1 expression.
Since βTrCP is the most well characterized F box component of the SCF complex, we analyzed primary protein sequence of the BMI1 for the presence of a βTrCP recognition motif (βTrCP degron) (Fig. 2A). Our analysis indicated the presence of a DSGsdkanS sequence, which conforms to the consensus βTrCP recognition motif DSG(X)2 + n(S) found in several SCF regulated proteins such as CDC25A, MCL1, SNAIL and β-catenin (Fig. 2A and right part). We further determined that the putative βTrCP degron site of BMI1 is highly conserved among various mammalian species (Fig. 2A left part). Interestingly βTrCP degron site is not present in Mel-18, which is highly homologus to BMI1, suggesting that Mel-18 is not regulated by βTrCP. Thus, the βTrCP degron site in BMI1 is likely to have functional relevance.
Figure 2.

βTrCP, an F box component of SCF complex regulates BMI1 proteolysis and BMI1-mediated H2A K119Ub activity of PRC1. (A) Sequence alignment of putative βTrCP degron site of BMI1 with known βTrCP degron sites of CDC25A, MCL1, Snail and β catenin (left part), and sequence alignment of βTrCP degron site of BMI1 from various mammalian species (right part). (B) Knockdown of βTrCP stabilizes BMI1. U2OS cells stably overexpressing HA tagged βTrCP were transiently transfected with shRNAs against βTrCP. After 48 hr, cells were harvested and βTrCP, BMI1 and α-tubulin were analyzed by western blot analysis. The quantitative densitometric values were obtained after BMI1 and TrCP HA levels were normalized to α-tubulin levels. (C) MCF10A cells were stably transfected with Vector (B0), HA-tagged TrCP full length protein (TrCP WT-HA) and F box deletion mutant of the TrCP (TrCPΔF-HA). After stable selection of cells, TrCP (HA), BMI1 and β-actin levels were analyzed by western blot analysis. The relative levels of BMI1 were determined by densitometric analysis. (D) The levels of BMI1 and H2A K119Ub were examined in vector control and TrCPΔF-HA expressing MCF10A cells (as indicated) by western blot analysis (left part). Similarly, the expression of BMI1, and H2A K119Ub levels were determined in control (Conti) and βTrCPi expressing 293T cells. 293T cells were transiently transfected with either 8 µg of conti-1 plasmid or with 4 µg each of TrCPi-1 and TrCPi-2 plasmids together with 100 ng of GFP-tubulin as a transfection efficiency control. Total cell lysates or total histones were prepared and analyzed by western blot analysis using respective antibodies.
The presence of βTrCP degron in BMI1 prompted us to investigate whether βTrCP regulates BMI1 proteolysis via its ubiquitination and degradation. First, we examined the role of βTrCP in BMI1 degradation using RNA interference (RNAi) approach. U2OS cells overexpressing HA tagged wild-type βTrCP were transiently transfected with plasmids expressing a control shRNA, and two specific shRNA (TrCPi-1 and TrCPi-2), which have been shown to target βTrCP.22 Our data showed a modest increase of BMI1 levels after transient expression of shRNAs targeting βTrCP (Fig. 2B). Next, using a retroviral expression system, we overexpressed HA tagged full length wild-type βTrCP (TrCPWT-HA) and a DN mutant of it (TrCPΔF-HA) in MCF10A cells, and analyzed the levels of BMI1 in cells expressing wild-type or DN mutant TrCP proteins. Our results indicated that cells overexpressing wild-type βTrCP had decreased levels of BMI1 as compared to control cells, while cells overexpressing ΔF box mutant had increased levels of BMI1 protein (Fig. 2C). The biochemical activity associated with the BMI1 containing polycomb repressive complex 1 (PRC1) is monoubiquitination of K119 residue of the histone 2A (H2A K119Ub).23 Accordingly, BMI1 overexpression has been shown to increase H2A K119Ub activity of the PRC1. Hence, we determined whether upregulation of BMI1 by βTrCP RNAi approach or exogenous expression of a DN mutant of βTrCP resulted in increase in H2A K119Ub levels. Our results indicated that indeed overexpression of DN mutant of βTrCP or downregulation of βTrCP by RNAi approach resulted in a modest increase in H2A K119 levels (Fig. 2D), suggesting that βTrCP is a physiological regulator of BMI1.
βTrCP promotes ubiquitination of BMI1.
Next, we determined whether βTrCP regulates BMI1 degradation via ubiquitination of BMI1. MCF10A cells overexpressing TrCPWT-HA and TrCPΔF-HA along with vector control cells were treated with 10 µM MG132 for 5 hr followed by immunoprecipitation with a BMI1 antibody. The immunoprecipitated samples were probed with an anti-ubiquitin antibody. The whole cell extracts were also probed for BMI1, and βTrCP using anti-HA antibody. The results of immunoblot assays indicated that overexpression of wild-type βTrCP increased ubiquitination of BMI1 as compared to control cells, while the DN mutant (βTrCPΔF) decreased relative ubiquitination of BMI1 (Fig. 3A). Based on these data, we conclude that the βTrCP promotes ubiquitination of BMI1.
Figure 3.

βTrCP promotes BMI1 ubiquitination and βTrCP degron mutant of BMI1 is expressed at higher levels. (A) Whole cell extracts (WCEs) from MCF10A-derived B0, TrCPWT-HA and TrCPΔF-HA cells were subjected to immunoprecipitation (IP) with anti-BMI1 antibody and analyzed by immunoblotting for the indicated proteins. (B) Expression levels of wild-type and mutant (βTrCP degron site mutant) BMI1 proteins in U2OS and MCF10A cells were determined by the western blot analysis. Relative expression of wild-type and mutant BMI1 were determined by densitometry and normalized to control α-tubulin levels. The expression level of wild-type BMI1 was considered 1. The expression level of endogenous BMI1 was not relevant and thus not determined (ND). (C) Whole cell extracts (WCEs) from MCF10A derived BMI1 WT-FLAG and BMI1 Mut-FLAG cells were subjected to IP with anti-FLAG antibody in the presence of solvent control or MG132 and analyzed by immunoblotting for ubiquitinated BMI1 and FLAG (input). (D) U2OS cells stably overexpressing wild-type BMI1 and either TrCPWT HA or TrCPΔF HA were treated with 10 µM MG132 for 5 hr. WCEs were subjected to IP with anti-HA and anti-BMI1 antibodies and analyzed by immunoblotting for the indicated proteins. (E) U2OS cells stably overexpressing TrCPWT-HA and BMI1 WT-FLAG or BMI1 Mut-FLAG were treated with 10 µM MG132 for 5 hr. WCEs were subjected to IP with anti-HA and anti-Flag antibodies and analyzed by immunoblotting for the indicated proteins.
To further examine the role of βTrCP in BMI1 ubiquitination, we constructed a BMI1 mutant in which the important βTrCP degron motif residues DSGS were changed to AAAA using site directed mutagenesis. We reasoned that this mutant of BMI1 (here in referred to as BMI Mut-FLAG) should be partially defective in ubiquitination. The Flag tagged wild-type (BMI1 WT-FLAG) and mutant BMI1 (BMI1 Mut-FLAG) were overexpressed in MCF10A and U2OS cells. The western blot analysis of wild-type and mutant BMI1 in U2OS and MCF10A cells suggested that the mutant BMI1 is expressed at higher levels than the wild-type BMI1 (Fig. 3B), possibly due to its inability to undergo βTrCP-mediated ubiquitination and degradation. To examine this possibility, we further studied ubiquitination of exogenously expressed wild-type and mutant BMI1 in MCF10A cells by immunoprecipitation using FLAG antibody followed by immunoblot analysis of ubiquitin using an anti-ubiquitin antibody. Our results indicated that indeed the mutant BMI1 is partially defective in ubiquitination (Fig. 3C).
βTrCP interacts with wild-type BMI1.
We reasoned that defective ubiquitination of mutant BMI1 protein may result due to inability of the βTrCP to interact with it and recruit SCF ubiquitination machinery. To examine this possibility, first we determined whether the wild-type BMI1 interacts with βTrCP. We developed U2OS cells stably overexpressing wild-type BMI1 and HA tagged wild-type βTrCP (TrCP WT-HA) or mutant βTrCP (TrCPΔF-HA). The interaction of BMI1 with the wild-type or mutant βTrCP was studied by co-immunoprecipitation (co-IP) assays. The cells were treated with 10 µM MG132 for 5 hr followed by IP of BMI1 and βTrCP using anti BMI1 and anti HA antibodies respectively. The immunoblot analysis of the IPs indicated that BMI1 co-IPed both wild-type βTrCP and ΔF mutant of βTrCP, and conversely βTrCP co-IPed BMI1 (Fig. 3D). Thus our data indicated that BMI1 physically interacts with βTrCP in an F-box-independent manner. These data are consistent with published reports, which have shown that βTrCP interacts with its substrates via WD repeats and not the F box.24,25
Next, we examined whether wild-type BMI1 but not the mutant BMI1 with an altered βTrCP degron motif interacts with βTrCP. For this purpose, we exogenously expressed HA-tagged wild-type βTrCP in U2OS cells expressing FLAG tagged wild-type or mutant BMI1 protein, and carried co-IP assays using anti FLAG and anti HA antibodies. The results indicated that βTrCP specifically co-IPed wild-type but not the mutant BMI1 protein, and conversely wild-type BMI1 but not the mutant BMI1 co-IPed βTrCP (Fig. 3E). Thus, our data indicate that the βTrCP interacts with BMI1 via its recognition motif present in BMI1.
Mutant BMI1 with an altered βTrCP degron motif exhibits increased oncogenic activity.
As the mutant BMI1 with an altered βTrCP degron motif does not interact with βTrCP, and is partially defective in ubiquitination and degradation, we reasoned that the mutant BMI1 should be biologically more active than the wild-type BMI1. We have previously shown that BMI1 overexpression leads to increased proliferation and upregulation of AKT and ERK activities in HMECs.3,8,11 To determine whether mutating βTrCP degron motif of BMI1 augments proliferative activity of BMI1, we examined the expression of AKT, GSK and ERK in control, and wild-type and mutant BMI1 expressing MCF10A cells. MCF10A-derived cells were made quiescent by EGF and serum deprivation for 72 hr, and cells were then stimulated by EGF for 20 min. Consistent with the role of AKT, GSK and ERK in cell signaling, expression of P-ERK (phospho-ERK) was very low whereas P-AKT (phospho-AKT) was undetectable in quiescent MCF10A cells (Fig. 4A). EGF stimulation of cells caused substantial upregulation of P-AKT, P-GSK (Phospho-GSK), and P-ERK in all control as well wild-type and mutant BMI1 expressing MCF10A cells. Furthermore, compared to wild-type BMI1, mutant BMI1 expression always led to a modest increase of P-ERK, P-GSK and P-AKT (Fig. 4A). We also examined the rate of proliferation of control MCF10A, and wild-type- and mutant BMI1-expressing cells. Our results indicated that MCF10A cells expressing mutant BMI1 proliferated more rapidly than control or wild-type BMI1 expressing cells (Fig. 4B).
Figure 4.

Mutant BMI1 exhibits increased pro-oncogenic activities. (A) MCF10A cells overexpressing BMI1 WT and BMI1 Mut along with B0 control were growth factor deprived using D3 medium for 72 hr and stimulated for 30 min by addition of D medium containing 12.5 ng/ml epidermal growth factor (EGF). Western blot analysis of phospho-AKT, total AKT (AKT1 and AKT2), phospho-ERK, total ERK, phospho-GSK and total GSK in MCF10A B0 and MCF10A-derived cells (as indicated) was done and densitometric quantifications were done for each phosphorylated signaling intermediate after normalizing it with the levels of their respective total proteins. (B) Short term proliferation assay of MCF10A cells stably overexpressing BMI1 WT and BMI1 Mut along with B0 (vector) control. (C) Invasive potential of MCF 10A-derived B0 (vector), BMI1 WT and BMI1 Mut cells towards EGF was determined as described in Materials and Methods. Columns indicate mean of triplicates and bars indicate ±SD. (D) The migration potential of MCF10A-derived B0, BMI1 WT and BMI1 Mut was determined by wound-healing assay as described in the Materials and Methods. The values were plotted as indicated (right part).
To determine whether the mutation in βTrCP degron motif of BMI1 impacts its oncogenic potential in HMECs, we performed invasion and wound healing assays. First, we studied invasion potential of MCF10A-derived cells using transwell invasion assay. The results suggested a modest but significant increase in invasion potential of MCF10A cells expressing mutant BMI1 compared to wild-type BMI1 expressing cells (Fig. 4C). Next, we determined the migration potential of control, and wild-type and mutant BMI1 expressing MCF10A cells using wound healing assay. The relative migration of cells was computed by measuring the average distance remaining free of cells in each case. The result showed a significant increase in migration potential of mutant BMI1 expressing cells compared to wild-type BMI1 expressing and control MCF10A cells (Fig. 4D).
The mutant BMI1 promotes proliferation and bypass senescence in HDFs more efficiently than the wild-type BMI1.
Next, we determined whether the overexpression of wild-type and mutant BMI1 resulted in increased H2A K119Ub activity of PRC1. The levels of H2A K119Ub in control, and wild-type and mutant BMI1 overexpressing IMR90 fibroblasts were analyzed by western blot analysis. The results indicated a modest two fold increase in H2A K119Ub levels in cells expressing mutant BMI1 as compared to wild-type BMI1 expressing IMR90 cells (Fig. 5A). Next, we determined whether the increased stability of the mutant BMI1 is reflected in increased binding of it to the known targets of BMI1, and that whether the mutant BMI1 expressing fibroblasts proliferated more rapidly as compared to the wild-type BMI1 expressing and control fibroblasts. Using chromatin-immunoprecipitation linked PCR (ChIP) assay, we examined the binding of wild-type and mutant BMI1 to two well known PcG targets-p16INK4a,26 and HOXA9.23 The results of ChIP assay showed that compared to wild-type BMI1, the mutant BMI1 immunoprecipitated two-fold more p16INK4a chromatin (Fig. 5B). The mutant BMI1 also exhibited increased ChIP activity when primers for HOXA9 promoter were chosen for the ChIP assay (not shown). As expected, increased binding of the mutant BMI1 to p16INK4a locus resulted in proportionally decreased expression of p16INK4a mRNA as determined by the qRT-PCR (Fig. 5C), as well as decreased levels of p16INK4a as determined by the western blot analysis (Fig. 5D).
Figure 5.

Mutant BMI1 promotes proliferation and bypasses senescence more efficiently than the wild-type BMI1. (A) IMR90-derived B0, BMI1 WT and BMI1 Mut cells were analyzed for the expression of H2A K119Ub and total H2A by western blot analysis. (B) ChIP analysis of BMI1 binding to p16INK4a promoter. Left part shows ethidium bromide stained agarose gel picture showing the amplified product of p16INK4a after ChIP by anti-BMI1 antibody or control IgG as indicated. The values obtained from qPCR performed in triplicates were plotted as indicated (right part). (C) Relative mRNA levels of p16INK4a were analyzed by qRT PCR from IMR90-derived B0, BMI1 WT and BMI1 Mut cells as indicated. (D) IMR90-derived B0, BMI1 WT and BMI1 Mut cells were analyzed by western blot analysis for expression of BMI1, p21, p53, p16, pRb and β-actin (loading control). The densitometric values were obtained after normalizing the value of the indicated proteins with β-actin. *For determining the relative expression of wild-type and mutant BMI1, only two lower bands of BMI1 were considered as the mutant BMI1 lacks slow migrating top band(s). The expression level of wild-type BMI1 was considered 1. (E) Short term proliferation rate of IMR90 cells stably overexpressing BMI1 WT and BMI1 Mut along with B0 (vector) control was determined in triplicates as described in the Materials and Methods. (F) SA-β-gal assay was performed on IMR90-derived B0, BMI1 WT and BMI1 Mut cells. The left part shows the staining pictures taken at 10× magnification. The % senescence (fraction of SA-β-gal positive cells) was plotted as indicated (right part).
Next, we determined whether the mutant BMI1 expressing cells proliferated more rapidly. A short term proliferation assay indicated that the mutant BMI1 expressing HDFs proliferated more rapidly compared to wild-type BMI1 expressing cells, which also proliferated better than the control vector infected cells (Fig. 5E). Because p16INK4a is an important determinant of cellular senescence, and its downregulation by BMI1 overexpression has been shown to bypass senescence,12,14 we determined whether the mutant BMI1 bypasses senescence more efficiently. Senescence bypass in IMR90 cell was studied using SA-β-gal marker.27 We compared the number of SA-β-gal positive (senescent) cells in control, and wild-type and mutant BMI1 expressing cells at similar passage number. The results indicated that mutant BMI1-expressing IMR90 culture has the least number of senescent cells (Fig. 5F). Thus our data indicate that the mutant BMI1 protein is much more efficient in bypassing senescence in HDFs.
Discussion
The role of PcG proteins in cancer is well documented. These proteins are often overexpressed in human cancers, and their expression is thought to facilitate mechanisms that evade favorable therapy response.2,28 PcG proteins are also key determinants of cellular senescence.12,26,29,30 Consistent with their pro-proliferative role, expression of two key PcG proteins BMI1 and EZH2 has been shown to be downregulated in senescent human cells,12,30 and knockdown of BMI1 and/or EZH2 inhibits growth of tumor cells via induction of senescence or apoptosis. Thus, the PcG proteins are attractive molecular targets for cancer therapy. In order to develop therapies to target PcG proteins, a proper understanding of the regulatory pathways, in particular, the mechanisms of overexpression of PcG proteins in cancer cells is essential.
Although the most common mechanism of overexpression of oncogenes is transcriptional, many oncoproteins including cell cycle regulatory proteins and key signaling molecules are posttranslationally regulated by E3 ubiqutin ligases, which target proteins for proteasome-mediated degradation.19 Indeed, we have shown that BMI1 is positively regulated by c-Myc,14 and that in some cases overexpression of BMI1 correlates with c-Myc overexpression, however, it is possible that in many cancer cells overexpression of BMI1 is independent of c-Myc.
Here, we studied a possible transcriptional regulation-independent mechanism of BMI1 expression in human cells. Multiple lines of evidence suggest that BMI1 is posttranslationally regulated by the SCF complex. First, a DN mutant of Cullin1 upregulated steady state levels of BMI1. Second, overexpression of βTrCP, which is a rate-limiting component of the SCF complex, resulted in decreased expression of BMI1, and overexpression of a DN mutant of βTrCP increased expression of BMI1. Accordingly, knockdown of βTrCP also resulted in decreased proteolysis of BMI1. Finally, substitution of DSGS residues of the βTrCP degron motif of BMI1 protein sequence into AAAA residues reduced its ubiquitination and abolished interaction of BMI1 with βTrCP. When measured the biological activity of the mutant BMI1 using ChIP assays, we found that the mutant BMI1 was much more active in HDFs. Increased activity of mutant BMI1 could reflect its increased steady state levels due to its slower turnover. Alternatively, it is also possible that the mutant BMI1 is somehow more active not merely because of increased levels but due to its increased affinity to DNA or its differential interaction with other PRC1 components. Although such possibility remains to be further explored, our studies point toward the functional importance of βTrCP degron present in BMI, and its regulation by βTrCP containing ubiquitin liagse complex.
Cancer is a multigenic disease, and its development results due to a multistep process.31 The very first step often is abrogation or bypass of senescence.10,32 It is well established that the PcG protein BMI1 contributes to oncogenesis by repressing p16INK4a, which results in bypass of senescence. In this context, it is worthwhile noting that the abrogation of βTrCP binding to BMI1 results in its increased binding activity to p16INK4a locus and decreased expression of p16INK4a. Because p16INK4a is a major regulator of senescence, as expected abrogation of βTrCP mediated regulation of BMI1 results in efficient bypass of senescence in HDFs. In addition to repression of p16INK4a locus, BMI1 can also positively regulate growth regulatory signaling pathways such as AKT and ERK, which results in increased proliferation and induction of invasive phenotype in HMECs and possibly in other cell types. Accordingly, we noticed that compared to wild-type BMI1, the mutant BMI1 exhibited increased activity with respect to upregulation of AKT and ERK, and that it also increased proliferation of HMECs as well as HDFs. In summary, our data strongly implicate βTrCP and SCF ubiquitin ligase complex in regulating BMI1 proteolysis.
βTrCP is one of the most versatile component of SCF complex; its substrates include potential oncoproteins as well as tumor suppressors, and accordingly it can function either as a tumor suppressor or as an oncogene.33 Addition of a new oncogenic substrate BMI1 to the list of the targets of βTrCP adds to the intriguing nature of this versatile protein. By targeting BMI1, it could promote tumor suppressor function of βTrCP, whereby it could suppress BMI1-dependent oncogenesis. On the other hand, by negatively regulating BMI1, it could also inhibit self-renewal/proliferation of stem cells, and promote senescence in normal human cells. Importantly, BMI1 has been shown to regulate age-related pathologies in mouse model.34,35 Thus, by regulating BMI1, βTrCP could indirectly regulate stem cell phenotype and senescence, and contribute to aging phenotypes and age-related pathologies in higher organisms.
Materials and Methods
Cells and cell culture methods.
Immortal HMEC strain MCF10A and a human osteosarcoma cell line U2OS were obtained from the American Type Culture Collection (ATCC) (Manassas, VA USA). The MCF10A cells were cultured as described previously in reference 36. IMR90 strain of HDFs was obtained from the National Institute on Aging (NIA), Aging Cell Repository, Coriell Institute for Medical Research, Camden, NJ. HDFs and U2OS were cultured in DMEM containing 10% FCS, 2 mM L-glutamine at 37°C under 10% CO2 and 90% atmosphere as described in reference 27.
Expression vectors and expression methods.
A retroviral vector overexpressing wild-type BMI1 has been described earlier in reference 11 and 12. The cDNA of βTrCP degron site mutant of BMI1 was generated by PCR, sequence verified and cloned into pBabe-puro and pMSCV-neo retroviral vectors using standard cloning methods. Where indicated FLAG or HA epitopes were added to cDNA of interest using PCR. For βTrCP knockdown experiments, shRNA vectors pU6-shRNA-βTrCP1-1, pU6-shRNA-βTrCP1-2 and pU6-shRNA-hHOS (mutant control) were obtained from Dr. Serge Fuchs (Univ. of Pennsylvania, PA). To express human βTrCP1, its cDNA was amplified from Flag-β TrCP1 vector (Addgene, Cambridge, MA) and cloned into pBabe-puro vector. An F-box deletion mutant of βTrCP1 was also generated by PCR and cloned into pBabe-puro vector. The DN mutant of Cullin 1 was also obtained from Addgene (Cambridge, MA).
Retroviruses were generated by transfecting retroviral vectors together with a packaging plasmid pIK into tsa54 cells and collecting supernatant as described in reference 37. Stable cell lines expressing gene(s) of interest were generated by infection of the retroviral vector(s) expressing a particular gene and selecting cells in either puromycin or G418 as described in reference 11, 14 and 37. For transient expressions, U2OS cells were transfected using different concentrations of plasmids using lipofectamine-2000 (Invitrogen, Carlsbad, CA).
Chromatin immunoprecipitation and quantitative real time RT-PCR assays.
Chromatin immunoprecipitation-linked PCR (ChIP) assays were performed as described in reference 30 and 38. Briefly, cells were treated with 1% formaldehyde for 20 min at room temperature. The cross-linked chromatin was isolated and sonicated to yield 200–500 bp fragments. Immunoprecipitations (IPs) were carried out using a custom made rabbit polyclonal antibody raised against BMI1, and a control IgG. The BMI1- and IgG-bound chromatin were amplified using p16INK4a-specific primers by quantitative real time PCR (qPCR) in a Rotor-Gene 6000 series PCR machine (Corbett Life Sciences-Qiagen Inc., Valencia, CA). The primers used for amplification of p16INK4a were- Forward (F) 5′-GGG CTC TCA CAA CTA GGA AAG-3′ and Reverse (R) 5′-GGT GTT TGG TGT CAT AGG G-3′. To perform quantitative real time reverse transcript PCR (qRT-PCR), total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA), and cDNA synthesis and PCR amplification were carried out using SensiMix Plus SYBR kit (Quantace Inc.). The primers used for p16INK4a cDNA amplification were- F-5′-CCC CTT GCC TGG AAA GAT AC-3′ and R-5′-AGC CCC TCC TCT TTC TTC CT-3′. Real time PCR data were analyzed using Rotor-Gene Q software package (Corbett Life Sciences-Qiagen Inc., Valencia, CA).
Antibodies, western blot analyses and protein half life determination.
BMI1 was detected either using 1H6B10G7 monoclonal antibody (mAb) (Invitrogen, Carlsbad, CA), or F6 mAb (Millipore (Billerica, MA). For the analysis of AKT pathway, phospho-AKT 1/2/3 (Ser-473) and total AKT-1/2/3 were obtained from Santa Cruz Biotech (Santa Cruz, CA). Antibodies against pRb, phospho-ERK, ERK, p16INK4a, p21 and α-tubulin were obtained from Santa Cruz Biotech (Santa Cruz, CA). Among other antibodies, Phospho-GSK3β (Ser 9) and GSK antibodies were obtained from Cell Signaling Technologies (Danvers, MA), and β-actin, HA and FLAG mAbs were obtained from Sigma-Aldrich (St. Louis, MO). For immunostaining, a pAb against H2AK119Ub were obtained from Millipore (Billerica, MA). Recombinant protein A- and protein G-sepharose beads were obtained from GE Healthcare Biosciences (Pittsburgh, PA).
When required, MCF10A cells were growth factor deprived using D3 medium for 72 hrs and/or stimulated for 20 min by addition of D medium, which contains 12.5 ng/ml epidermal growth factor (EGF) as described in reference 36. Western blot analyses of total cell extracts from control and wild-type or mutant BMI1 expressing cells, and different combinations of BMI1 and βTrCP were performed using respective antibodies as described in reference 14. For the analysis of H2A ubiquitination, total histones were prepared using acid extraction method as described in reference 39. The half life of wild-type and mutant BMI1 protein was determined using cycloheximide (CHX) treatment of MCF10A or U2OS cells expressing BMI1 proteins as described in reference 14.
Immunoprecipitation and immunostaining methods.
For immunoprecipitation (IP) cells were scraped off and lysed in IP buffer (20 mM Tris HCl pH 8, 137 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, 1 mM PMSF, 1 ug/ml Aprotinin, 1 mM NaF, 1 mM sodium orthovanadate, x100 diluted protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). IPs were performed using specific antibodies and protein A- or Protein G-sepharose as described in reference 36. The immunoprecipitates were analyzed for interacting proteins using western blot analyses as described above. Immunostaining assays were performed as described in reference 36. After staining, cells were photographed (40x) using a Nikon Eclipse 80i confocal microscope.
Senescence, proliferation, invasion and wound healing assays.
Senescence induction or bypass assays in HDFs expressing wild-type or mutant BMI1 proteins was determined using histochemical staining for senescence-associated beta galactosidase (SA-β-gal) marker as described in reference 12, 14, 27 and 37. Similarly, proliferation assays were done as described in reference 12, 14, 27 and 37. Invasion and migration assays were performed as described in references 8 and 36.
Acknowledgements
We thank Prasad Rote for technical assistance, and Dr. Ajay K. Yadav for helpful discussions. This work was supported in part by RO1 CA094150 from the National Cancer Institute, NIH to G.P.D. M.D. gratefully acknowledges support from Breast and Ovarian Research Program of the NorthShore University HealthSystem, Evanston, IL.
References
- 1.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 2.Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115:1503–1521. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo WJ, Zeng MS, Yadav A, Song LB, Guo BH, Band V, et al. Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and downregulating Akt activity in breast cancer cells. Cancer Res. 2007;67:5083–5089. doi: 10.1158/0008-5472.CAN-06-4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim JH, Yoon SY, Jeong SH, Kim SY, Moon SK, Joo JH, et al. Overexpression of Bmi-1 oncoprotein correlates with axillary lymph node metastases in invasive ductal breast cancer. Breast. 2004;13:383–388. doi: 10.1016/j.breast.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Pardal R, Molofsky AV, He S, Morrison SJ. Stem cell self-renewal and cancer cell proliferation are regulated by common networks that balance the activation of proto-oncogenes and tumor suppressors. Cold Spring Harb Symp Quant Biol. 2005;70:177–185. doi: 10.1101/sqb.2005.70.057. [DOI] [PubMed] [Google Scholar]
- 6.Pietersen AM, Evers B, Prasad AA, Tanger E, Cornelissen-Steijger P, Jonkers J, et al. Bmi1 regulates stem cells and proliferation and differentiation of committed cells in mammary epithelium. Curr Biol. 2008;18:1094–1099. doi: 10.1016/j.cub.2008.06.070. [DOI] [PubMed] [Google Scholar]
- 7.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008 doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datta S, Hoenerhoff MJ, Bommi P, Sainger R, Guo WJ, Dimri M, et al. Bmi-1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res. 2007;67:10286–10295. doi: 10.1158/0008-5472.CAN-07-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoenerhoff MJ, Chu I, Barkan D, Liu ZY, Datta S, Dimri GP, et al. BMI1 cooperates with H-RAS to induce an aggressive breast cancer phenotype with brain metastases. Oncogene. 2009;28:3022–3032. doi: 10.1038/onc.2009.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimri GP. What has senescence got to do with cancer? Cancer Cell. 2005;7:505–512. doi: 10.1016/j.ccr.2005.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dimri GP, Martinez JL, Jacobs JJ, Keblusek P, Itahana K, Van Lohuizen M, et al. The Bmi-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 2002;62:4736–4745. [PubMed] [Google Scholar]
- 12.Itahana K, Zou Y, Itahana Y, Martinez JL, Beausejour C, Jacobs JJ, et al. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 2003;23:389–401. doi: 10.1128/MCB.23.1.389-401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guney I, Wu S, Sedivy JM. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a) Proc Natl Acad Sci USA. 2006;103:3645–3650. doi: 10.1073/pnas.0600069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo WJ, Datta S, Band V, Dimri GP. Mel-18, a polycomb group protein, regulates cell proliferation and senescence via transcriptional repression of Bmi-1 and c-Myc oncoproteins. Mol Biol Cell. 2007;18:536–546. doi: 10.1091/mbc.E06-05-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowak K, Kerl K, Fehr D, Kramps C, Gessner C, Killmer K, et al. BMI1 is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. Nucleic Acids Res. 2006;34:1745–1754. doi: 10.1093/nar/gkl119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yadav AK, Sahasrabuddhe AA, Dimri M, Bommi PV, Sainger R, Dimri GP. Deletion analysis of BMI1 oncoprotein identifies its negative regulatory domain. Mol Cancer. 2010;9:158. doi: 10.1186/1476-4598-9-158. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;49:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- 18.Nakayama KI, Nakayama K. Ubiquitin ligases: cell cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 19.Skaar JR, Pagano M. Control of cell growth by the SCF and APC/C ubiquitin ligases. Curr Opin Cell Biol. 2009;21:816–824. doi: 10.1016/j.ceb.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuchs SY, Spiegelman VS, Kumar KG. The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene. 2004;23:2028–2036. doi: 10.1038/sj.onc.1207389. [DOI] [PubMed] [Google Scholar]
- 21.Skaar JR, D'Angiolella V, Pagan JK, Pagano M. SnapShot: F Box Proteins II. Cell. 2009;137:1358. doi: 10.1016/j.cell.2009.05.040. [DOI] [PubMed] [Google Scholar]
- 22.Tang W, Li Y, Yu D, Thomas-Tikhonenko A, Spiegelman VS, Fuchs SY. Targeting beta-transducin repeat-containing protein E3 ubiquitin ligase augments the effects of antitumor drugs on breast cancer cells. Cancer Res. 2005;65:1904–1908. doi: 10.1158/0008-5472.CAN-04-2597. [DOI] [PubMed] [Google Scholar]
- 23.Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc Natl Acad Sci USA. 1999;96:6273–6278. doi: 10.1073/pnas.96.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, et al. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998;1:565–574. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 26.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 27.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glinsky GV. “Stemness” genomics law governs clinical behavior of human cancer: implications for decision making in disease management. J Clin Oncol. 2008;26:2846–2853. doi: 10.1200/JCO.2008.17.0266. [DOI] [PubMed] [Google Scholar]
- 29.Agherbi H, Gaussmann-Wenger A, Verthuy C, Chasson L, Serrano M, Djabali M. Polycomb mediated epigenetic silencing and replication timing at the INK4a/ARF locus during senescence. PloS one. 2009;4:5622. doi: 10.1371/journal.pone.0005622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 32.Dimri G, Band H, Band V. Mammary epithelial cell transformation: insights from cell culture and mouse models. Breast Cancer Res. 2005;7:171–179. doi: 10.1186/bcr1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chatoo W, Abdouh M, David J, Champagne MP, Ferreira J, Rodier F, et al. The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J Neurosci. 2009;29:529–542. doi: 10.1523/JNEUROSCI.5303-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. 2009;58:1312–1320. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimri M, Naramura M, Duan L, Chen J, Ortega-Cava C, Chen G, et al. Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: c-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res. 2007;67:4164–4172. doi: 10.1158/0008-5472.CAN-06-2580. [DOI] [PubMed] [Google Scholar]
- 37.Dimri GP, Itahana K, Acosta M, Campisi J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol Cell Biol. 2000;20:273–285. doi: 10.1128/mcb.20.1.273-285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kia SK, Gorski MM, Giannakopoulos S, Verrijzer CP. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol Cell Biol. 2008;28:3457–3464. doi: 10.1128/MCB.02019-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dimri M, Bommi PV, Sahasrabuddhe AA, Khandekar JD, Dimri GP. Dietary omega-3 polyunsaturated fatty acids suppress expression of EZH2 in breast cancer cells. Carcinogenesis. 2010;31:489–495. doi: 10.1093/carcin/bgp305. [DOI] [PMC free article] [PubMed] [Google Scholar]