

Abstract
This article considers the fidelity of DNA replication performed by eukaryotic DNA polymerases involved in replicating the nuclear genome. DNA replication fidelity can vary widely depending on the DNA polymerase, the composition of the error, the flanking sequence, the presence of DNA damage and the ability to correct errors. As a consequence, defects in processes that determine DNA replication fidelity can confer strong mutator phenotypes whose specificity can help determine the molecular nature of the defect.
Keywords: Fidelity, DNA polymerase, DNA replication, Mutator phenotypes, Microsatellite instability
1. Introduction
The fidelity with which genetic information is replicated depends on the ability of DNA polymerases to select correct nucleotides rather than incorrect or damaged nucleotides for incorporation, without adding or deleting nucleotides. Polymerase selectivity is a major determinant of fidelity both at the replication fork and during synthesis to repair DNA damage. Mismatches generated during DNA chain elongation can be removed by 3′-exonuclease activity of the major replicative polymerases, thereby enhancing fidelity. If a mismatch escapes proofreading, DNA mismatch repair (MMR) can excise the replication error in the nascent strand and replace it with the correct sequence. Under normal circumstances, nucleotide selectivity, proofreading and MMR operate in series to replicate the genome with very high fidelity, thereby contributing to low spontaneous mutation rates (Fig. 1). Consequently, defects in any of these three processes can increase mutation rates, and this mutator phenotype can potentially be a driving force for cancer.
Fig. 1.

Contribution of nucleotide selectivity, proofreading and DNA mismatch repair to DNA replication fidelity. The figure depicts the wide-ranging contributions of three major processes that act in series to determine DNA replication fidelity. The colored brackets illustrate the investment each process makes to the overall fidelity. Conditions that comprise each process are shown on the right.
When lesions in DNA generated by endogenous cellular metabolism or exposure to chemical or physical insults from the external environment are not removed prior to replication, they sometimes distort the DNA helix and impede replication fork progression. In such circumstances, cell survival can be enhanced by several different DNA transactions. One such transaction is translesion synthesis (TLS), a process that allows lesions to be tolerated until they can be repaired. TLS is catalyzed by highly specialized, exonuclease-deficient DNA polymerases whose catalytic properties, including fidelity, are quite different than those that perform the bulk of replication. This review focuses on the eukaryotic replicative and TLS DNA polymerases that have critical roles in accurately and efficiently replicating the nuclear genome. Readers interested in DNA synthesis catalyzed in mitochondria, or in additional DNA polymerases that function in DNA repair, can consult other reviews [1–4]. Emphasis here is on the fidelity of replication, how it can be reduced to result in mutator phenotypes associated with cancer, and differences in replication error specificity that may have diagnostic value when considering replication infidelity as a source of point mutations arising in tumor cells.
2. Multiple DNA polymerases and their functions
DNA polymerases are classified by sequence homology into seven families (A, B, C, D, X, Y and RT). Most organisms encode multiple polymerases, often including several members of the same family. The human genome encodes 16 DNA polymerases, and more than half are involved in replicating the nuclear genome (Table 1). Four (Pols α, δ, ε and ζ) are B family members, four (Pols η, κ, ι and Rev1) are Y family members; two (Pols θ and ν) are A family members. These enzymes can have multiple functions, and some uncertainty remains as to exactly where and when they operate in vivo. Nonetheless, their primary functions are currently thought to be in replication and TLS (Table 1).
Table 1.
Human DNA template-dependent DNA polymerases.
| DNA polymerase | Family | Catalytic subunit
|
3′ to 5′ Exonuclease | Proposed Primary function | |||
|---|---|---|---|---|---|---|---|
| Mass (kDa) | Human gene | Location | Yeast (Sc) gene | ||||
| α (alpha) | B | 165 | POLA | Xp22.1–p21.3 | POL1 (CDC17) | No (Primase) | Okazaki fragment initiation |
| δ (delta) | B | 125 | POLD 1 | 19q13.3 | POL3 (CDC2) | Yes | Lagging strand replication |
| ε (epsilon) | B | 255 | POLE | 12q24.3 | POL2 | Yes | Leading strand replication |
| ξ (zeta) | B | 353 | POLZ (REV3) | 6q21 | REV3 | No | Translesion synthesis, SHM |
| η (eta) | Y | 78 | POLH (RAD30, RAD30A, XPV) | 6p21.1 | RAD30 | No | Translesion synthesis |
| SHM | |||||||
| ι (iota) | Y | 80 | POLI (RAD30B) | 18q21.1 | none | No | Translesion synthesis, SHM? |
| κ (kappa) | Y | 76 | POLK (DINB1) | 5q13 | none | No | Translesion synthesis |
| ν (nu) | A | 100 | POLN | 4p16.3 | none | No | Translesion synthesis? |
| θ (theta) | A | 198 | POLNQ | 3q13.3 | none | No | Translesion synthesis, SHM? |
2.1. The major replicative DNA polymerases
Pols α, δ and ε perform the vast majority of nuclear DNA replication. Several models have been put forth for their division of labor at the nuclear DNA replication fork [5,6]. These models all share the idea that initiation of replication at origins and of each Okazaki fragment on the lagging strand template requires the RNA primase activity associated with Pol α to synthesize RNA primers of ~10 bases (Fig. 2A). Pol α then extends these primers by incorporating about 20–30 deoxynucleotides. Once DNA chains are initiated, current evidence [5,7] suggests that the leading strand template may be copied primarily by Pol ε, a highly processive enzyme with an associated proofreading exonuclease activity. Pol δ copies primarily the lagging strand template, generating a series of Okazaki fragments of approximately 200–300 base pairs each that are ultimately processed to remove RNA primers and permit ligation [8].
Fig. 2.
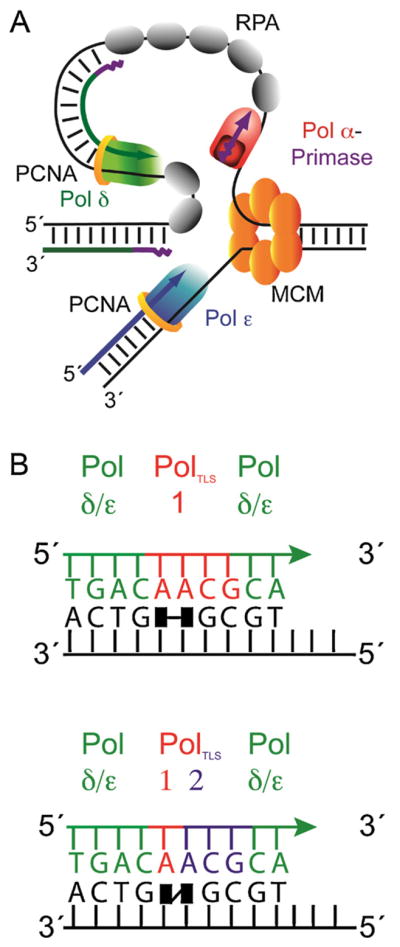
Models for the replication fork and for translesion synthesis. (A) Simplified schematic depicting one current model for the roles of the three major replicative DNA polymerases at the eukaryotic DNA replication fork. (B) The 1-TLS polymerase model (left) is illustrated for bypass of a thymine–thymine dimer (black squares with horizontal line). This model shows that one polymerase is responsible for bypass of the dimer, including insertion opposite the lesion and extension of the primer terminus (red bases) followed by synthesis by a replicative DNA polymerase (green). The 2-TLS polymerase model (right) is illustrated for bypass of thymine–thymine 6–4 photoproduct (black squares with diagonal line). In this example, one polymerase inserts a nucleotide opposite the 3′ T and a second polymerase extends the primer-terminus by three nucleotides, to then allow continuation of synthesis by a major replicative polymerase (green).
2.2. The TLS polymerases
Although the above situation may apply to replicating undamaged DNA, enzymology becomes more complicated when the fork encounters lesions that distort the DNA helix (Fig. 2B). Family B members like Pols α, δ and ε depend heavily on normal helix geometry for efficient and accurate synthesis [1,9], such that helix distorting lesions can slow or prevent replication fork progression. To solve this problem, a stalled replication fork can elicit one or more switches among DNA polymerases capable of bypassing lesions. The specialized TLS polymerases include Pols ζ, η, κ, ι and Rev1 (Y family) and Pols θ and ν (A family). These polymerases can incorporate nucleotides opposite lesions and/or extend the resulting primer terminus for one or more nucleotides, thus creating primer-templates that can be used by the major replication enzymes [10,11]. The number of switches involved in TLS, and the identity and number of polymerases required, can vary depending on the lesion, of which there are many types that differ by composition and structure [12]. The number of switches also depends on the substrate preferences of the various polymerases, which also vary widely and can partially overlap. The location and timing of TLS may vary, sometimes occurring at the fork during ongoing DNA replication, sometimes occurring during post-replication gapfilling synthesis, e.g., catalyzed by Pol ζ [13], or perhaps occurring during excision repair (e.g., Pol κ). In fact, eukaryotic DNA polymerases other than those listed in Table 1 also have been implicated in TLS, including two family X members Pol β and Pol λ that fill short gaps during base excision repair and non-homologous end joining of double strand breaks in DNA. Pol β has been shown to be involved with bypass of an abasic site and a d(GpG)-cisplatin adduct [14,15] and PCNA was found to efficiently stimulate the bypass of an abasic site by Pol λ [2,16,17]. Readers interested in the substrate specificities of the TLS polymerases and mechanisms of polymerase switching involving post-translational modifications of polymerases and their accessory proteins can consult any of several recent reviews on this topic [4,18,19].
3. The fidelity of DNA synthesis
The potential of replication infidelity to drive cancer via a mutator phenotype may depend heavily on the enzymatic source and the specificity of DNA biosynthetic errors. Studies of DNA synthesis fidelity in vitro reveal wide variations in error rates for the two main types of errors that DNA polymerases generate, single base pair substitutions and single base deletions (Fig. 3). These rates reflect nucleotide selectivity, which prevents errors from forming, and also exonucleolytic proofreading, which corrects mismatches during ongoing replication.
Fig. 3.

Eukaryotic DNA polymerase error rates for single base mutations. (A) Error rates for Homo sapiens Pol δ and S. cerevisiae polymerases α, δ and ε, for single base substitutions (BS; light grey bars) and one base deletions (dark grey). (B) Error rates for Homo sapiens polymerases η, κ, θ and ν and cerevisiae Pol ζ. Note the difference in scales between panels A and B. Error rates for each polymerase were obtained with the lacZ-α forward mutation assay. The assay measures error made when copying a template in a gapped DNA substrate in vitro that contains the wildtype lacZ-α complementation sequence. The assay [89] scores all 12 single base–base mismatches and different single-base deletion mismatches in numerous different sequence contexts. [85,87,90,91].
3.1. Nucleotide selectivity/error prevention
Most DNA polymerases lack intrinsic 3′-exonuclease activity to excise errors. Therefore, their fidelity depends on the ability to prevent incorporation of incorrect dNTPs that lead to base substitutions, and to prevent incorporation involving misaligned substrates that leads to loss or addition of nucleotides. Amazingly, DNA polymerase selectivity can vary over a million-fold range (Fig. 1).
3.1.1. Major replicative polymerases
Pols α, δ and ε nearly always insert correct dNTPs onto properly aligned primer-templates. This is illustrated by the low single base substitution and deletion error rates of Pols α, δ and ε. These error rates are for a polymerase that naturally lacks proofreading activity (Pol α) and for polymerases where their intrinsic proofreading exonucleases are intentionally inactivated (Pols δ and ε) (Fig. 3A). This high nucleotide selectivity reflects the fact that the nascent base pair binding pocket of accurate DNA polymerases, whose assembly is induced by dNTP binding, snugly accommodates base pairs with correct Watson–Crick geometry. The induced fit mechanism establishes the active site geometry needed for rapid phosphodiester bond formation. Deviations from correct geometry resulting from incorrect dNTP binding or primer-terminal mismatches strongly reduce the probability of incorporation.
Geometric distortions can also result from strand misalignments that generate one or more unpaired bases, either in the primer strand (leading to additions) or in the template (leading to deletions). Ideas to account for how these misalignments initiate and are stabilized for continued synthesis include classical DNA strand slippage proposed by Streisinger, misinsertion followed by primer relocation, and misalignment of a nucleotide at the active site [20]. Extensive biochemical and structural support for each of these models exists [6,21]. Typically, single base deletion error rates are substantially higher than are single base addition error rates. It is also worth noting that the base substitution and deletion error rates in Fig. 3 are “average” values, with wide variations in rates observed among the 12 possible single base–base mismatches, the composition of deletion and insertion mismatches, and the local sequences flanking these mismatches. For example, while the average base substitution error rate of Pol α is about 10−4, the rate at which it forms C–C mismatches can be much lower, e.g., 10−6. Other examples of such variability are considered below.
3.1.2. TLS polymerases
The TLS polymerases lack proofreading activity and have lower nucleotide selectivity than the major replicative polymerases, as indicated by their higher error rates for base substitutions and deletions (Table 1 and Fig. 3B). The extreme case is for Pol ι. This remarkable enzyme actually prefers to insert dGTP more often than correct dATP opposite template T, i.e., the error rate for this event approaches one ([22] and references therein). Structural studies suggest that the generally low fidelity of Y family members ability is partly due to relaxed geometric selectivity in the nascent base pair binding pocket, which is more open and solvent-accessible than those of more accurate DNA polymerases [23]. For example, recent work shows that Pol η can accommodate two nucleotides in the active site [24,25]. Pols η, κ, θ and ν are not only error-prone for base substitutions, but also for deletions (Table 1 and Fig. 3B). While not the focus of this review, elegant work on Y family polymerases also has been performed using bacterial TLS polymerases, whose functions and properties are reviewed elsewhere [26–28].
The B-family member Pol ζ also has critical roles in TLS. When copying undamaged DNA, Pol ζ has somewhat higher fidelity than the Y-family polymerases, but lower fidelity than the other B-family members (Table 1). The ability of Pol ζ to generate single base mismatches at relatively high rates is consistent with its known contribution to spontaneous mutagenesis, as well as mutagenesis induced by a variety of DNA damaging agents [29,30]. Pol ζ’s high base substitution error rate clearly demonstrates that it has low nucleotide selectivity, consistent with a possible direct role in mutagenic misinsertion of dNTPs in vivo. Also relevant are kinetic studies demonstrating that Pol ζ efficiently extends terminal mismatches [31]. This is true for undamaged DNA as well as for extending damaged termini, the latter being consistent with a role for Pol ζ in the extension step of TLS in a 2-polymerase model (Fig. 2B). A similar role also has been proposed for Pol κ, which like Pol ζ, is promiscuous for mismatch extension. There are numerous reports describing the ability of TLS and fidelity of various Y-family (and other) polymerases when encountering a wide range of structurally diverse lesions. These studies [11,23,32–34] show that for a given lesion, the bypass efficiency and fidelity is highly polymerase specific.
3.2. Proofreading
Once an incorrect dNTP is incorporated into DNA, the mismatched primer terminus is more difficult to extend than a correctly paired and properly aligned primer terminus. The delay in extension caused by a mismatch allows the primer terminus to fray and travel (switch) from the polymerase to the exonuclease active site for excision of the error [35].
3.2.1. Intrinsic proofreading by Pols δ and ε
Among the many eukaryotic DNA polymerases, only the three polymerases responsible for the bulk of chain elongation during replication (Pols κ, ε and the mitochondrial replicase Pol γ) contain intrinsic 3′-exonucleolytic proofreading activity. The contribution of proofreading to base substitution fidelity is illustrated by the high fidelity of the exonuclease-proficient Pols δ and ε compared to their exonuclease-deficient derivatives (Fig. 3A). A variety of studies in vitro indicates that proofreading improves replication fidelity by factors ranging from a few-fold to more than 100-fold, depending on the mismatch, the sequence context and the polymerase [1,35]. Proofreading corrects mismatches at the terminus itself, and it also corrects more internal insertion and deletion mismatches resulting from misalignments.
3.2.2. Extrinsic proofreading
Given a base substitution error rate of ~10−4 (Fig. 3A) and its role in initiating Okazaki fragments, Pol α may generate many thousands of mismatches during each replication cycle. This leads to the question of whether a separate exonuclease may edit Pol α’s errors. This possibility, which we refer to as extrinsic proofreading, has been examined both biochemically [36] and genetically [37]. The results suggest that the 3′-exonuclease of Pol δ may indeed proofread errors generated by Pol α during initiation of Okazaki fragments. Extrinsic proofreading could be relevant to other DNA transactions that involve exonuclease-deficient polymerases, such as base excision repair and TLS [38].
4. DNA mismatch repair
Replication errors that escape proofreading are corrected by DNA mismatch repair (MMR). Eukaryotic MMR (reviewed in [39–41]) requires recognition of mismatches by MutS proteins, followed by binding of MutL proteins. Both these proteins bind and hydrolyze ATP to undergo conformational changes that help coordinate the multiple protein partnerships and reactions needed to find the strand-discrimination signal (still unknown in eukaryotes), incise the nascent strand, excise the replication error, correctly synthesize new DNA and ligate the nascent strand. Generally, MMR improves replication fidelity by about 100- to 1,000-fold (Fig. 1), with exceptions wherein MMR efficiency can be negligible (e.g., some C–C mismatches), or much higher (deletion mismatches in mononucleotide runs, discussed below). MMR is subdivided into multiple “subpathways” that differ by substrate specificity and protein requirements, in ways that are only partly understood. MMR proteins also can modulate recombination, participate in repair of double-strand DNA breaks and signal apoptosis in response to DNA damage. As a consequence, loss of MMR is associated with elevated mutation rates and altered survival in response to DNA damage.
5. The many causes of replication infidelity
There are multiple ways to perturb each of the three major mechanisms that determine replication fidelity (Fig. 1, right).
5.1. Reduced nucleotide selectivity
Nucleotide selectivity can be reduced by mutations in polymerase genes that alter amino acids comprising the active site or the nascent base pair binding pocket, or even more distant amino acids that indirectly influence selectivity [42] (for further review please see chapter by Sweasy). For the major replicative polymerases whose polymerase activities are critical for cell viability, the probability that such defects will be associated with cancer is likely to be small, because most such mutations reduce polymerase activity and therefore reduce proliferative potential. The probability that a dNTP will be misinserted also depends on the relative concentrations of each of the four dNTPs available for DNA synthesis. For example, an abnormally high concentration of dTTP relative to dCTP will promote misinsertion of dTMP opposite template guanine which, if uncorrected, will yield G–C to T–A substitutions. Interestingly, the four dNTP concentrations are not equal even in normal cells (e.g., the dGTP concentration is typically lower than the other three), and they are highly regulated throughout the cell cycle as well as in response to environmental stress. Any deviation from the normal dNTP ratios has the potential to decrease replication fidelity and generate a mutator phenotype. This is illustrated by the elevated mutation rates in cells containing mutations in genes that regulate dNTP concentrations, e.g., ribonucleotide reductase [43–45]. dNTP pool imbalances induced by means other than genetic defects, e.g., by environmental stress, also have the potential to elevate mutation rates. This could occur in a transient manner that would contribute only to a subset of the multiple mutations needed for tumor formation. dNTP pool imbalances that result from increasing one or more dNTP concentrations may be more relevant to cancer than imbalances resulting from decreasing dNTP concentrations below normal levels, because the latter can limit proliferative potential [44].
5.2. Reduced proofreading efficiency
The efficiency of proofreading depends on a balance between polymerization from terminal mismatches and 3′-exonucleolytic excision of the error. Thus, defects to either activity can reduce the contribution of proofreading to replication fidelity (Fig. 1). The exonuclease can be inactivated by active site mutations. It can be reduced by the presence of dNMPs, which are the end products of excision that can bind to the exonuclease active site and inhibit activity. Proofreading can be reduced by mutations that alter the ability of the primer terminus to switch from the polymerase to the exonuclease active site. Proofreading can be suppressed by mutations in the polymerase active site that promote mismatch extension, or by dNTP pool changes that increase the concentration of the correct dNTPs needed to extend mismatches.
Proofreading potential can also be affected by DNA damage. A fine example involves the common oxidative lesion, 8-oxo-guanine. In its syn conformation, 8-oxo-G pairs with adenine and largely escapes proofreading because it mimics the shape of a correct base pair within the nascent base pair binding pocket of the polymerase. Finally, even some undamaged mismatches are simply poor substrates for normal proofreading. The best example involves inefficient proofreading of insertion and deletion mismatches in repetitive sequences. Efficiency decreases as the length of a repetitive sequence increases, because the unpaired base can reside upstream of the terminus, where it can be protected from excision by intervening correct base pairs [21]. In principle, any of these mechanisms could be relevant to a mutator phenotype associated with cancer, because proofreading is dispensable for eukaryotic cell viability.
5.3. Reduced MMR efficiency
MMR is also dispensable for eukaryotic cell viability, yet critical for replication fidelity. Like proofreading, MMR efficiency can be reduced by mutations in genes whose products are required for MMR (Fig. 1). The degree to which these genetic defects elevate mutation rates varies considerably, depending on the nature of the gene mutation, the function of the particular gene product in MMR and the type and location of the replication error considered. Another way to reduce MMR efficiency is to change the expression of MMR genes. MMR can be inactivated by promoter hypermethylation to silence expression of the essential human MMR gene MLH1, a mechanism strongly associated with spontaneous colon cancer. The level of MMR proteins needed to correct replication errors under normal conditions may be insufficient when replication errors accumulate at a higher rate. For example, studies have shown that MMR capacity can be saturated by rapid error-prone replication in Escherichia coli, or by the presence of DNA lesions to which MMR proteins can bind. In yeast, over-expression of two MMR genes, MLH1 and MSH3, reduces MMR efficiency, presumably by perturbing normal protein–protein partnerships required for MMR. Exposure of budding yeast to cadmium, a metal that is ubiquitously present in the environment and is a known human carcinogen, can inhibit MMR and strongly elevate the mutation rate [46]. There potentially may be other environmental exposures that inhibit MMR. Reduced MMR due to altered expression, saturation, or cadmium inhibition is reversible; again potentially generating transient mutator phenotypes.
6. Cancer consequences
Defects in the cellular machinery required for accurate DNA replication have been correlated strongly with mutator phenotypes and increased cancer susceptibility. A particularly well-understood example involves Pol η, which is highly efficient at bypassing cyclobutane pyrimidine dimers, one of the major lesions generated by exposure to ultraviolet light. Cells lacking Pol η have an elevated frequency of UV-light-induced mutations, and patients lacking Pol η have increased susceptibility to sunlight-induced skin cancer [47–49]. This effect has been recapitulated in mouse models [50,51], and additional studies in mice also reveal increased cancer susceptibility due to defects in two other TLS polymerases, Pol ι [52,53] and Pol ζ [54,55]. Defects in the major replicative polymerases, and in polymerases involved in repair synthesis, also are associated with cancer susceptibility. These studies are reviewed elsewhere [56] (for further reading please see Sweasy, Hoffman, Preston and Loeb chapters in this volume), allowing us to turn final attention to the potential value of replication error specificity for identifying the causes of mutator phenotypes.
7. Replication error signatures as biomarkers
Finally, we consider examples of how replication error signatures are, or might, be used to probe the mutator hypothesis.
7.1. Microsatellite instability
A highly successful current example is microsatellite instability (MSI), the loss or gain of base pairs in short (1–6 base) repetitive DNA sequences. These mutations result from strand slippage during replication to generate one or more unpaired bases. When slippage occurs in repetitive sequences, the extra base(s) comprising the mismatch can be present in otherwise duplex DNA, well upstream of the primer terminus and stabilized by intervening correct base pairs whose numbers increase as the number of base pairs in the repetitive sequence increase [57–59]. As a consequence, slipped mismatch intermediates are not only frequently generated during replication, they are also more readily extended than terminal mismatches and therefore inefficiently proofread. For these reasons, MMR is the major guardian of genome stability against MSI, as illustrated by the observation that a MMR defect can increase the rate of certain single base deletions by 100,000-fold [60,61]. This is why MSI is now a widely used “replication error signature” that is diagnostic of MMR defective tumors [62–66] (for further review see chapter by Salk and Horwitz). MSI may be a useful biomarker for causes of mutator phenotypes other than MMR defects. For example, a recent study reported that if ribonucleotides incorporated into DNA during replication by yeast Pol ε are not removed by RNase H2-dependent repair, they result in 2–5 base pair deletions in repetitive sequences [67].
7.2. Somatic hypermutation
Another clear example of a useful error signature, albeit one not directly related to cancer per se, involves a base substitution signature for somatic hypermutation (SHM) of immunoglobulin genes. SHM generates mutations in the variable regions of immunoglobulin genes at an extraordinary rate, thereby contributing to affinity maturation of antibodies [68]. Initial studies of the error specificity of Pol η in vitro demonstrated that human and mouse Pol η preferentially generate mismatches at A–T base pairs, and preferentially in certain sequence contexts [69]. This motivated subsequent studies (e.g. [70–73]) that now strongly support the idea that Pol η is indeed responsible for much of the SHM occurring in vivo at A–T base pairs. Table 2 lists other point mutation signatures of replication infidelity that eventually may have diagnostic relevance for mutator phenotypes and cancer.
Table 2.
DNA synthesis-related error signatures as potential “biomarkers”.
| Mutational Event | Origin of mutations |
|---|---|
| Microsatellite Instability | Strand-slippage |
| Inefficient proofreading | |
| Unrepaired rNMPs in DNA | |
| DNA damage (e.g., ROS) | |
| Somatic hypermutation | Deamination by AID |
| Pol η, Pol ζ, Pol ι?, Pol θ? | |
| Tandem double substitutions | TLS Pols (η > ζ > κ ≫> α/δ/ε) |
| DNA damage (e.g., dipyrimidine lesions) | |
| “Clustered complex” mutations | Pol ζ |
| Multiple “regional” mutations | Pol ζ when copying of ssDNA tracts |
| Unusual error specificities of normal DNA polymerases | |
| Misinsertions opposite pyrimidines | Pol ι (e.g., T-dGTP error rate approaches 1) |
| G-dTTP error rate | Pol ν (unusually high proportion) |
| One-base addition error rate | Pol θ (highest rate among human Pols) |
| Biased error specificity of mutator polymerases | |
| T-dTTP ≫ A-dATP | Yeast M644G Pol ε |
| T-dGTP ≫ A-dCTP; G-dTTP ≫ C-dATP | Yeast L612Pol δ and human L606 Pol δ |
| Genome-wide mutable motifs | Yeast L612Pol δ |
7.3. Tandem double base substitutions
Forming a tandem double base substitution by DNA synthesis requires misinsertion, followed by another misinsertion onto a mismatched terminus, and then followed by multiple extensions of a doubly mismatched terminus. Each of these steps alone is normally rare, and therefore accurate replicative DNA polymerases have rarely been seen to generate tandem double base substitutions. However, Pol κ [74], Pol ζ [75] and especially Pol η [69,72,76], generate tandem double base substitutions at readily detectable rates when copying undamaged DNA. In fact, the accuracy of Pol η is so low that it also generates tandem triple substitutions and mutants with substitutions separated by 1–3 base pairs. Tandem double base substitutions are also a characteristic feature of mutagenesis resulting from exposure to UV light, e.g., CC to TT mutations that are likely generated during TLS of UU dimers resulting from deamination of CC dimers. For these reasons, tandem double base substitutions are a biomarker for involvement of TLS polymerases.
7.4. Clustered-complex and multiple mutations generated by Pol ζ
When copying undamaged DNA in vitro, Pol ζ generates multiple substitutions, deletions and/or insertions within a few base pairs of each other. Pol ζ also generates such complex errors in vivo [77,78], more so in genetic backgrounds defective in processing DNA damage. Highly mutagenic DNA synthesis by yeast Pol ζ also has recently been observed when copying long tracts of damaged single stranded DNA [79]. Such long tracts can be generated during the end-resection phase of DNA double strand break repair [80], or by uncoupling leading and lagging strand replication. This Pol ζ-dependent mutagenesis can introduce multiple mutations spread over thousands of base pairs. Whether tandem, clustered or spread out, the concerted introduction of multiple mutations in a single DNA transaction provides an opportunity for selection in tumor progression, or in evolution, that differs from the selective advantages of single point mutations introduced in series over multiple cell generations (for further discussion, see [79,81,82] and references therein).
7.5. Unusual error signatures of other DNA polymerases
Other TLS polymerases also have unusual error specificities (Table 2). These include Pol ι, which frequently inserts incorrect dNTPs opposite template pyrimidines [83,84], Pol ν, which forms T-dGTP mismatches at an unusually high rate [85,86], and Pol θ, which generates single base additions at a higher rate than other polymerases [87]. In addition, mutant alleles of the three major replicative polymerases have high error rates for some mismatches but not others. These biases have been useful for assigning the roles of Pol δ and Pol ε in replicating the leading and lagging strand templates in yeast [7,8], and for identifying potential mutable sequence motifs for Pol δ errors in a whole yeast genome sequencing study [88].
The above examples of replication error specificity illustrate the potential value of testing the mutator hypothesis by not only looking for increases in spontaneous and stress-induced mutation rates and frequencies, but also actually using replication error signatures as clues to the molecular nature of the defects that initiate and/or promote tumor formation.
Acknowledgments
We thank Katarzyna Bebenek and Jessica Williams for thoughtful comments on the manuscript. The research conducted in Thomas Kunkel’s laboratory is supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Projects Z01 ES065070 and Z01 ES065089).
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest.
References
- 1.Bebenek K, Kunkel TA. Functions of DNA polymerases. Adv Protein Chem. 2004;69:137–65. doi: 10.1016/S0065-3233(04)69005-X. [DOI] [PubMed] [Google Scholar]
- 2.Moon AF, Garcia-Diaz M, Batra VK, Beard WA, Bebenek K, Kunkel TA, et al. The X family portrait: structural insights into biological functions of X family polymerases. DNA Repair (Amst) 2007;6:1709–25. doi: 10.1016/j.dnarep.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Copeland WC. The mitochondrial DNA polymerase in health and disease. Subcell Biochem. 2010;50:211–22. doi: 10.1007/978-90-481-3471-7_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA repair and mutagenesis. 2. ASM Press; 2006. [Google Scholar]
- 5.Kunkel TA, Burgers PM. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 2008;18:521–7. doi: 10.1016/j.tcb.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunkel TA. Evolving views of DNA replication (in)fidelity. Cold Spring Harb Symp Quant Biol. 2009;74:91–101. doi: 10.1101/sqb.2009.74.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–30. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Division of labor at the eukaryotic replication fork. Mol Cell. 2008;30:137–44. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–67. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 10.McCulloch SD, Kokoska RJ, Kunkel TA. Efficiency, fidelity and enzymatic switching during translesion DNA synthesis. Cell Cycle. 2004;3:580–3. [PubMed] [Google Scholar]
- 11.Takata K, Wood RD. Bypass specialists operate together. EMBO J. 2009;28:313–4. doi: 10.1038/emboj.2008.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Livneh Z, Ziv O, Shachar S. Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle. 2010;9:729–35. doi: 10.4161/cc.9.4.10727. [DOI] [PubMed] [Google Scholar]
- 13.Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010;465:951–5. doi: 10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Efrati E, Tocco G, Eritja R, Wilson SH, Goodman MF. Abasic translesion synthesis by DNA polymerase beta violates the “A-rule”. Novel types of nucleotide incorporation by human DNA polymerase beta at an abasic lesion in different sequence contexts. J Biol Chem. 1997;272:2559–69. doi: 10.1074/jbc.272.4.2559. [DOI] [PubMed] [Google Scholar]
- 15.Hoffmann JS, Pillaire MJ, Maga G, Podust V, Hubscher U, Villani G. DNA polymerase beta bypasses in vitro a single d(GpG)-cisplatin adduct placed on codon 13 of the HRAS gene. Proc Natl Acad Sci U S A. 1995;92:5356–60. doi: 10.1073/pnas.92.12.5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maga G, Villani G, Ramadan K, Shevelev I, Tanguy Le Gac N, Blanco L, et al. Human DNA polymerase lambda functionally and physically interacts with proliferating cell nuclear antigen in normal and translesion DNA synthesis. J Biol Chem. 2002;277:48434–40. doi: 10.1074/jbc.M206889200. [DOI] [PubMed] [Google Scholar]
- 17.Blanca G, Villani G, Shevelev I, Ramadan K, Spadari S, Hubscher U, et al. Human DNA polymerases lambda and beta show different efficiencies of translesion DNA synthesis past abasic sites and alternative mechanisms for frameshift generation. Biochemistry. 2004;43:11605–15. doi: 10.1021/bi049050x. [DOI] [PubMed] [Google Scholar]
- 18.Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73:134–54. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo C, Kosarek-Stancel JN, Tang TS, Friedberg EC. Y-family DNA polymerases in mammalian cells. Cell Mol Life Sci. 2009;66:2363–81. doi: 10.1007/s00018-009-0024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Streisinger G, Okada Y, Emrich J, Newton J, Tsugita A, Terzaghi E, et al. Frameshift mutations and the genetic code. This paper is dedicated to Professor Theodosius Dobzhansky on the occasion of his 66th birthday. Cold Spring Harb Symp Quant Biol. 1966;31:77–84. doi: 10.1101/sqb.1966.031.01.014. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Diaz M, Kunkel TA. Mechanism of a genetic glissando: structural biology of indel mutations. Trends Biochem Sci. 2006;31:206–14. doi: 10.1016/j.tibs.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Bebenek K, Tissier A, Frank EG, McDonald JP, Prasad R, Wilson SH, et al. 5′-Deoxyribose phosphate lyase activity of human DNA polymerase iota in vitro. Science. 2001;291:2156–9. doi: 10.1126/science.1058386. [DOI] [PubMed] [Google Scholar]
- 23.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc Natl Acad Sci U S A. 2007;104:15591–8. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biertumpfel C, Zhao Y, Kondo Y, Ramon-Maiques S, Gregory M, Lee JY, et al. Structure and mechanism of human DNA polymerase eta. Nature. 2010;465:1044–8. doi: 10.1038/nature09196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silverstein TD, Johnson RE, Jain R, Prakash L, Prakash S, Aggarwal AK. Structural basis for the suppression of skin cancers by DNA polymerase eta. Nature. 2010;465:1039–43. doi: 10.1038/nature09104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jarosz DF, Beuning PJ, Cohen SE, Walker GC. Y-family DNA polymerases in Escherichia coli. Trends Microbiol. 2007;15:70–7. doi: 10.1016/j.tim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 27.Sutton MD. Coordinating DNA polymerase traffic during high and low fidelity synthesis. Biochim Biophys Acta. 2010;1804:1167–79. doi: 10.1016/j.bbapap.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baynton K, Fuchs RP. Lesions in DNA: hurdles for polymerases. Trends Biochem Sci. 2000;25:74–9. doi: 10.1016/s0968-0004(99)01524-8. [DOI] [PubMed] [Google Scholar]
- 29.Lawrence CW. Cellular functions of DNA polymerase zeta and Rev1 protein. Adv Protein Chem. 2004;69:167–203. doi: 10.1016/S0065-3233(04)69006-1. [DOI] [PubMed] [Google Scholar]
- 30.Gan GN, Wittschieben JP, Wittschieben BO, Wood RD. DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res. 2008;18:174–83. doi: 10.1038/cr.2007.117. [DOI] [PubMed] [Google Scholar]
- 31.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–53. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 32.Diaz M, Lawrence C. An update on the role of translesion synthesis DNA polymerases in Ig hypermutation. Trends Immunol. 2005;26:215–20. doi: 10.1016/j.it.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 33.Fuchs RP, Fujii S, Wagner J. Properties and functions of Escherichia coli: Pol IV and Pol V. Adv Protein Chem. 2004;69:229–64. doi: 10.1016/S0065-3233(04)69008-5. [DOI] [PubMed] [Google Scholar]
- 34.Prakash S, Prakash L. Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev. 2002;16:1872–83. doi: 10.1101/gad.1009802. [DOI] [PubMed] [Google Scholar]
- 35.Reha-Krantz LJ. DNA polymerase proofreading: Multiple roles maintain genome stability. Biochim Biophys Acta. 2010;1804:1049–63. doi: 10.1016/j.bbapap.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 36.Perrino FW, Loeb LA. Hydrolysis of 3′-terminal mispairs in vitro by the 3′–5′ exonuclease of DNA polymerase delta permits subsequent extension by DNA polymerase alpha. Biochemistry. 1990;29:5226–31. doi: 10.1021/bi00474a002. [DOI] [PubMed] [Google Scholar]
- 37.Pavlov YI, Frahm C, Nick McElhinny SA, Niimi A, Suzuki M, Kunkel TA. Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Curr Biol. 2006;16:202–7. doi: 10.1016/j.cub.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 38.Nick McElhinny SA, Pavlov YI, Kunkel TA. Evidence for extrinsic exonucleolytic proofreading. Cell Cycle. 2006;5:958–62. doi: 10.4161/cc.5.9.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 40.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–23. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 41.Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391–407. doi: 10.1016/j.mad.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 43.Kunz BA, Kohalmi SE, Kunkel TA, Mathews CK, McIntosh EM, Reidy JA. International commission for protection against environmental mutagens and carcinogens. Deoxyribonucleoside triphosphate levels: a critical factor in the maintenance of genetic stability. Mutat Res. 1994;318:1–64. doi: 10.1016/0165-1110(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 44.Kumar D, Viberg J, Nilsson AK, Chabes A. Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res. 2010;38:3975–83. doi: 10.1093/nar/gkq128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar D, Abdulovic AL, Viberg J, Nilsson AK, Kunkel TA, Chabes A. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucl Acids Res. 2010 doi: 10.1093/nar/gkq829. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin YH, Clark AB, Slebos RJ, Al-Refai H, Taylor JA, Kunkel TA, et al. Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat Genet. 2003;34:326–9. doi: 10.1038/ng1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;285:263–5. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 48.Yamada A, Masutani C, Iwai S, Hanaoka F. Complementation of defective translesion synthesis and UV light sensitivity in xeroderma pigmentosum variant cells by human and mouse DNA polymerase eta. Nucleic Acids Res. 2000;28:2473–80. doi: 10.1093/nar/28.13.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masutani C, Hanaoka F, Ahmad SI. Xeroderma pigmentosum variant. XP-V: its product and biological roles. Adv Exp Med Biol. 2008;637:93–102. doi: 10.1007/978-0-387-09599-8_10. [DOI] [PubMed] [Google Scholar]
- 50.Lin Q, Clark AB, McCulloch SD, Yuan T, Bronson RT, Kunkel TA, et al. Increased susceptibility to UV-induced skin carcinogenesis in polymerase eta-deficient mice. Cancer Res. 2006;66:87–94. doi: 10.1158/0008-5472.CAN-05-1862. [DOI] [PubMed] [Google Scholar]
- 51.Ohkumo T, Kondo Y, Yokoi M, Tsukamoto T, Yamada A, Sugimoto T, et al. UV-B radiation induces epithelial tumors in mice lacking DNA polymerase eta and mesenchymal tumors in mice deficient for DNA polymerase iota. Mol Cell Biol. 2006;26:7696–706. doi: 10.1128/MCB.01076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang M, Devereux TR, Vikis HG, McCulloch SD, Holliday W, Anna C, et al. Pol iota is a candidate for the mouse pulmonary adenoma resistance 2 locus, a major modifier of chemically induced lung neoplasia. Cancer Res. 2004;64:1924– 31. doi: 10.1158/0008-5472.can-03-3080. [DOI] [PubMed] [Google Scholar]
- 53.Dumstorf CA, Clark AB, Lin Q, Kissling GE, Yuan T, Kucherlapati R, et al. Participation of mouse DNA polymerase iota in strand-biased mutagenic bypass of UV photoproducts and suppression of skin cancer. Proc Natl Acad Sci U S A. 2006;103:18083–8. doi: 10.1073/pnas.0605247103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diaz M, Verkoczy LK, Flajnik MF, Klinman NR. Decreased frequency of somatic hypermutation and impaired affinity maturation but intact germinal center formation in mice expressing antisense RNA to DNA polymerase zeta. J Immunol. 2001;167:327–35. doi: 10.4049/jimmunol.167.1.327. [DOI] [PubMed] [Google Scholar]
- 55.Wittschieben JP, Patil V, Glushets V, Robinson LJ, Kusewitt DF, Wood RD. Loss of DNA polymerase zeta enhances spontaneous tumorigenesis. Cancer Res. 2010;70:2770–8. doi: 10.1158/0008-5472.CAN-09-4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Loeb LA, Monnat RJ., Jr DNA polymerases and human disease. Nat Rev Genet. 2008;9:594–604. doi: 10.1038/nrg2345. [DOI] [PubMed] [Google Scholar]
- 57.Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435–45. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 58.Kelkar YD, Strubczewski N, Hile SE, Chiaromonte F, Eckert KA, Makova KD. What is a microsatellite: a computational and experimental definition based upon repeat mutational behavior at A/T and GT/AC repeats. Genome Biol Evol. 2010;2:620–35. doi: 10.1093/gbe/evq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Subramanian S, Mishra RK, Singh L. Genome-wide analysis of microsatellite repeats in humans: their abundance and density in specific genomic regions. Genome Biol. 2003;4:R13. doi: 10.1186/gb-2003-4-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tran HT, Gordenin DA, Resnick MA. The prevention of repeat-associated deletions in Saccharomyces cerevisiae by mismatch repair depends on size and origin of deletions. Genetics. 1996;143:1579–87. doi: 10.1093/genetics/143.4.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol. 1997;17:2859–65. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Umar A, Kunkel TA. DNA-replication fidelity, mismatch repair and genome instability in cancer cells. Eur J Biochem. 1996;238:297–307. doi: 10.1111/j.1432-1033.1996.0297z.x. [DOI] [PubMed] [Google Scholar]
- 63.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–8. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57. [PubMed] [Google Scholar]
- 65.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76:1–18. doi: 10.1111/j.1399-0004.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Subramanian S, Madgula VM, George R, Mishra RK, Pandit MW, Kumar CS, et al. Triplet repeats in human genome: distribution and their association with genes and other genomic regions. Bioinformatics. 2003;19:549–52. doi: 10.1093/bioinformatics/btg029. [DOI] [PubMed] [Google Scholar]
- 67.Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, Lundstrom EB, et al. Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol. 2010;6:774–81. doi: 10.1038/nchembio.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Diaz M, Casali P. Somatic immunoglobulin hypermutation. Curr Opin Immunol. 2002;14:235–40. doi: 10.1016/s0952-7915(02)00327-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuda T, Bebenek K, Masutani C, Rogozin IB, Hanaoka F, Kunkel TA. Error rate and specificity of human and murine DNA polymerase eta. J Mol Biol. 2001;312:335–46. doi: 10.1006/jmbi.2001.4937. [DOI] [PubMed] [Google Scholar]
- 70.Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat Immunol. 2001;2:530–6. doi: 10.1038/88732. [DOI] [PubMed] [Google Scholar]
- 71.Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. DNA polymerase eta is an A–T mutator in somatic hypermutation of immunoglobulin variable genes. Nat Immunol. 2001;2:537–41. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 72.Pavlov YI, Rogozin IB, Galkin AP, Aksenova AY, Hanaoka F, Rada C, et al. Correlation of somatic hypermutation specificity and A–T base pair substitution errors by DNA polymerase eta during copying of a mouse immunoglobulin kappa light chain transgene. Proc Natl Acad Sci U S A. 2002;99:9954–9. doi: 10.1073/pnas.152126799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Masuda K, Ouchida R, Yokoi M, Hanaoka F, Azuma T, Wang JY. DNA polymerase eta is a limiting factor for A:T mutations in Ig genes and contributes to antibody affinity maturation. Eur J Immunol. 2008;38:2796–805. doi: 10.1002/eji.200838502. [DOI] [PubMed] [Google Scholar]
- 74.Ohashi E, Bebenek K, Matsuda T, Feaver WJ, Gerlach VL, Friedberg EC, et al. Fidelity and processivity of DNA synthesis by DNA polymerase kappa, the product of the human DINB1 gene. J Biol Chem. 2000;275:39678–84. doi: 10.1074/jbc.M005309200. [DOI] [PubMed] [Google Scholar]
- 75.Zhong X, Garg P, Stith CM, Nick McElhinny SA, Kissling GE, Burgers PM, et al. The fidelity of DNA synthesis by yeast DNA polymerase zeta alone and with accessory proteins. Nucleic Acids Res. 2006;34:4731–42. doi: 10.1093/nar/gkl465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Matsuda T, Bebenek K, Masutani C, Hanaoka F, Kunkel TA. Low fidelity DNA synthesis by human DNA polymerase-eta. Nature. 2000;404:1011–3. doi: 10.1038/35010014. [DOI] [PubMed] [Google Scholar]
- 77.Harfe BD, Jinks-Robertson S. DNA polymerase zeta introduces multiple mutations when bypassing spontaneous DNA damage in Saccharomyces cerevisiae. Mol Cell. 2000;6:1491–9. doi: 10.1016/s1097-2765(00)00145-3. [DOI] [PubMed] [Google Scholar]
- 78.Sakamoto AN, Stone JE, Kissling GE, McCulloch SD, Pavlov YI, Kunkel TA. Mutator alleles of yeast DNA polymerase zeta. DNA Repair (Amst) 2007;6:1829–38. doi: 10.1016/j.dnarep.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang Y, Sterling J, Storici F, Resnick MA, Gordenin DA. Hypermutability of damaged single-strand DNA formed at double-strand breaks and uncapped telomeres in yeast Saccharomyces cerevisiae. PLoS Genet. 2008;4:e1000264. doi: 10.1371/journal.pgen.1000264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang Y, Gordenin DA, Resnick MA. A single-strand specific lesion drives MMS-induced hyper-mutability at a double-strand break in yeast. DNA Repair (Amst) 2010;9:914–21. doi: 10.1016/j.dnarep.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang J, Gonzalez KD, Scaringe WA, Tsai K, Liu N, Gu D, et al. Evidence for mutation showers. Proc Natl Acad Sci U S A. 2007;104:8403–8. doi: 10.1073/pnas.0610902104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006;40:209–35. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- 83.Vaisman A, Woodgate R. Unique misinsertion specificity of poliota may decrease the mutagenic potential of deaminated cytosines. EMBO J. 2001;20:6520–9. doi: 10.1093/emboj/20.22.6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tissier A, Frank EG, McDonald JP, Iwai S, Hanaoka F, Woodgate R. Misinsertion and bypass of thymine–thymine dimers by human DNA polymerase iota. EMBO J. 2000;19:5259–66. doi: 10.1093/emboj/19.19.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Arana ME, Takata K, Garcia-Diaz M, Wood RD, Kunkel TA. A unique error signature for human DNA polymerase nu. DNA Repair (Amst) 2007;6:213–23. doi: 10.1016/j.dnarep.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Takata K, Shimizu T, Iwai S, Wood RD. Human DNA polymerase N (POLN) is a low fidelity enzyme capable of error-free bypass of 5S-thymine glycol. J Biol Chem. 2006;281:23445–55. doi: 10.1074/jbc.M604317200. [DOI] [PubMed] [Google Scholar]
- 87.Arana ME, Seki M, Wood RD, Rogozin IB, Kunkel TA. Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic Acids Res. 2008;36:3847–56. doi: 10.1093/nar/gkn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Larrea AA, Lujan SA, Nick McElhinny SA, Mieczkowski PA, Resnick MA, Gordenin DA, et al. Genome-wide model for the normal eukaryotic DNA replication fork. Proc Natl Acad Sci U S A. 2010;107:17674–9. doi: 10.1073/pnas.1010178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bebenek K, Kunkel TA. Analyzing fidelity of DNA polymerases. Methods Enzymol. 1995;262:217–32. doi: 10.1016/0076-6879(95)62020-6. [DOI] [PubMed] [Google Scholar]
- 90.Schmitt MW, Matsumoto Y, Loeb LA. High fidelity and lesion bypass capability of human DNA polymerase delta. Biochimie. 2009;91:1163–72. doi: 10.1016/j.biochi.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nick McElhinny SA, Pursell ZF, Kunkel TA. Mechanisms for high fidelity DNA replication. Cambridge: Royal Society of Chemistry; 2009. [Google Scholar]