

Abstract
Adenoviruses with deletion of E1b have been used in clinical trials to treat cancers that are resistant to conventional therapies. The efficacy of viral replication within cancer cells determines the results of oncolytic therapy, which remains poorly understood and requires further improvement. In this report, we show that adenoviruses induce autophagy by increasing the conversion of LC3-I to LC3-II and the formation of the Atg12-Atg5 complex. Inhibition of autophagy with 3-methyladenine (3MA) resulted in a decreased synthesis of adenovirus structural proteins, and thereby a poor viral replication; promotion of autophagy with rapamycin increased adenovirus yield. This study indicates that adenovirus-induced autophagy correlates positively with virus replication and oncolytic cell death, and that autophagy may generate nutrients that can be used for building viral progeny particles. These results further suggest that chemotherapeutic agents that increase cancer cell autophagy may improve the efficacy of oncolytic virotherapy.
Keywords: autophagy, adenovirus, cancer, oncolysis, virotherapy
Introduction
Adenoviruses (Ads) with E1b deletion can selectively replicate in cancer cells and cause oncolysis. Oncolytic viruses have been used in clinical trials and cancer treatments (Crompton and Kirn, 2007; Garber, 2006; Koski et al., 2010); however, as single agents, the antitumoral effects of oncolytic Ads have been somewhat disappointing in preclinical and clinical trials (Kirn et al., 2001; McCormick, 2003; Yamamoto and Curiel, 2010; Zhao et al., 2003). We previously reported that intratumoral injection of viruses could efficiently inhibit small human tumor growth in an animal model, but could not efficiently repress large tumors (Zhao et al., 2003). Many clinical studies have shown that the oncolytic approach alone could not efficiently destroy tumors in patients, especially for large tumors (Kirn et al., 2001; McCormick, 2003). Thus, oncolytic virus replication and spread are likely restricted in the larger tumor mass. This restriction may be a major hurdle limiting the efficacy of virotherapy. We believe that increasing the efficacy of viral replication within cancer cells will improve the results of oncolytic therapy.
Autophagy is a regulated process of degradation and recycling of cellular constituents, this process is important in organelle turnover and the bioenergetics management of starvation (Levine and Klionsky, 2004; Ohsumi, 2001). Autophagy starts with formation of double-membrane vesicles containing cytoplasm and organelles, a structure known as autophagosome. Ultimately, autophagosomes fuse with lysosomes where the bulk cytoplasmic content undergoes degradation, resulting in the liberation of amino acids and fatty acids that can be reused by cells (Baehrecke, 2005; Klionsky, 2008; Klionsky and Emr, 2000). During autophagy, several autophagy (Atg) proteins have been implicated in autophagosome formation. Atg5 and Atg12 form a complex that is required to recruit other proteins to the autophagosomal membrane. LC3 is the mammalian equivalent of yeast Atg8. The precursor form of LC3 is post-modified into two forms, LC3-I and LC3-II. LC3-I is localized in the cytosol and LC3-II in autophagosomal membranes. LC3-II can be used to estimate the abundance of autophagosomes before they are destroyed through fusion with lysosomes. Thus, Atg5/Atg12 complex formation, and the LC3-II conversion and integration into autophagosomal membrane, are the major hallmarks of activation of the autophagic process.
Some reports have suggested that autophagy has dual roles, acting as a survival mechanism and as a caspase-independent form of programmed cell death (Gozuacik and Kimchi, 2007; Wang et al., 2006; Yu et al., 2004). Autophagy is also involved in combating infection by breaking down pathogens (Gutierrez et al., 2004). However, some viruses subvert the autophagic pathway to promote their own replication (Jackson et al., 2005). The role of autophagy in Ad-mediated oncolytic cell death has not been well characterized. Kondo’s group has reported that hTERT-Ad, a conditionally replicating Ad regulated by the human telomerase reverse transcriptase promoter, killed cancer cells by inducing autophagy instead of apoptosis, and proposed that autophagy is an antitumoral effect, not a host defensive mechanism against viral replication (Ito et al., 2006). Their study was supported by a report showing that brain tumor cells were susceptible to Ad-mediated cell death via autophagy in vitro and in vivo (Jiang et al., 2007). In contrast, Baird et al (Baird et al., 2008) have reported that autophagy acts as a cell survival response to Ad infection in ovarian cancer, as an inhibitor of autophagy decreased cell survival. Thus, the autophagic functions in viral oncolytic replication need further investigation.
In this report, we have applied wild-type (wt) Ad5, Adhz60 (with deletion of E1b) (Rao et al., 2004), and AdlacZ (with deletion of both E1a and E1b, and replication defective) to study Ad-induced autophagy. We observed that Ad5 and Adhz60 can induce autophagy, but AdlacZ cannot. Ad5 and Adhz60 promote the conversion of LC3-I to LC3-II and formation of the Atg12-Atg5 complex. However, these processes were more pronounced in cells infected with Ad5, which expresses both E1a and E1b, suggesting that the E1b gene deleted in Adhz60 is not essential for autophagy but enhances it. We show that inhibition of autophagy decreases viral protein production and virus-induced autophagy correlates positively with virus replication. Our results suggest that chemotherapeutic agents that increase cancer autophagy may improve the efficacy of Ad-mediated oncolysis.
Results
Adenoviruses induce autophagy activation
To study Ad-triggered autophagy in lung cancer cells, we first applied the Gaussia luciferase (GLUC)-based assays to monitor the autophagic signaling pathway (Ketteler and Seed, 2008). This autophagy assay specifically measures the proteolytic cleavage of LC3 by the autophagy-activated protease Atg4B. In this system, the LC3 gene is linked with β-actin and GLUC genes to form the tripartite Actin-LC3-GLUC in the plasmid pEAK12-Actin-LC3-dNGLUC (Ketteler and Seed, 2008). During autophagy, activation of Atg4B cleaves at LC3 in the tripartite sensor protein Actin-LC3-GLUC and releases GLUC, which is secreted to the culture medium. The levels of released GLUC indicate the degree of autophagy activation (Ketteler and Seed, 2008). In our experiment, A549 lung cancer cells were transfected with pEAK12-Actin-LC3-dNGLUC following infection with Ad5 (wt) or Adhz60 (deleted-E1b) at a multiplicity of infection (MOI) value of 4 or AdlacZ (deleted-E1a and E1b) at an MOI of 10. We used a higher MOI for AdlacZ to compensate for the fact that this virus does not replicate. We observed that both Ad5 and Adhz60 increased GLUC activity in a time-dependent manner, and Ad5 induced significantly higher GLUC activity in comparison with that of Adhz60. At 36 h post-infection, Ad5 increased the Relative Luminescence Units (RLU) to 230,000, but Adhz60 increased to only 80,000 RLUs. The GLUC activity levels from AdlacZ-infected cells were similar to that of mock treatment (Fig. 1). Similar results were obtained when AdlacZ was used at either at an MOI value of 4 or 20 (data not shown). These results were important in that both Ad5 and Adhz60 were able to trigger the activation of the autophagic pathway, and that the E1b gene (deleted in Adhz60) may be involved in enhancing this activity.
Fig. 1. Quantification of autophagy activation.
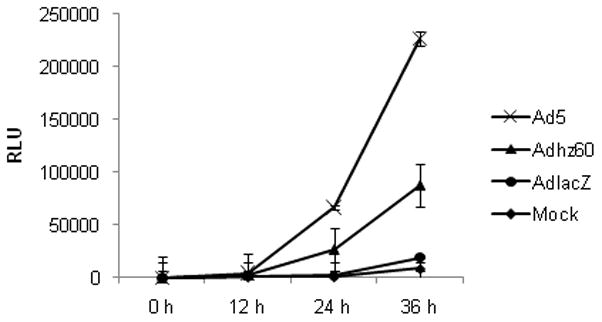
A549 cells were transfected with pEAK12-Actin-LC3-dNGLUC followed by no infection (mock) or infection with Ad5, Adhz60 (MOI=4), or AdlacZ (MOI=10). AdlacZ with deletion of E1 gene is used as a negative control vector incapable of replication. Secretion of LC3/GLUC was monitored over the time with the Renilla Luciferase kit (Promega). Each point represents the mean of three independent experiments ± standard deviation (SD; bars).
Adenoviruses induce autophagosome formation in lung cancer cells
One hallmark of autophagy activation is the formation of cellular autophagosome puncta containing LC3-II. LC3-II is the only known mammalian protein that stably binds to the autophagosomal membrane after the cytoplasmic form LC3-I is converted to LC3-II by site-specific proteolysis during autophagy. To investigate LC3-II incorporation into autophagosome, we used the GFP-tagged LC3 plasmid, pEGFP-LC3 (Kabeya et al., 2000). A549 lung cancer cells were transfected with pEGFP-LC3, and then infected with Ads as described above. Conversion of cytoplasm-diffuse GFP to membrane-associated GFP puncta was observed at 36 h by fluorescence microscopy (Fig. 2A), indicating that there was GFP-LC3-II incorporation into the autophagosomes. Ad5 and Adhz60 induced a significant number of cells with the GFP punctual pattern. The percentage of cells with the GFP punctuate pattern was calculated as punctuate GFP cell per total GFP cell percentage (Fig. 2B). Ad5 induced a higher percentage of cells (76%) with the GFP punctual pattern than Adhz60 (58%). In contrast, cells infected with AdlacZ exhibited a diffuse cytoplasmic pattern or low percentage (6%) of cells with this punctual pattern.
Fig. 2. Autophagy activation and integration of LC3 into autophagosome.
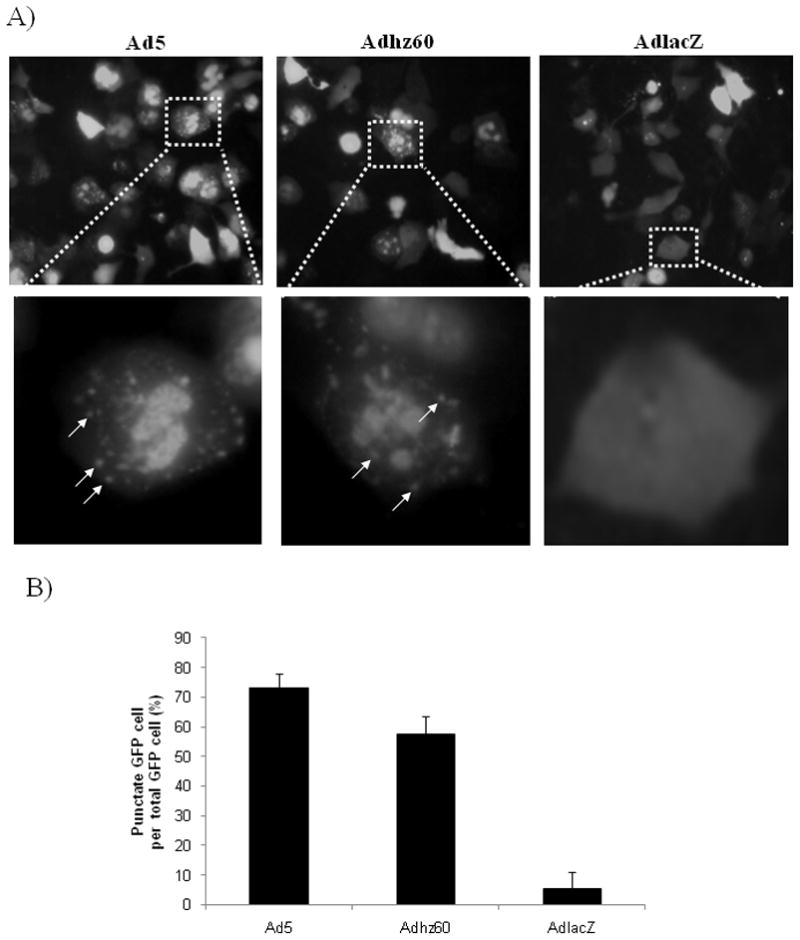
A549 cells were transfected with pEGFP-LC3 followed by infection with Ad5, Adhz60, or AdlacZ. (A) Integration of GFP-LC3 into the autophagosome is depicted by punctate structures (indicated by arrows) and was analyzed by fluorescence microscopy at 36 h p.i. Images were taken with Kodak MDS 290 software (magnification ×40). (B) The percentage of cells with punctate GFP-LC3 fluorescence was calculated relative to all GFP-LC3-positive cells. A representative experiment is shown from three performed.
Ad E1a and E1b activate LC3 conversion and Atg12-Atg5 complex formation
In the above experiments, we found that Ad5 induced higher autophagy activation and more autophagosome formation than Adhz60. At the protein level, we investigated whether E1b deletion in Adhz60 may result in a less efficient conversion of LC3-1 to LC3-II and the formation of the Atg12-Atg5 complex, which are other hallmarks of autophagy. To study this, A549 cells were infected with Ads, and cellular proteins were collected at 0-, 24-, 48-, and 72-h post-infection. An immunoblotting assay revealed the two active species of LC3, the upper band corresponding to LC3-I and lower band corresponding to LC3-II (Kabeya et al., 2000). Significant conversion of LC3-I to LC3-II was readily detected after Ad5 infection; nearly all LC3-I was converted to LC3-II after 48 h post-infection (Fig. 3). There was partial conversion from LC3-I to II in cells infected with Adhz60. For AdlacZ-treated cells, there was no detectable change of relative amounts of LC3-I and LC3-II. The apparent molecular weight of the LC3-I and LC3-II proteins are different to the all known values of adenoviral proteins.
Fig. 3. Expression of the classical autophagy hallmarks.
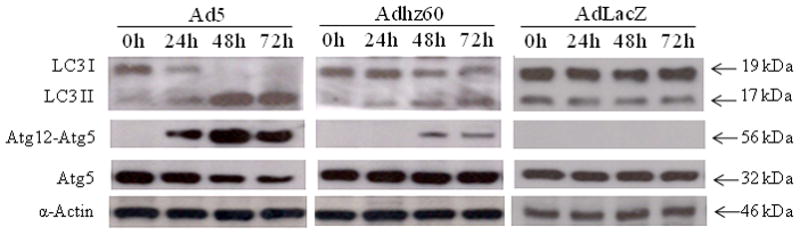
A549 cells were infected with Ad5, Adhz60, or AdLacZ. LC3 and Atg5 expression and modification were analyzed in a time-course assay. LC3-I and LC3-II were detected with a pAb anti-LC3. To detect Atg5 or Atg12-Atg5 complex, a pAb anti-Atg5 was used.
We next analyzed Atg5 expression and its conjugation with Atg12. Conjugation of Atg5 with Atg12 is essential in the early stage of autophagy for the elongation of the isolation membrane during autophagosome formation (Mizushima et al., 2001). We observed that Ad5 induced a considerable increase of Atg12-Atg5 complex after infection (Fig. 3). Infection with Adhz60 also caused the formation of the Atg12-Atg5 complex, but at lower levels compared with Ad5. There was no change of these proteins and complexes in AdlacZ-infected cells (Fig. 3). These data suggest that the lack of E1b resulted in a less efficient conversion of LC3-1 to LC3-II and formation of the Atg12-Atg5 complex.
Pharmacological inhibition of autophagy decreases the yield of adenoviruses
Autophagy may be an intracellular host defensive mechanism against viruses, or it can be used by viruses for their own benefit of efficient replication (Colombo, 2005). We examined the effect of autophagy on Ad replication. Since 3MA inhibits autophagy by suppressing the formation of the pre-autophagosomal structure (Kondo et al., 2005; Shintani and Klionsky, 2004), A549 cells were infected with Ad5 or Adhz60 in the absence or presence of 3MA at a concentration of 10 mM. Viruses were harvested after infection and titered on HEK-293 cells. We observed that the autophagy inhibitor 3MA reduced the viral yields about 10-fold for both Ad5 and Adhz60 (Fig. 4A). The differences were statistically significant, as shown by the Student t-test.
Fig. 4. Effect of 3-methyladenine (3MA) on adenovirus yield and structural protein production.
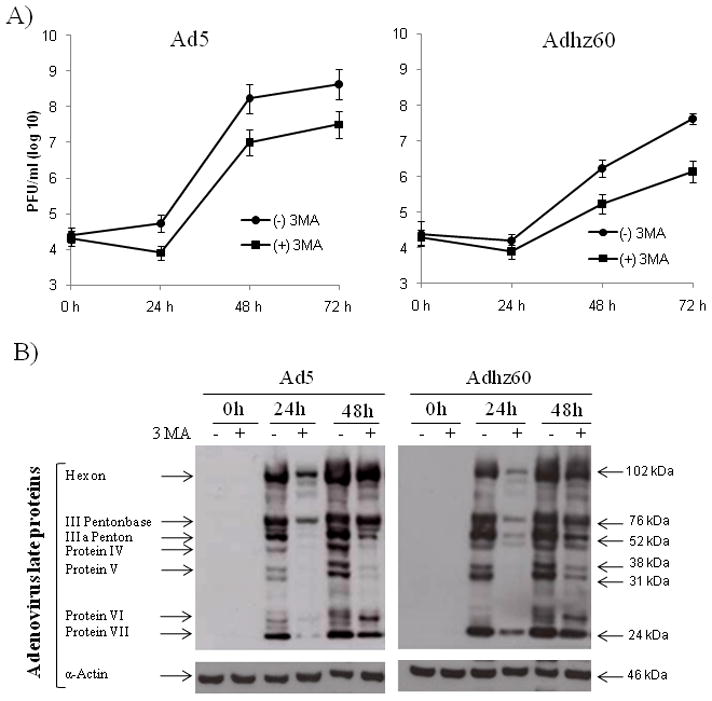
A549 cells were infected with Ad5 or Adhz60 in absence or presence of 3MA. Cells were harvested at indicated time points. (A) For titering, supernatants were serially diluted to determine the titers, plaque forming unit (PFU), in HEK-293 cells. Each point represents the mean of three independent experiments ± standard deviation (SD; bars; P < 0.05). (B) Ad structural proteins were detected with rabbit-antihuman Ad protein virions. One experiment is shown from three performed.
It is well known that autophagy degrades intracellular components, such as organelles and long-lived proteins, to liberate amino acids and fatty acids that can be reused within cells, especially under starvation condition (Baehrecke, 2005; Klionsky, 2008; Klionsky and Emr, 2000). Therefore, it is possible that Ads may promote autophagy to provide elements for viral particle production. If so, inhibition of autophagy should decrease or delay viral protein synthesis. We analyzed the production of viral structure proteins in cells treated with the autophagy inhibitor 3MA. The production of viral structural proteins of both Ad5 and Adhz60 at 24 h was strongly inhibited in cells treated with 3MA (Fig. 4B). The decreased viral protein production, caused by 3MA treatment, correlates with inefficient virus replication as shown in Figure 4A. These results suggest that autophagy plays a critical role in viral structure protein synthesis, likely by degrading intracellular components necessary for building progeny virus particles.
Treatment of Ad5-infected cells with 3MA prevents autophagosome formation and inhibits the conversion of LC3-I to LC3-II
We investigated whether 3MA could prevent the conversion of GFP-LC3-1 to GFP-LC3-II induced by Ad5. A549 cells were transfected with pEGFP-LC3 followed by infection with Ad5. The treated cells were then cultured in the absence or presence of 3MA at a concentration of 10 mM. We found that 3MA repressed the conversion of LC3-I to LC3-II, resulting in a diffuse GFP-LC3 pattern (Fig. 5A). We further investigated whether 3MA might inhibit conversion of LC3-I to LC3-II at the protein level. We analyzed the effect of 3MA in cells infected with Ad5, because this virus induced significant conversion of LC3-I to LC3-II. A549 cells were infected with Ad5 in the presence of 3MA (Fig. 5C). The conversion of LC3-I to LC3-II was significantly inhibited by 3MA in Ad5-infected cells compared to the result in the absence of 3MA (Fig. 3). The results indicate that inhibition of autophagy with 3MA results in decreased viral replication.
Fig. 5. Inhibition or induction of autophagosome formation and inhibition of LC3-I to LC3-II conversion.
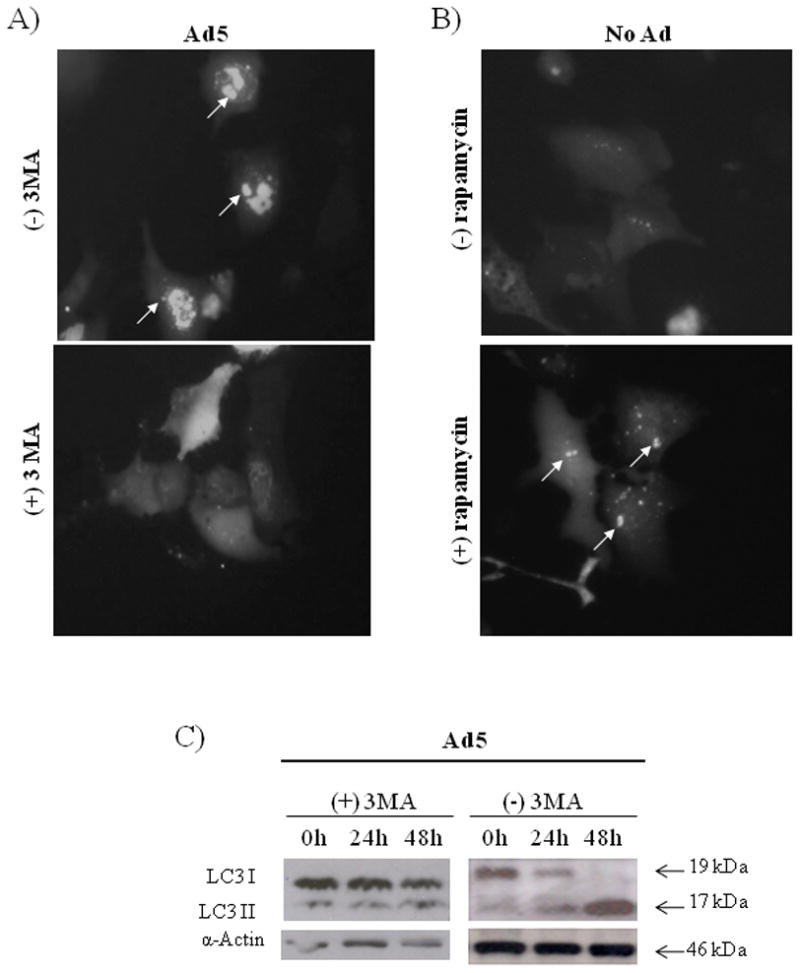
(A) A549 cells were transfected with pEGFP-LC3 following infection with Ad5 (MOI=4) in absence or presence of 3MA (10 mM). (B) A549 cells were transfected with pEGFP-LC3 followed by no treatment or treatment with rapamycin (50 nM). Integration of GFP-LC3 into the autophagosome is depicted by punctate structures (indicated by arrows) and was analyzed by fluorescence microscopy at 36 h p.i. Images were taken with Kodak MDS 290 software (magnification, ×40). (C) A549 cells were infected with Ad5 in presence of 3MA (10 mM). LC3 expression was analyzed in a time-course assay. LC3-I and LC3-II were detected with a pAb anti-LC3. A representative experiment is shown from three performed.
Role of autophagy in adenovirus replication and oncolysis
To further investigate the role of autophagy in oncolytic therapy, we compared the effect of 3MA (autophagy inhibitor) and rapamycin (autophagy inducer) on virus replication and cancer cell viability. First, we verified the autophagy induction function of rapamycin in our experimental conditions. Rapamycin induces autophagy by inhibiting the mammalian target of rapamycin (mTOR), an autophagy-associated molecule (Kondo et al., 2005; Shintani and Klionsky, 2004). A549 cells were transfected with pEGFP-LC3 followed by treatment with rapamycin at a concentration of 50 nM. Conversion of the cytoplasm-diffused pEGFP-LC3-I to membrane-associated pEGFP-LC3-II puncta was observed by fluorescence microscopy at 36 h (Fig. 5B), indicating that rapamycin indeed induced autophagy in A549 cells.
A549 cells were infected in the presence or absence of 3MA or rapamycin. The virus progeny produced in A549 cells were determined at day 3 after infection. In the presence of 3MA, there was significant inhibition of Ad5 and Adhz60 yields: 30- and 10-fold decrease, respectively (Fig. 6A). We also observed that rapamycin treatment increased the virus titers of Ad5 and Adhz60 2- to 3-fold higher than that of untreated cells (Fig. 6A). The results, as well as the results shown in Figs. 4 and 5, suggest that autophagy has a positive effect on Ad replication.
Fig. 6. Effect of pharmacological inhibition and stimulation of autophagy on adenovirus replication and oncolysis.

(A) A549 cells were infected with Ad5 or Adhz60 in the presence or absence of 3MA (10 mM) or rapamycin (50 nM). Seventy-two h later, titers were determined by serial dilutions of supernatants and titers were measured by standard infection units. Cytotoxicity was analyzed by a MTT assay at 72 h after 3MA (B) or rapamycin (C) treatment. Each point represents the mean of three independent experiments ± (SD; bars).
Next, MTT assay was used to determine the relative cell survival. This assay showed that cells infected with Ad5 or Adhz60 at an MOI of 4 resulted in 28% and 63% viability at 72 h, respectively. Treatment with 3MA prevented cell death and increased the cell viability to 48% for Ad5 and 85% for Adhz60 (Fig. 6B). Unlike 3MA, rapamycin did not significantly affect the cell survival rate (Fig. 6C). Thus, inhibition of autophagy significantly reduces virus replication and decreases the oncolytic effect.
Oncolytic adenovirus induces activation of autophagy in different types of cancer cells
Since the ability of oncolytic Ads to induce autophagy in A549 lung cancer cells was demonstrated, we studied whether these Ads could induce the activation of autophagy in other types of cancer cell lines. Therefore, Hela cervical cancer cells, HCT116, and RKO colon cancer cells, or H1299 lung cancer cells were transfected with pEAK12-Actin-LC3-dNGLUC followed by no infection (mock) or infection with Ad5 (wt) or Adhz60 (deleted-E1b) at an MOI of 4. GLUC activity was measured 36 h after infection; mock level was used as a baseline. It was observed that both Ad5 and Adhz60 induced activation of autophagy in Hela, RKO, and H1299 cells; H1299 showed the highest autophagy activation (Fig. 7). Ad5 also induced autophagy activation in HTC116 cells. Even though our experiment did not attempt to address optimal conditions for induction of autophagy by virus infection for each of the cell lines, the results indicate that Ads can generally induce autophagy in cancer cells.
Fig. 7. Activation of autophagy in different types of cancer cells.
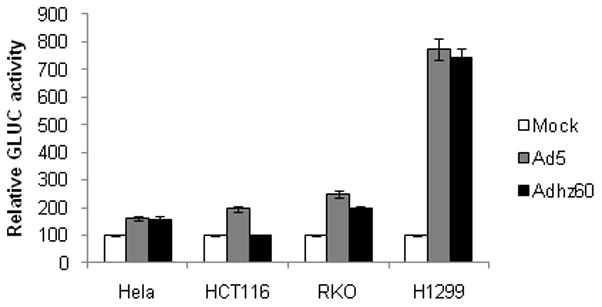
Hela, HCT116, RKO, or H1299 cancer cell lines were transfected with pEAK12-Actin-LC3-dNGLUC followed by no infection (mock) or infection with Ad5 or Adhz60 (MOI=4). Secretion of LC3/GLUC was monitored at 36 h after infection with the Renilla Luciferase kit (Promega). Each point represents the mean of three independent experiments ± standard deviation (SD; bars).
Discussion
Ads have been genetically modified for selectively replicating within cancer cells and causing oncolysis. The advantage of virotherapy is that the released progeny viruses further spread infection until all cancer cells are destroyed. This is true for cancer cells in tissue culture. Even when less than 1 percent of the cancer cells are initially infected, all cancer cells are eventually killed in a few days. However, animal experiments and clinical trials have indicated that virus replication and spread in larger tumor masses are restricted, and that the restricted replication and spread may be major hurdles limiting the efficacy of virotherapy (Kirn, 2001; McCormick, 2003; Yamamoto and Curiel, 2010; Zhao et al., 2003). Recent studies suggest that Ads have the ability to induce autophagy (Baird et al., 2008; Ito et al., 2006; Jiang et al., 2007; Jiang et al., 2008). Thus, autophagy may be used to promote virus replication and improve oncolysis.
In this study, we have applied wild-type (wt) Ad5, Adhz60 (with deletion of E1b) (Rao et al., 2004), and AdlacZ (with deletion of both E1a and E1b) to study Ad-induced autophagy. We observed that autophagy is induced by replication-competent Ad5 and Adhz60, but not by AdlacZ. We also found that Ad5 and Adhz60 promote the conversion of LC3-I to LC3-II and the formation of the Atg12-Atg5 complex two—hallmarks of autophagy. These processes were more pronounced in cells infected with Ad5, which expresses both E1a and E1b, than in cells infected with the E1b-deleted Adhz60. Thus, autophagy induction appears to be dependent on the E1a gene and viral replication, and the E1b gene may be involved in enhancing autophagy. We do not know the exact mechanisms by which Ad infection activates autophagy. Ad infection may initially trigger cellular autophagic responses that can be taken over by Ads for the benefit of viral replication. It is also possible that a virus-associated or activated protease activity may directly contribute to the conversion of LC3-I to LC3-II and activate the autophagy pathway.
To study the role of autophagy in Ad replication, we have applied 3MA to inhibit autophagy and rapamycin to promote the process. Both 3MA and rapamycin have been broadly used in autophagy studies (Kondo et al., 2005; Shintani and Klionsky, 2004). It is known that 3MA inhibits autophagy by suppressing the formation of the pre-autophagosomal structure. Rapamycin inhibits the mammalian target of rapamycin (mTOR), an autophagy-associated molecule, and is used in model studies to induce autophagy. Although we cannot exclude the possibility that 3MA and rapamycin may also affect other processes in the Ad-infected cells, we have shown that 3MA partially represses Ad-induced autophagy, and rapamycin alone can induce autophagy in treated cancer cells in our experimental set (Fig. 5). We observed that 3MA significantly reduced Ad5 and Adhz60 yields in A549 cancer cells and partially prevented Ad-caused cell death (Fig. 6A and B). Rapamycin increased Ad5 and Adhz60 titers 2- to 3-fold (Fig. 6A). Since Ad oncolytic replication is restricted in large tumor masses, promotion of autophagy with chemotherapeutic agents may have the potential to improve the efficacy of virotherapy.
To the best of our knowledge, this study shows, for the first time, that 3MA represses Ad structure protein synthesis (Fig. 4). Autophagy is a catabolic process involving the degradation of cellular components through the lysosomal machinery. Autophagy is a major mechanism by which a starving cell reallocates nutrients from unnecessary processes to more-essential processes, although this process also plays a number of other roles in normal cell growth, development, and homeostasis (Levine and Klionsky, 2004; Ohsumi, 2001). We have shown that replication-competent Ads strongly induce autophagy in cancer cells; inhibition of autophagy with 3MA partially represses viral protein production and decreases virus titers, and the autophagy inducer, rapamycin, increases virus replication. These results have indicated that virus-induced autophagy correlates positively with virus replication. Since autophagy has a major function in the generation of nutrients for starving cells, and inhibition of autophagy specifically represses viral structural protein synthesis (Fig. 4), we hypothesize that autophagy may play a critical role in viral replication by degrading intracellular components for building progeny virus particles.
We also observed that replicative Ads can activate autophagy in other cancer cell lines, Hela, HCT116, RKO, and H1299, but the levels are different from cell to cell. This may reflect the different sensitivities of cancer cells to Ad-induced autophagy or that optimal conditions are not selected for each of the cell lines, such as transfection efficacy of plasmid and autophagy peaking time points. A new publication has shown that human adenovirus type 5 induces autophagy in glioma cells (Jiang et al., 2011). Thus, it is likely that oncolytic Ads are generally able to induce the activation of autophagy.
In summary, we found that wt Ad5 and E1b-deleted oncolytic Adhz60 induced autophagy in cancer cells, the conversion of LC3-I to LC3-II and the complex formation of Atg12-Atg5. Autophagy correlated positively with virus replication and oncolytic cell death. Based on these findings, we suggest that chemotherapeutic agents that increase cancer cell autophagy may promote the efficacy of oncolytic virotherapy. These findings may have clinical relevance in the development of oncolytic virotherapy.
Material and methods
Cell line and culture conditions
Human lung cancer cell lines A549 and H1299 were cultured in MEM-Alpha medium; human cervical cancer cell line Hela and human colon cancer cell line RKO were cultured in RPMI medium; and human colon cancer cell line HCT116 was cultured in McCoy’s medium at 37°C in a humid incubator with 5% CO2. All media were supplemented with 10% heat-inactivated fetal bovine serum (FBS) and penicillin/streptomycin (100 U/ml). All cell culture reagents were obtained from Gibco BRL (Bethesda, MD). Adherent cells were collected after trypsin treatment, and cell numbers were determined using a Neubauer cell counting chamber. All experiments were repeated at least three times.
Plasmids
The pEAK12-Actin-LC3-dNGLUC (autophagy sensor) was kindly provided by Dr. Brian Seed (Ketteler and Seed, 2008) and was used for transfection, as well as GFP-LC3 plasmid, which was generously donated by Dr. Tamotsu Yoshimori (Kabeya et al., 2000). Cells grown to 70% confluency were transfected with indicated plasmids using Lipofectamine 2000 (Invitrogen, 11668-019), according to the manufacturer’s protocol. For the luciferase assay and the GFP-LC3 puncta assay, cells were seeded at a density of 1.5 × 105 cells in 12-well tissue culture plates.
Adenoviral vectors
AdCMV-lacZ (AdlacZ) has E1 deletion and has the lacZ gene as a reporter. Adhz60 is an Ad vector with deletion of E1b gene (Rao et al., 2004). Ad5 (Adwt) was obtained from ATCC and does not have any deletions. Cells were infected with wt Ad5, Adhz60 (at an MOI of 4) or AdlacZ (at an MOI of 10) in FBS-free medium for 1 h. Infected cells were then untreated or treated with 3MA at 10 mM or rapamycin at 50 nM. We used a higher MOI of AdlacZ for compensation of its lack of replication ability. The autophagy results are the same when AdlacZ was used at MOIs of 4, 10, or 20. The cultures were collected at indicated time points. Ad titers were determined as follow: cells went through three cycles of freezing and thawing to release viruses from the cells. Lysates were serially diluted to determine the titers in HEK-293 cells by standard infective unit (IFU) measurement.
Gaussia luciferase assay
After transfection with the pEAK12-Actin-LC3-dNGLUC, supernatant samples were collected at indicated time points. GLUC activity was determined using the Renilla Luciferase kit (Promega, E2810). To avoid harvesting luciferase activity from detached cells, supernatants were spun at 14,000 rpm for 5 minutes. Ten microliters of supernatant were diluted (1:10) in 100 μl 1 × renilla lysis buffer. Twenty microliters of this mixture was added to 100 μl of renilla substrate prior to analysis in a luminescence plate reader.
GFP-LC3 puncta
Plasmid vector containing green fluorescent protein (GFP) linked to microtubule-associated protein 1 light chain 3 (LC3) (pGFP-LC3) was used to detect autophagosome formation in A549 cells caused by Ad5 or Adhz60. After infection with Ad5 or Adhz60, no infected or Ad-infected cells were cultured in the presence or absence of 3MA (10 mM) or rapamycin (50 nM). 36 h later, cells were fixed with 4% formaldehyde and then observed under a fluorescence microscope. Cells were classified as having a predominantly diffuse GFP stain or having numerous punctate structures representing autophagosomes. Images were taken with Kodak MDS 290 software with the 40X objective. The percentage of cells with GFP punctuate pattern was calculated as punctuate GFP cell per total GFP cell percentage.
Western blot analysis
After respective treatment, the cells were harvested and lysed in a radio immunoprecipitation assay (RIPA) buffer. Cell lysates were centrifuged, and protein concentration was determined by BIO-RAD DC protein assay (BIO-RAD, Hercules, CA). Equal amounts (50 ug) of total protein were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to Hybond-Polyvinylidene Difluoride (PVDF) membranes (Amersham, Arlington Heights, IL). The membranes were blocked with 5% dry milk in Tris-buffered saline–Tween 20 (TBST), probed with the following primary antibodies: rabbit polyclonal anti-LC3 (Novus Biologicals, NB100-2331); rabbit polyclonal anti-Atg5 (C-terminal) (Sigma-Aldrich, A0713); rabbit- antihuman Ad proteins virion (abcam, ab6982); and rabbit-antihuman-α-actin pAb to ensure equal loading of proteins. Next, the membranes were incubated with anti-mouse immunoglobulin (Ig) or anti-rabbit Ig, peroxidase-linked, species-specific whole antibody (Amersham, Piscataway, NJ). Chemiluminescent detection was performed with ECL reagents, according to the manufacturer’s instructions (Amersham).
MTT assay
Cell proliferation was assessed at three days after respective treatments by measuring the conversion of the tetrazolium salt (MTT) to formazan, according to the manufacturer’s instructions (Boehringer Mannheim, Indianapolis, IN). The supernatant from each plate was collected for measurement of absorbance at a wavelength of 570 nm. The results were expressed as the percentage of live cells.
Statistical Analysis
The results of the in vitro assays were analyzed with the Student t test for unpaired data using a one-way ordinary parametric analysis of variance. P < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by Award Numbers R01CA129975 (HSZ) from the National Cancer Institute, GMB081410 (KMM & HSZ) from the Kentucky Lung Cancer Research Program and Lung Cancer Research Foundation (JGG). AGG is recipient of a scholarship from the National Council of Science and Technology (CONACYT) of Mexico.
We are grateful to Dr. Brian Seed (Harvard Medical School, Boston, MA) for kindly providing the pEAK12-Actin-LC3-dNGLUC and pEAK12-Actin-flagDEVDG2-dNGLUC plasmids and Dr. Tamotsu Yoshimori (Osaka University, Japan) for kindly providing the EGFP-LC3 plasmid. We thank Margaret A. Abby and Nancy Alsip for editing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- Baird SK, Aerts JL, Eddaoudi A, Lockley M, Lemoine NR, McNeish IA. Oncolytic adenoviral mutants induce a novel mode of programmed cell death in ovarian cancer. Oncogene. 2008;27:3081–3090. doi: 10.1038/sj.onc.1210977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo MI. Pathogens and autophagy: subverting to survive. Cell Death Differ. 2005;12(Suppl 2):1481–1483. doi: 10.1038/sj.cdd.4401767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton AM, Kirn DH. From ONYX-015 to armed vaccinia viruses: the education and evolution of oncolytic virus development. Curr Cancer Drug Targets. 2007;7:133–139. doi: 10.2174/156800907780058862. [DOI] [PubMed] [Google Scholar]
- Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98:298–300. doi: 10.1093/jnci/djj111. [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A. Autophagy and cell death. Curr Top Dev Biol. 2007;78:217–245. doi: 10.1016/S0070-2153(06)78006-1. [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Ito H, Aoki H, Kuhnel F, Kondo Y, Kubicka S, Wirth T, Iwado E, Iwamaru A, Fujiwara K, Hess KR, Lang FF, Sawaya R, Kondo S. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J Natl Cancer Inst. 2006;98:625–636. doi: 10.1093/jnci/djj161. [DOI] [PubMed] [Google Scholar]
- Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F, Xu J, Kondo Y, Bekele BN, Colman H, Lang FF, Fueyo J. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007;99:1410–1414. doi: 10.1093/jnci/djm102. [DOI] [PubMed] [Google Scholar]
- Jiang H, White EJ, Gomez-Manzano C, Fueyo J. Adenovirus’s last trick: you say lysis, we say autophagy. Autophagy. 2008;4:118–120. doi: 10.4161/auto.5260. [DOI] [PubMed] [Google Scholar]
- Jiang H, White EJ, Rios-Vicil CI, Xu J, Gomez-Manzano C, Fueyo J. Human adenovirus type 5 induces cell lysis through autophagy and autophagy-triggered caspase activity. J Virol. 2011 doi: 10.1128/JVI.02032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketteler R, Seed B. Quantitation of autophagy by luciferase release assay. Autophagy. 2008;4:801–806. doi: 10.4161/auto.6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: what have we learned? Gene Ther. 2001;8:89–98. doi: 10.1038/sj.gt.3301377. [DOI] [PubMed] [Google Scholar]
- Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat Med. 2001;7:781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740–743. doi: 10.4161/auto.6398. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, Diaconu I, Nokisalmi P, Raki M, Rajecki M, Guse K, Ranki T, Oksanen M, Holm SL, Haavisto E, Karioja-Kallio A, Laasonen L, Partanen K, Ugolini M, Helminen A, Karli E, Hannuksela P, Joensuu T, Kanerva A, Hemminki A. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther. 2010;18:1874–1884. doi: 10.1038/mt.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- McCormick F. Cancer-specific viruses and the development of ONYX-015. Cancer Biol Ther. 2003;2:S157–160. [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- Rao XM, Tseng MT, Zheng X, Dong Y, Jamshidi-Parsian A, Thompson TC, Brenner MK, McMasters KM, Zhou HS. E1A-induced apoptosis does not prevent replication of adenoviruses with deletion of E1b in majority of infected cancer cells. Cancer Gene Ther. 2004;11:585–593. doi: 10.1038/sj.cgt.7700739. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Curiel DT. Current issues and future directions of oncolytic adenoviruses. Mol Ther. 2010;18:243–250. doi: 10.1038/mt.2009.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Lenardo MJ, Baehrecke EH. Autophagy and caspases: a new cell death program. Cell Cycle. 2004;3:1124–1126. [PubMed] [Google Scholar]
- Zhao T, Rao XM, Xie X, Li L, Thompson TC, McMasters KM, Zhou HS. Adenovirus with insertion-mutated E1A selectively propagates in liver cancer cells and destroys tumors in vivo. Cancer Res. 2003;63:3073–3078. [PubMed] [Google Scholar]