

Abstract
Background
Graft arterial disease (GAD) limits long-term solid organ allograft survival. The thickened intima in GAD contains smooth muscle-like cells (SMLC), leukocytes, and extracellular matrix. The intimal SMLC in mouse GAD lesions differ from medial smooth muscle cells in their function and phenotype. While intimal SMLC may originate by migration and modulation of donor medial cells or by recruitment of host-derived precursors, the mechanisms underlying localization within grafts and the factors that drive these processes remain unclear.
Methods and Results
This study of aortic transplantation in mice demonstrated an important function for chemokines beyond their traditional role in leukocyte recruitment and activation. Intimal SMLC, but not medial smooth muscle cells (SMC), express functional CC chemokine receptor-1 (CCR1), and respond to RANTES by increased migration and proliferation. Although RANTES infusion in vivo promotes inflammatory cell accumulation in the adventitia of aortic allografts of wild-type as well as CCR1-deficient recipients, RANTES infusion increases GAD intimal thickening with SMLC proliferation in only the wild-type hosts. Aortic allografts transplanted into CCR1-deficient mice after wild-type bone marrow transplantation did not develop intimal lesions, indicating that CCR-1 bearing inflammatory cells do not contribute to intimal lesion formation. Moreover, RANTES induces SMLC proliferation in vitro, but does not promote medial SMC growth. Blockade of CCR5 attenuates RANTES-induced T cell and monocyte/macrophage proliferation, but does not affect RANTES-induced SMLC proliferation, consistent with a larger role of CCR1-binding chemokines in SMLC migration and proliferation and GAD development.
Conclusions
These studies provide a novel mechanistic insight regarding the formation of vascular intimal hyperplasia and suggest a novel therapeutic strategy for preventing allograft arteriopathy.
Keywords: chemokine, chemokine receptor, smooth muscle cell, cytokine, chronic rejection, arteriosclerosis, pathogenesis, aortic transplantation, mitogen-activated protein kinase (MAPK)
Introduction
Despite contemporary therapies that effectively limit acute rejection, solid-organ allografts can still develop severe and diffuse arterial intimal hyperplastic lesions (graft arterial disease, GAD) that constitute a major obstacle to long-term success. GAD lesions consist primarily of smooth muscle-like cells (SMLC) and extracellular matrix, admixed with infiltrating mononuclear leukocytes 1, 2, and can lead to progressive organ ischemia.
GAD occurs predominantly as a consequence of immune-mediated allospecific vascular injury in grafts.1 Host inflammatory cells initially interact with allografts at the vascular wall interface. Alloresponses induce the expression of cytokines, chemokines, adhesion molecules, and growth factors from activated donor endothelial cells (EC) and medial smooth muscle cells (SMC) as well as from host inflammatory cells. The inflammatory events subsequently induce the accumulation and proliferation of intimal cells bearing smooth muscle cell markers — “modulated” smooth muscle cells3 — eventually obstructing the arteries of the allografted organ.
Because numerous important functional differences exist between intimal and medial SMC, we refer to the intimal smooth muscle α-actin positive cells as SMLC 4. SMLC have a spindle-shape, oval nucleus and cytoplasmic filaments as do medial SMCs; they also express SMC markers, including smooth muscle α-actin, calponin, and smooth muscle specific myosin heavy chain type 1 (SM1).4 However, these intimal SMLC differ distinctly from medial SMCs. While medial SMCs typically contain abundant myofilaments and proliferate slowly, if at all, intimal SMLC more closely resemble those within developing embryonic vessels.
Since the 1960s, various groups have investigated the origins of SMLC in vascular hyperplastic lesions.5 Ross et al. hypothesized that during atherogenesis, platelet products and (in later formulations) local inflammatory events can activate medial SMC, which then migrate into the intima, proliferate, and elaborate extracellular matrix6. However, more recent studies indicate that some SMLC in GAD lesions can derive from the host.4, 5, 7 In non-immunosuppressed animal aortic and cardiac allografts, host-derived SMLCs form the majority of intimal cells. In contrast, studies of human transplant recipients treated with immunosuppressant agents demonstrated that arterial allograft lesions have a substantially smaller proportion of host-derived SMLC; in human heart GAD the population of SMLC of recipient origin ranges between 2.6% through 30%.5 Regardless, the SMLC comprising the intimal lesions likely have diverse sources: donor medial SMCs (donor); fibroblast-myofibroblast transdifferentiation (donor, host); adjacent host medial SMCs (host); endothelial-mesenchymal transdifferentiation (donor, host); bone marrow-derived SM precursor (host); non-marrow-derived SMC precursor (host) 5. Bone marrow (BM)-derived cells comprise only 10–15% of the intimal SMLC in mouse GAD4, making precursor cell characterization difficult. Over all, the multiple diverse origins of the SMLC imply important differences in lineage that can figure mechanistically in the pathogenesis of GAD. Notably, BM-derived SMLC also populate human atherosclerotic lesions in BM transplant recipients.8
Regardless of whether the intimal SMLC derive from donor medial SMC or host precursor cells, the growing intima must first recruit them. Moreover, the intimal SMLC proliferate and increase the intimal lesion size, while medial SMCs do not contribute. These observations suggest that intimal SMLC selectively express chemokine and/or growth factor receptors that induce their recruitment and proliferation.
Classically, adhesion molecules and chemokines direct the recruitment of neutrophils, T cells, monocytes/macrophages, and other leukocytes into grafts and lead to vascular cell activation and tissue injury. Chemokines are 8–10-kDa peptides organized into CXC, CC, C, and CX3C subfamilies based on the position of conserved cysteine residues. All chemokine receptors belong to a superfamily of G-protein coupled receptors, whose differential expression on cells leads to selective chemokine function 9. In vitro characterization suggests substantial redundancy and promiscuity in chemokine receptor interactions; each receptor binds multiple chemokines and most chemokines bind to more than one receptor. 9, 10 Nevertheless, in vivo experiments show that individual ligand or receptor deficiency can have striking effects.11, 12
Clinical and animal studies demonstrate the presence of many chemokines in allografts during acute rejection and also during the development of GAD lesions13. Moreover, interruption of specific chemokine-receptor interactions reduces leukocyte recruitment into allografts and attenuates subsequent intimal hyperplasia 14. Rejecting allografts specifically contain high levels of chemokines that bind CCR1, CCR2, or CXCR3, such as Regulated upon Activation, Normal T-cell Expressed and Secreted (RANTES), MCP-1, macrophage inflammatory protein 1-α (MIP1-α), IP-10, or interferon-inducible T cell-chemoattractant (I-TAC); antibodies that block these mediators attenuate GAD 15–18. However, whether these chemokines exclusively influence leukocyte recruitment or can affect other cell populations remains uncertain. This study tested the hypothesis that SMLC bear functional chemokine receptors distinct from medial SMC that promote their recruitment into intimal lesions and/or in situ proliferation in the intima, contributing importantly to GAD formation.
Materials and Methods
Mice
BALB/c (B/c; H-2d) and C57BL/6 (B6; H-2b) mice were purchased from Jackson Laboratories (Bar Harbor, ME). CCR1-deficient mice on a C57BL/6 background were a kind gift from Dr. Craig Gerard (The Children’s Hospital, Boston, MA)19. Mice were maintained in accordance with the guidelines of the Committee on Animals of the Harvard Medical School and the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Resources, National Research Council.
Reagents
Recombinant-derived mouse tumor necrosis factor-α (TNF-α), interferon gamma (IFN-γ), regulated on activation, normal T expressed and secreted (RANTES), macrophage inflammatory protein 1 alpha (MIP-1α), macrophage inflammatory protein 1 gamma (MIP-1γ), mast cell activation-related chemokine (MARC), and MIP-related protein 1 (C10) were purchased from R&D Systems (Minneapolis, MN). Rabbit anti-CCR1 Ab was purchased from Santa Cruz (Santa Cruz, CA); Alexa 568-labeled anti-rabbit antibody and streptavidin-Alexa 568 were from Invitrogen (Carlsbad, CA); FITC-conjugated anti-SMα-actin was from Sigma Chemical Co. (St. Louis, MO); and biotinylated mouse anti-BrdU was obtained from Zymed Laboratories (South San Francisco, CA). CCR3 neutralizing rat anti-mouse monoclonal antibody (clone 83103) and isotype control IgG were purchased from R&D Systems (Minneapolis, MN) 20. Blocking antibody against mouse CCR5 (generated against amino acids 9–30 of mouse CCR5; rat IgG2c, clone: C34-3448) and isotype control were from BD pharmingen 21.
Aortic transplantation
B/c wild type, B6 wild type, and CCR1-deficient B6 male mice aged 8–12 w and weighing 20–25 g served as recipients or donors. Aortic transplantation was performed as described 4. No immunosuppression was used. Grafts that became occluded within the first 10 days were considered technical failures and excluded from further consideration. The graft success rate was >90%.
Recipient treatment with RANTES and BrdU
We implanted osmotic pumps subcutaneously to continuously treat B6 WT and/or B6CCR1−/− host animals with recombinant mouse RANTES (R&D) at 40 μg for 4 weeks, starting 4 weeks after aortic transplantation. RANTES administration was delayed for 4 weeks after aortic transplantation to allow resolution of lymphocyte accumulation in the donor aortas and to develop the early intimal lesions. Pyrogen-free PBS was administered to control mice. These recipient mice also received BrdU (Sigma) 50mg/kg/day for one week via osmotic pump prior to donor aorta harvest. ELISA assay showed that the serum RANTES levels at harvest were 96.7 ± 58.3 ng/ml in B6 WT receiving PBS (mean ±SD, n=8), and 80.2 ± 25.8 ng/ml in B6 CCR1−/− receiving PBS (n=8, p =0.54); 403.3 ± 333.2 ng/ml in B6 WT receiving RANTES (n=8), and 360.6 ± 212.0 ng/ml in B6 CCR1−/− receiving RANTES (n=8, p = 0.78).
Bone marrow transplantation (BMT)
BMT was performed as described previously.4 Briefly, 6–8 wk old male B6 wild-type or B6 CCR1−/− mice received a lethal dose of whole body irradiation (770 rads) from a cesium gamma source. Femurs and tibiae of male, 8–12 wk old B6 wild-type mice were excised and the marrow was flushed with RPMI using a syringe with a 30 gauge needle. Irradiated recipients received 1.0 × 107 bone-marrow cells in 0.2 ml RPMI by tail vein injection. Engraftment of transplanted marrow cells were confirmed by flowcytometry and routinely constituted 80–90% CCR1 positive cells in peripheral circulating mononuclear cells. Aortic transplantation from B/c into B6 wild-type or B6 CCR1−/− hosts was performed at least 4 wk after BMT.
Murine SMC Isolation
Descending thoracic aortas or aortic allografts were harvested from recipient WT B6 mice 8–12 weeks after transplantation using sterile microdissecting scissors under a dissecting microscope. After carefully removing the adventitia, aortas are soaked in 70% ethanol for 5 min, then placed in a 60-mm Petri dish, cut into pieces, and incubated in DMEM culture medium with 1 mg/mL type II collagenase (Worthington Biochemical Corporation) and 10% fetal bovine serum (FBS) for 1h at 37°C. Digested tissue and medium were transferred to a 50-ml conical polypropylene tube and centrifuged for 10 min; the medium was aspirated off and cells were resuspended in 40 ml fresh medium. After further centrifugation, cells were resuspended in fresh culture medium (DMEM with 10% FCS, 1% penicillin/Streptomycin (Life Technologies), 1% glutamine (BioWhittaker) and incubated in a 60-mm Petri dish. The purity of the SMLC and SMC cultures was >95%, as determined by immunofluorescence with anti-SM α-actin and SM1 Abs. SMC and SMLC of passages 4–8 were used for the study.
RNase Protection Assay
RNase protection assays (RPA) for chemokine receptor mRNA were conducted with multi-probe templates according to the manufacturer’s protocol (RiboQuant assay kit, BD-PharMingen). Gels were scanned and radioactive bands quantitated with a phosphoimager (Molecular Dynamics, Sunnyvale, CA), normalizing against the GAPDH housekeeping gene.
Fluorescence confocal microscopy
Frozen slide samples of human cardiac allografts or murine aortic allografts were fixed with 4% paraformaldehyde, blocked with 5% goat serum, and incubated with antibodies (anti-CCR1, SM alpha-actin or, BrdU) following appropriate secondary antibodies. For confocal microscope analysis, we used a Zeiss LSM 510 UV Meta laser scanning confocal microscope at ×63 magnification. Images were analyzed using Zeiss imaging software.
Co-localization analysis
We analyzed confocal microscope images for co-localization of SM α-actin, CCR1, or BrdU through ImageJ (W.S. Rasband, National Institutes of Health, Bethesda, Md., 1997–2004, http://rsb.info.nih.gov/ij/). Acquired images were analyzed as individual green and red channels corresponding to SMA (green) and anti-CCR1 (red) staining, or SMA (green) and BrdU (red) staining respectively. Background staining was corrected using the background subtraction function on ImageJ. Regions of interest were selected based on patching patterns in both red and green channels. Areas that contained staining artifacts, as indicated by intense regions of fluorescence in both channels, were excluded from analysis. Manders overlap coefficient, a measure of overlap of the red and green signals, was generated using the Manders coefficient plug-in 22, which is included in the WCIF version of ImageJ (http://www.uhnresearch.ca/facilities/wcif/imagej/), with regions of interest for analysis set for the green channel. Coefficients for 10 separate images were averaged.
SMC proliferation assay
SMC and SMLC were plated (1×104 cells/well) in quadruplicate to 96-well flat-bottom tissue culture plates (Costar, Cambridge, MA), maintained in DMEM with 10% FBS, and incubated at 37°C in a humidified atmosphere of 5% CO2. At confluence, cells were serum deprived for 48 h before treatment with chemokines with or without phosphatidylinositol 3-kinase (PI3K) inhibitor (wortmannin) or mitogen-activated protein kinase (MAPK) inhibitors (SB202190, SP600125 (JNK inhibitor), or PD98059). Cell proliferation was assessed by pulsing the wells for 6 h with 1 μCi of tritiated thymidine (New England Nuclear, Boston, MA). Proliferation was measured as incorporated radioactivity (cpm) using a Betaplate scintillation counter (LKA Pharmacia). Results were expressed as the mean ± SEM 23. For neutralizing CCR1, CCR3, or CCR5, we added 1 μg/mL of rabbit anti-mouse CCR1 (sc-7934, Santa cruz) polyclonal antibody, rat anti-mouse CCR3 (clone 83103, R&D systems), or mouse anti-mouse CCR5 (sc-17833, Santa Cruz) monoclonal antibodies, or control IgG (R&D Systems, Minneapolis, MN) to the culture medium20.
Intracellular Ca2+ influx measurement
Intracellular Ca2+ influx measurement was performed using Fluo-4NW (no wash) calcium assay kit (Molecular Probes). Cells were seeded on a 96-well black plate (Packard BioScience) in Dulbecco’s modified Eagle’s medium with 10% calf serum. After cells grew to 90% confluence, cells were starved with serum free medium for 48 h. Cells were loaded with 90 μl of 1 μM Ca2+ -sensitive fluorescent dye Fluo-4NW for 30 min at 37°C and 30 min at room temperature, and 10 μl of 10× various chemokine solutions were added to each well (n=4 for each chemokine) after 60 sec of reading. Fluorescence signals (excitation 485 nm, emission 525 nm) were measured by the fluorescence microplate reader for 10 min at sampling intervals of 3s. The subtraction of the basal fluorescence intensity from the maximum fluorescence intensity was calculated to estimate intracellular Ca2+.
SMC migration assay
Migration was measured using 48-well modified Boyden chambers24 with polycarbonate filter membranes, 8-μm pores (Nalge Nunc International, Rochester, NY). 50,000 SMLC or medial SMC in 0.2% FBS DMEM medium were seeded to the upper well of the Boyden chamber. Recombinant chemokines (100 ng/mL, RANTES or MIP1-α) were loaded in quadruplicate wells of the lower Boyden chamber with 0.2% FBS DMEM medium. 0.2% FBS in DMEM with or without PDGF-BB (20 ng/mL) served as a positive or negative control, respectively. After 48 h of incubation, cells were trypsinized, washed three times with PBS, and counted with a hemocytometer. Cell viability was determined by trypan blue exclusion. Values are expressed as the ratio of cells migrating in response to negative control (0.2% FBS in DMEM).
Western Blots
For CCR1 staining, we extracted proteins from cultured intimal SMLC, cultured medial SMC, wild-type C57BL6 spleen, or CCR1−/− spleen with RIPA buffers with proteinase inhibitors (Sigma). For JNK staining, intimal SMLC were treated for the indicated time with 100 ng/ml RANTES with or without MAPK inhibitors. Cell lysates with RIPA buffers with phosphatase inhibitors (Sigma) were loaded and separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Following transfer to Zeta-probe blotting membranes (Bio-Rad, Richmond, CA), blots were blocked overnight with 5% dry milk in TBS buffer (125 mM NaCl, 25 mM Tris pH8.0, 0.05% Tween 20) at 4° C and then probed with antibody (1 μg/ml) against CCR1 or unphosphorylated or phosphorylated JNK (Cell Signaling, Beverly, MA). Enhanced chemiluminescence (ECL) (Amersham, Piscataway, NJ) was used for detection.
Statistical Analysis
Numerical data with normal distribution are presented as mean ± SD. Comparison between 2 groups with normally distributed data with equal or unequal variance was performed using a one-way ANOVA or 2-sample t-test for unequal variance, respectively. Since the GAD lesion area was not normally distributed, the Mann-Whitney test was used to evaluate differences in intimal lesion area. Statview 4.5 for Macintosh, STATA 10, or GraphPad Prism 5 were used for all statistical calculation.
Results
Intimal SMLC express significantly higher levels of CCR-1 compared to medial SMC
To characterize the behavior and characteristics of intimal SMLC versus medial SMC, we developed a method for primary SMLC culture from murine aortic allografts. In 8- to 12-week-duration murine aortic allografts transplanted across major histocompatibility barriers (SI-1, Figure SI-1A), early vigorous rejection virtually eliminated the original donor medial smooth muscle cells (SMC)4 (Figure S2B). Consequently, culture of these 8- to 12-week aortic allografts permits isolation of a population highly enriched for intimal SMLC. We cultured medial SMC from native, uninjured recipient aortas. The cultured SMC from these two sources have comparable patterns of SM α-actin staining on confocal microscopy (Figure S3).
Demonstration that SMLC express chemokine receptors distinct from medial SMC entailed extracting total RNA from cultured intimal SMLC and medial SMC with or without prior cytokine stimulation, and performing RNase protection assay (RPA). Intimal SMLC but not SMC express CCR1 mRNA after treatment with IFN-γ and TNF-α (Figure 1A). Neither intimal SMLC nor medial SMC express mRNA for CCR2, CCR3, CCR4, CCR5, CXCR2, CXCR3, or CXCR4 (Figure 1A). Affymetrics gene® chip experiments (mouse gene® chip U74) using cDNA constructed from total RNA from cultured intimal SMLC and medial SMC after combined IFN-γ and TNF-α stimulation yield the same results that CCR1 expression was the only difference in chemokine receptor expression between intimal SMLC and medial SMC (Figure 1B).
Figure 1.


Chemokine receptor mRNA expression in medial SMCs vs intimal SMLCs. A, After culture for 24 hours in the presence of no cytokines (C), 500 U/mL interferon-γ (γ), 10 ng/mL tumor necrosis factor-α (α), or both cytokines (γ+α), cells were harvested and analyzed by RNase protection assay (RPA). Medial SMCs and nonstimulated SMLCs in culture did not express mRNA for any chemokine receptor (CC and CXCR RPA). Only intimal SMLCs showed chemokine receptor expression, and only after culture in TNF-α or TNF-α plus interferon-γ. m Indicates initial RPA probes. B, Fluorescence intensity values for chemokine receptor genes preferentially expressed in intimal SMLCs (open bars) and medial SMCs (solid bars) after culture for 24 hours in the presence of interferon-γ and TNF-α as measured by oligonucleotide microarrays. Error bars indicate variation in 2 hybridizations from duplicate preparations of the same sample. C, Western blot and densitometric analysis demonstrates higher expression of CCR1 by intimal SMLCs (iSMLC) than by medial SMCs (mSMC; a and b). B6 WT splenocytes served as positive control (pc), and B6 CCR1−/− splenocytes served as negative control (nc). D, Flow cytometry of intimal SMCs (iSMLC), medial SMCs (mSMC), and B6 CCR1−/− SMCs bearing SM myosin heavy chain. SM1, SMC marker, a through c; higher expression of CCR1 by intimal SMLCs than medial SMCs (d and e). B6 CCR1−/− SMCs served as negative control (c and f).
We used western blots of protein extracts from cultured intimal SMLC and medial SMC to determine CCR1 protein levels. Proteins extracted from spleens of wild-type C57BL/6 (B6, H-2b) mice served as positive controls, and those from B6 CCR1-deficient animals as negative controls. Western blot showed that cytokine-stimulated intimal SMLC have higher expression of CCR1 protein than either B6 or BALB/c (B/c, H-2d) medial SMC (Figure 1C, a and b). Flow cytometry also shows higher mean fluorescence intensity of CCR1 expression in intimal SMLC compared to B6 medial SMC (Figure 1D).
Intimal SMLC of GAD lesions of human cardiac allografts and murine aortic allografts express CCR-1
Confocal immunofluorescence microscopy identifies CCR1-positive cells co-localizing with SM α-actin-positive cells in the intima of GAD lesions in human cardiac allografts (Figure 2A, a–c). However, SM α-actin-positive cells in the medial SMC (Figure 2A, d–f) and normal aortic intimal cells (Figure 2A, g–i) do not express CCR1. A majority of SM α-actin-positive cells in GAD lesions are also CCR1-positive (64.6 ± 8.6%, n=10) as shown by fluorescent intensity spectrum analysis (Figure 2B, a and b) with a Manders coefficient of 0.86 ± 0.02 (n=10)22. Similarly, confocal immunofluorescence microscopy identified CCR1-positive cells that co-localized with SM α-actin-positive cells in the intimal lesions in BALB/c aortic allografts in C57BL6 host 8 weeks after transplantation. (Figure 2C, a–c), but not in the medial SMC (Figure 2C, d–f). A majority of SM α-actin-positive cells in murine GAD lesions are also CCR1-positive (57.2 ± 13.2%, n=10), while only 8.1 ± 0.9% (n=10, p < 0.0001) of medial SMC stain for CCR1 (Figure 2D, a and b), with a Manders coefficient in GAD lesions of 0.87 ± 0.02 (n=10). Thus, intimal SMLC have a pattern of chemokine receptor expression distinct from medial SMC in human and murine GAD.
Figure 2.

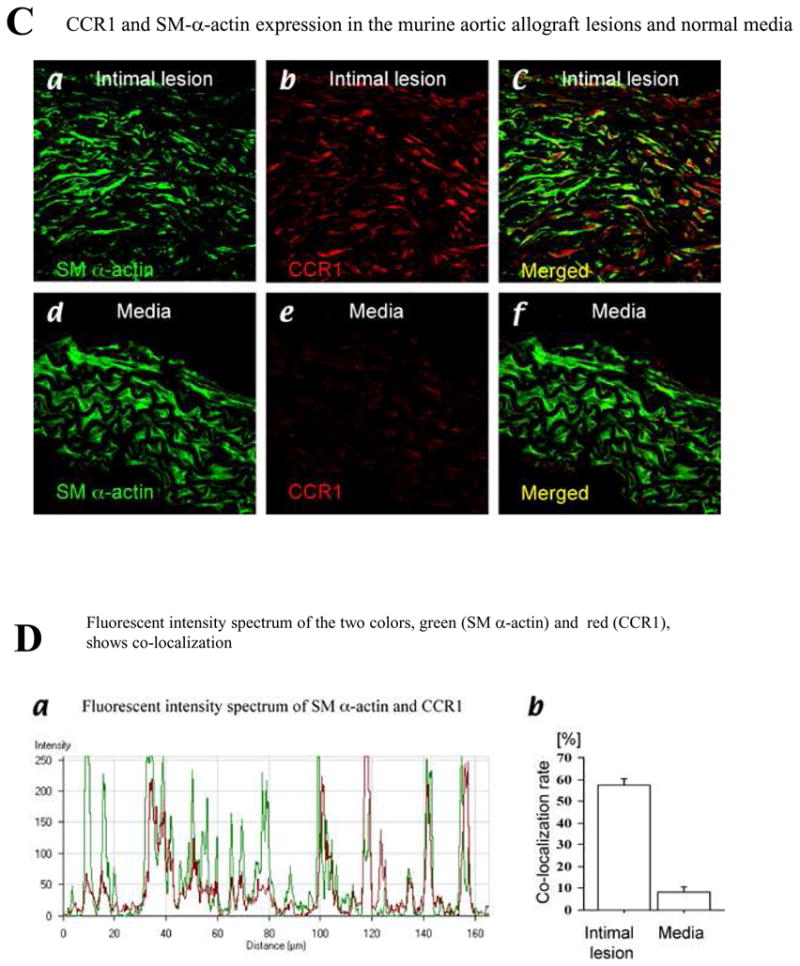
A, Confocal microscopic examination of coronary artery of human cardiac allograft (a through f) and human normal carotid arteries (g and h) stained with SM α-actin (a, d, and g) or CCR1 (b, e, and h) and merged image of SM α-actin and CCR1 (c, f, and i). B, Fluorescent intensity spectrum of SM α-actin (green) and CCR1 (red) of coronary artery of human cardiac allograft demonstrated colocalization of SM α-actin and CCR1 in intimal SMLCs (a); colocalization rate in intimal lesions and medial portions of coronary arteries of human cardiac allograft or normal intima of human carotid arteries (b). C, Confocal microscopic examination of murine aortic allografts (a through c) and murine normal aortas (d through f) stained with SM α-actin (a and d) or CCR1 (b and e) and merged image of SM α-actin and CCR1 (c and f). D, Fluorescent intensity spectrum of SM α-actin (green) and CCR1 (red) of murine aortic allografts demonstrated colocalization of SM α-actin and CCR1 in intimal SMLCs (a); colocalization rate in intimal lesions of murine aortic allografts and medial portions of murine normal aortas (b).
CCR-1 ligand RANTES induces intimal SMLC but not medial SMC proliferation
Murine CCR1 ligands include MIP1-α (CCL3), RANTES (CCL5), C10 (CCL6), mast cell activation-related chemokine (MARC/MCP-3/CCL7), MCP-2 (CCL8), and MIP1-γ (CCL9/10). In addition to CCR1, MIP1-α binds to CCR5, RANTES binds to CCR3 and 5, MARC binds to CCR2 and 3, and MCP-2 binds to CCR2, 3, and 5. C10/CCL6 shares substantial sequence similarity with a several chemokines, including MIP-1γ/CCL9/10, HCC-1/CCL14, and MIP-1δ/CCL15 25–27. In contrast to the prototypical CC chemokines, CCL14 and CCL15 have four exons, with a characteristic second exon that contributes importantly to their biologic effects 28. These homologous chemokines particularly attract T cells and monocytes 25.
To examine whether CCR1 ligation induces intimal SMLC and/or medial SMC proliferation, we assayed 3H-thymidine incorporation following treatment with various CCR1 ligands (Figure 3A). RANTES, C10, and MARC induce concentration-dependent intimal SMLC proliferation, but do not cause medial SMC proliferation. In contrast, MIP-1α and MIP-1γ do not stimulate proliferation of either intimal SMLC or medial SMC.
Figure 3.

A, 3H-thymidine incorporation of intimal SMLCs (iSMLC, open bars) and medial SMCs (mSMC, solid bars) stimulated with various chemokines. RANTES, C10, and MARC (CCR1 ligands) induced SMLC but not SMC proliferation in a dose-dependent manner. B, Intracellular Ca2+ influx measurement. Basal fluorescence intensity was subtracted from maximum fluorescence intensity to estimate intracellular Ca2+. Changes in fluorescence intensity were largest in RANTES-treated intimal SMLCs among various CC chemokines. Fluorescence intensities in RANTES-treated, C10-treated, and MARC-treated intimal SMLCs were significantly larger than in medial SMCs (543.2±262.6 in intimal SMLCs, 37.6±19.2 in medial SMCs, P<0.005; 272.2±73.3 in intimal SMLCs, 34.2±33.1 in medial SMCs, P<0.005; and 176.0±46.9 in intimal SMLCs, 31.2±15.3 in medial SMCs, P<0.0005, respectively). MIP-1α – treated and MIP-1γ – treated intimal SMLCs also showed significantly higher calcium influx responses than did medial SMCs (149.4±15.8 in intimal SMLCs, 42.2±20.4 in medial SMCs, P<0.0001; 116.4±34.6 in intimal SMLCs, 16.0±13.2 in medial SMCs, P<0.0003). PDGF-treated intimal SMLCs and medial SMCs showed comparably strong responses (630.4±68.9 in intimal SMLCs, 620.0±156.0 in medial SMCs, P=0.89). C, Migration assay of intimal SMLCs (open bars) and medial SMCs (solid bars) by modified Boyden chamber methods. RANTES induced higher rates of migration of intimal SMLCs than medial SMCs, but negative control, positive control (PDGF), and MIP-1α showed comparable migration rates between intimal SMLCs and medial SMCs. Ctrl indicates control. D, 3H-thymidine incorporation of intimal SMLCs treated with 1 μL/mL anti-CCR3 or anti-CCR5 blocking antibodies or isotype-matched control (ctrl) antibodies followed by stimulation with 100 ng/mL RANTES.
CCR-1 ligand RANTES induces significantly higher Ca2+ influx by the intimal SMLC compared to medial SMC
Ca2+ influx importantly regulates growth factor-mediated SMC proliferation 29, 30; platelet-derived growth factor (PDGF) also activates Ca2+ entry into vascular SMCs in the absence of intracellular Ca2+ mobilization 29. To examine the effects of CCR1 ligation on intracellular calcium influx, we labeled intimal SMLC and medial SMC with Fluo-4 NW (Molecular Probes) and stimulated them with various ligands; an increase in fluorescence signal corresponded to a rise in intracellular Ca2+ in the Fluo4-labeled SMC. Intimal SMLC treated with RANTES show the largest Ca2+ influx among the various CC chemokines tested (Figure 3B). RANTES, C10, or MARC elicit significantly more fluorescence in intimal SMLC compared to medial SMC (p < 0.005). Although MIP-1α and MIP-1γ do not affect SMC proliferation, MIP-1α-treated and MIP-1γ-treated intimal SMLC nevertheless show significantly higher calcium influx responses than do medial SMC (p=0.0003). Intimal SMLC and medial SMC treated with platelet-derived growth factor (PDGF) show comparably strong responses (630.4±68.9 in intimal SMLC, 620.0±156.0 in medial SMC, p=0.8949).
CCR-1 ligand RANTES induces intimal SMLC but not medial SMC migration
We then performed a migration assay using cultures of intimal SMLC or medial SMC with modified Boyden chamber methods. PDGF-BB served as a positive control; the absence of chemokines or PDGF-BB served as a negative control. Intimal SMLC cultured with RANTES show augmented migration rates compared to medial SMC (Figure 3C). In contrast, intimal SMLC and medial SMC cultured with MIP-1α, negative control, or PDGF-BB show comparable migration rates (Figure 3C). We also performed an alternative migration assay using cultures of intimal SMLC or medial SMC adjacent to HEK293 cells transfected with various chemokines (RANTES, C10, and MIP-1α); transfected PDGF-BB served as a positive control and transfected green fluorescent protein (GFP) served as a negative control. Intimal SMLC co-cultured with RANTES- or C10-transfected-HEK293 cells show augmented migration rates compared to medial SMC (Figure S4). In contrast, intimal SMLC and medial SMC co-cultured with MIP-1α-, control (GFP)-, or PDGF-transfected-HEK293 cells show comparable migration rates. The concentration of C10, RANTES, MIP-1α, and PDGF-BB in the co-culture medium ranged from 20–60ng/ml.
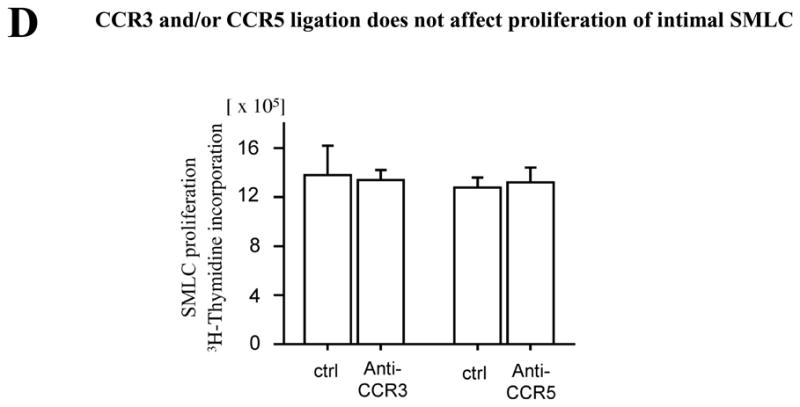
CCR3 and/or CCR5 ligation does not affect intimal SMLC proliferation
RANTES can bind CCR1, CCR3, and CCR5. Several investigators reported that SMC express CCR1 as well as CCR3 and CCR5 14, 31, 32, although in our hands RNase protection assay did not detect CCR3 or CCR5 mRNA expression by intimal SMLC. To examine whether RANTES can ligate CCR3 or CCR5 and induce SMLC proliferation, we treated SMLC with RANTES and assayed 3H-thymidine incorporation with anti-CCR3 or -CCR5 blocking antibody versus isotype control monoclonal antibodies. Intimal SMLC cultured with 100 ng/mL of RANTES and 1 μg/mL of anti-CCR3 or -CCR5 blocking antibody or control IgG showed comparable 3H-thymidine incorporation (Figure 3D), indicating that CCR3 and/or CCR5 ligation does not affect proliferation of intimal SMLC. Of note, the absence of neutralizing antibodies or small molecules to inhibit CCR1 binding by its ligands in rodents prevented us from performing comparable CCR1-blocking experiments.
JNK signaling pathways involve RANTES-induced SMLC proliferation
Phosphatidylinositol 3-kinase (PI3K) and/or mitogen-activated protein kinase (MAPK) pathways can participate in SMC migration and proliferation.33, 34 To assess the roles of the MAPK and PI3K pathways in chemokine-induced SMLC proliferation, RANTES- or C10-stimulated SMLC (100 ng/mL) were incubated with SB202190 (p38 inhibitor), SP600125 (JNK inhibitor), wortmannin (PI3K inhibitor), or PD98059 (MEK1 inhibitor). PD98059 and SP600125 significantly reduce RANTES- and C10-induced SMLC proliferation (Figure 4A), while treatment with either the PI-3 kinase inhibitor wortmannin or the p38 inhibitor SB202190 does not affect SMLC proliferation (Figure 4A). RANTES also induces phosphorylation of ERK and JNK in SMLC (Figure 4B). Consistent with an important role for JNK signaling pathways in RANTES-induced SMLC proliferation, JNK siRNA significantly reduces JNK expression (Figure 4C) as well as RANTES-induced 3H-thymidine incorporation in SMLC (Figure 4D).
Figure 4.

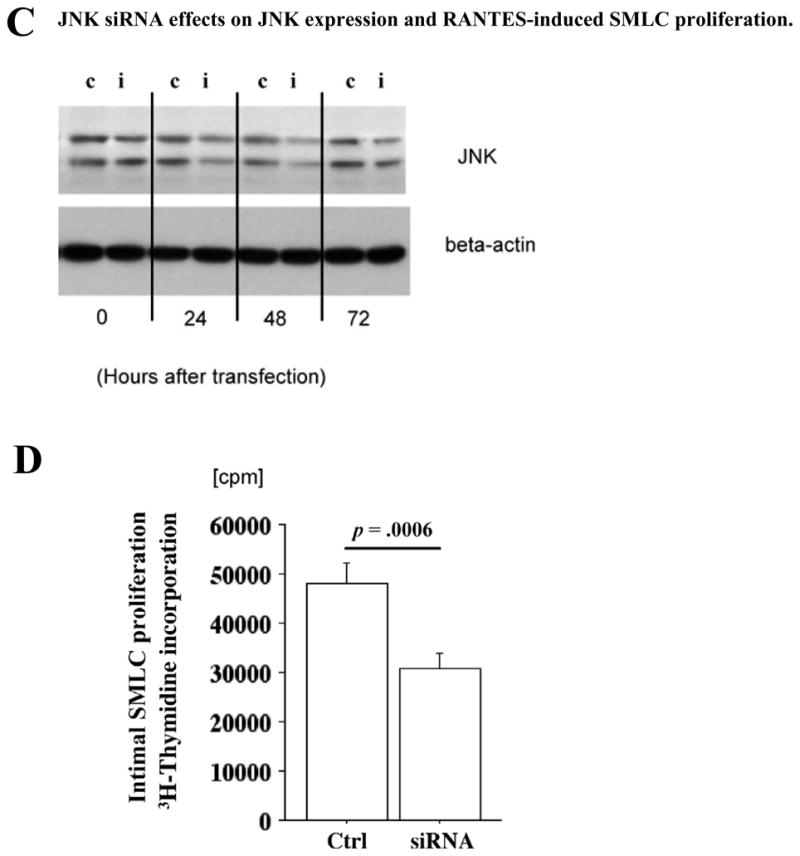
A, Proliferation of SMLCs (3H-thymidine incorporation) stimulated with 100 ng/mL RANTES or C10 in the presence or absence of various kinase inhibitors: SB202190 (p38 inhibitor, 5 μmol/L), SP600125 (JNK inhibitor, 10 μmol/L), wortmannin (phosphatidylinositol 3-kinase inhibitor, 1 μmol/L), and PD98059 (MEK1 inhibitor, 50 μmol/L). 3H-thymidine incorporation of RANTES-stimulated SMLCs decreased significantly with PD98059 (10.1±0.3×104 cpm, n=4, P<0.001) and with SP600125 (7.2±0.5×104 cpm, P<0.0001) compared with control cells (ctrl; 15.6±1.7×104 cpm, n=4). 3H-thymidine incorporation of C10-stimulated SMLCs also significantly decreased with PD98059 (8.7±0.7×104 cpm, n=4, P<0.0001) and with SP600125 (6.0±0.5×104 cpm, n=4, P<0.0001) compared with control cells (13.9±0.6×104 cpm, n=4). B, Western blot demonstrated that RANTES induced phosphorylation of extracellular signal-regulated kinase (ERK) and JNK in SMLCs. C, Western blot demonstrated that JNK small interfering RNA (siRNA) reduced JNK expression. c Indicates control; i, siRNA treatment. D, 3H-thymidine incorporation of RANTES-stimulated SMLCs decreased significantly with JNK siRNA (3.0±0.3×104, P=0.0006) compared with RANTES-stimulated SMLCs treated with RNA interference–negative controls (4.9±0.5×104 cpm, n=4). Ctrl indicates control.
Host CCR-1 depletion attenuates aortic allograft GAD
To evaluate whether CCR1 signaling attenuates GAD in vivo, we performed aortic transplantation using a fully allomismatched (major histocompatibility complex (MHC) class I and class II mismatched) combination of B/c (H-2d) and B6 (H-2b) mice. Thoracic aortic donor grafts were anastomosed into host infrarenal abdominal aortas and harvested eight weeks after transplantation. No immunosuppressive agent was administered to avoid confounding effects on inflammatory cell accumulation. Aortic allografts in B6 CCR1-deficient hosts show significantly reduced GAD lesions compared to allografts in wild-type (B6 WT) recipients (Figure 5A, p<0.001). To ensure that the effect of host CCR1 deficiency did not derive from diminished inflammatory cell recruitment, we examined aortic allografts one week after transplantation, at the peak of acute rejection4. Aortic allografts in B6 CCR1-deficient hosts show comparable numbers of CD4+ and CD8+ cells as well as CD11b+ macrophages relative to WT recipient allografts (Figure 5B); moreover, subsequent harvest at 8 weeks shows complete loss of donor medial SMC (Figure 5A), indicating a comparable degree of allograft rejection. This result indicates that CCR1 depletion alone does not affect T cell and macrophage migration, accumulation, or activity in vivo, and suggests that redundant pathways allow inflammatory cell recruitment in the absence of CCR1.
Figure 5.
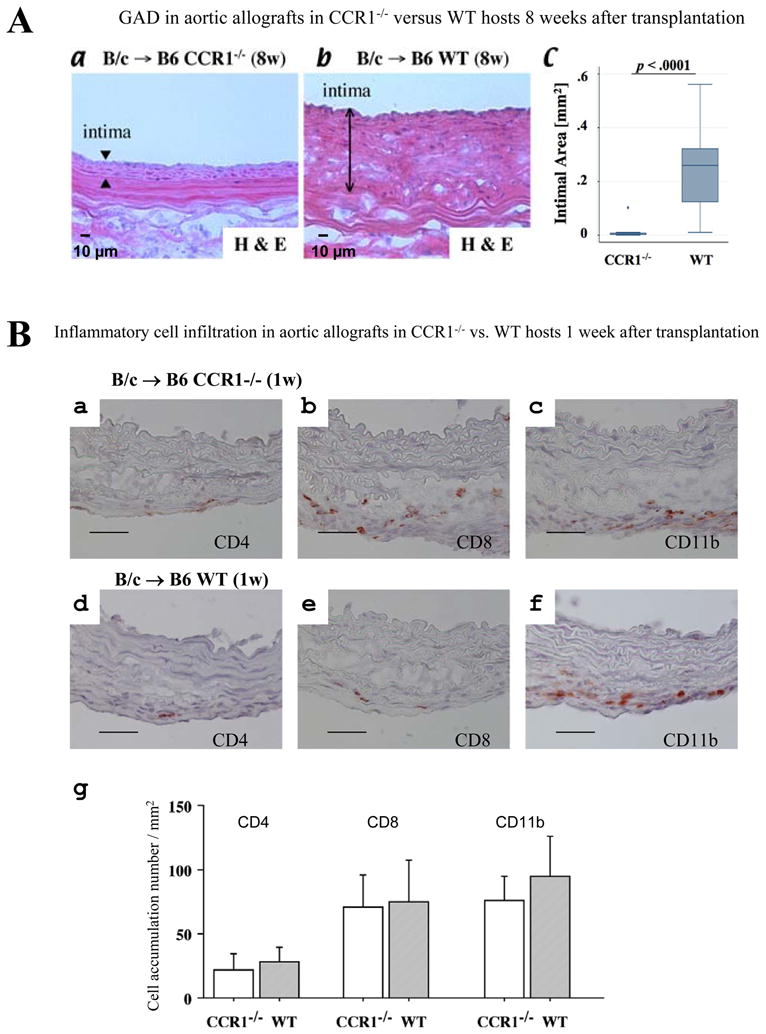
A, Hematoxylin-and-eosin (H&E) staining of intimal lesions of aortic allografts in CCR1- deficient recipients (a) vs WT recipients (b). Bar=10 μm. c, Allografts in CCR1-deficient hosts showed significantly reduced intimal area of GAD lesions (median [interquartile range] 0.002 [0.001 to 0.007] mm2, n=10, P<0.001) compared with allografts in WT recipients (0.26 [0.12 to 0.32] mm2, n=12). B, Immunohistochemistry of total allogeneic aortic allografts. BALB/c aorta transplanted into B6CCR1−/− (a, b, and c) or WT B6 (d, e, and f) 1 week after transplantation; staining with CD4 (a and d), CD8 (d and e), and CD11b (c and f). Bar=20 μm. g, Graft accumulation number of inflammatory cells per section of the aortic allografts of CCR1−/− (n=6) and WT (n=6) recipients was counted under the microscope. 8w Indicates 8 weeks; 1w, 1 week.
RANTES induces intimal SMLC proliferation in vivo in aortic allografts
To evaluate whether CCR1 ligation induces intimal SMLC proliferation and intimal thickening in vivo, we performed aortic transplantation followed by continuous RANTES or PBS (control) infusion for 4 weeks (Figure 6A). Each aortic allograft recipient also received BrdU infusion for 4 weeks before harvest of the donor aortas to assess in vivo DNA replication in intimal SMLC35. Neither RANTES nor PBS infusion induce intimal thickening in B6 CCR1−/− recipient allografts (Figure 6B, a and b), whereas RANTES infusion induces significantly more severe intimal thickening (the median with interquartile range, 0.34, 0.31–0.36 mm2, n=9) with greater luminal stenosis (92.5, 89.8–93.2%, n=9) in B6 WT recipient allografts than does PBS infusion (0.21, 0.17–0.25 mm2, n=12, p<0.0001; 36.7, 32.3–46.8%, n=12, p<0.0001, respectively) (Figure 6B, c–f).
Figure 6.

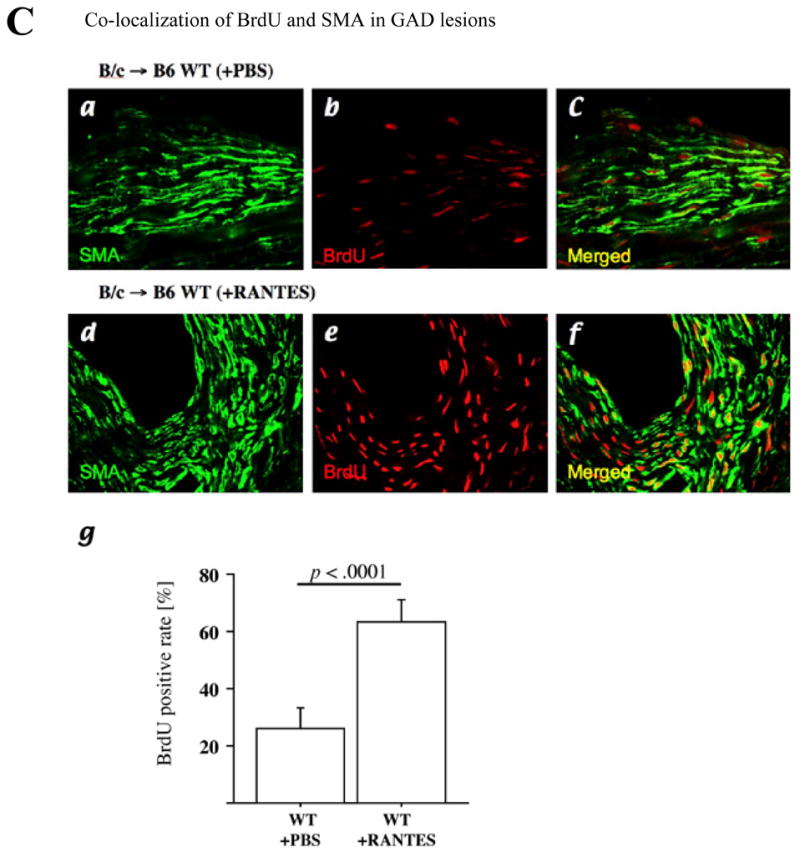
A, Experimental scheme of RANTES infusion in vivo experiment. A miniaturized osmotic pump in the dorsal lower pocket delivered recombinant RANTES (10 μg per week) for 4 weeks (4w) to B6 CCR1−/− or B6 WT recipients of B/c aortic allografts 4 weeks after aortic transplantation (each n=6). PBS served as negative control (a and c; each n=6). B, Hematoxylin-and-eosin staining of aortic allografts of CCR1−/− (a, with PBS; b, with RANTES) or B6 WT recipients (c, with PBS; d, with RANTES) 8 weeks after transplantation. Bar=200 μm. RANTES infusion did not affect intimal area (e) or luminal stenosis (f) of CCR1-deficient host allografts, but RANTES infusion significantly augmented intimal area of GAD lesions compared with PBS infusion. C, Confocal microscopic pictures of BrdU (red) incorporation in intimal SM α-actin (SMA; green)–positive cells responding to RANTES infusion; upper panels, aortic allograft intimal lesions of B6 WT recipients receiving PBS, and lower panels, B6 WT recipients receiving RANTES. Confocal microscopic analysis showed that RANTES infusion significantly increased BrdU (red) incorporation in intimal SM α-actin (green)–positive cells in aortic allografts compared with control PBS treatment group (g).
The intimal lesions of B6 WT recipients receiving RANTES infusion also contain significantly more BrdU-labeled nuclei than those of B6 recipients receiving PBS (Figure 6C, a–f). The labeling index calculated from the percentage of BrdU-labeled cells relative to the total number of SM α-actin positive cells in the intimal lesions shows marked augmentation of DNA replication in RANTES-treated recipient allografts (63.2 ±7.8%, n=9, versus 26.3 ±7.2% in PBS-treated B6WT recipient allografts; p < 0.0001, n=12) (Figure 6, g). These results demonstrate that intimal SMLC, but not medial SMC, express functional CCR1, and that CCR1 signaling promotes SMLC proliferation as well as GAD development in vivo.
RANTES infusion increases inflammatory cell accumulation in CCR1−/− recipient allografts, but does not drive GAD development
RANTES can ligate both CCR1 and CCR5 expressed on T cells and monocyte/macrophages and thereby stimulate these inflammatory cells. RANTES infusion possibly stimulates inflammatory cells, subsequently and indirectly inducing SMLC proliferation. To examine whether RANTES infusion modulates inflammatory cell accumulation in the aortic allografts, we performed immunohistochemistry (Figure 7A). Indeed, RANTES infusion significantly increases CD4+ (T cell) and MAC3+ (macrophage) cell accumulation in the aortic allografts of WT hosts compared to PBS infusion. By the same token, RANTES infusion significantly increases inflammatory cell accumulation in CCR1−/− recipient allografts (Figure 7B) even though such infusion does not drive GAD development (Figure 6). The study indicates that RANTES can modulate inflammatory cell accumulation in the GAD lesions (presumably via CCR5), but that SMLC requires CCR1 ligation for SMLC proliferation and intimal lesion formation.
Figure 7.

A, Immunohistochemistry of 8-week aortic allografts; BALB/c aorta transplanted into B6CCR1−/− with PBS (a and b) or RANTES (c and d) treatment or WT B6 with PBS (e and f) or RANTES (g and h) treatment; staining with CD4 (a, c, e, and g) and Mac3 (b, d, f, and h). Bar=20 μm. B, Graft accumulation number of inflammatory cells per section of the aortic allografts of CCR1−/− and WT recipients treated with PBS or RANTES (each n=6) was counted under the microscope.
Aortic allografts of B6 CCR1−/− host receiving B6 WT bone marrow do not develop intimal lesions
Since inflammatory cells also express CCR-1 and can conceivably contribute to GAD by driving the extent of allograft rejection, we specifically asked whether CCR1-bearing inflammatory cells contribute to the intimal lesion formation. We therefore transplanted B6 WT bone marrow into either irradiated B6 WT or B6 CCR1 deficient mice (bone marrow transplantation, BMT); 2 weeks later we performed aortic transplantation into these bone marrow transplanted hosts. If CCR1-bearing inflammatory cells contribute importantly to the intimal lesion formation, aortic allografts in B6 CCR1−/− hosts receiving B6 WT bone marrow should have induced intimal lesion formation. However, this was not the case (Figure 8). Allografts in CCR1-deficient hosts show significantly reduced intimal area due to GAD (the median and interquartile range, −0.005, 0–0.03 mm2, n=4, p=0.001) compared to allografts in WT recipients (0.19, 0.14–0.265 mm2, n=4). B6 CCR1−/− host allografts following B6 WT BMT did not develop intimal lesions, indicating that CCR1+ inflammatory cells do not importantly contribute to intimal lesion formation.
Figure 8.

A, Experimental protocol for combined BMT and aortic transplantation. 2w Indicates 2 weeks; 4w, 4 weeks. B, Immunohistochemistry from 8-week aortic allografts after BMT; B6 WT(a) and/or B6 CCR1−/− hosts (b) received B6 WT BMT. BALB/c aortas were transplanted 2 weeks after BMT and evaluated 8 weeks later. Bar=20 μm.
Discussion
This study demonstrates a number of important findings: intimal SMLC but not medial SMC express higher levels of CCR1 in human and murine GAD lesions; CCR1 ligation by RANTES induces intimal SMLC migration and proliferation in vitro; aortic allografts in CCR1-deficient recipients show significantly reduced GAD development despite the recruitment of graft-infiltrating lymphocytes comparable to WT hosts; and RANTES accelerates GAD lesion formation with intimal SMLC proliferation in murine aortic allotransplantation.
A previous study by Gao, et al. demonstrated attenuation of GAD in cardiac allografts transplanted into CCR1-deficient hosts. The administration of anti-CD4 antibody and cyclosporine confounded these studies36, and precluded the conclusion that the effect resulted from blocking the recruitment of inflammatory cells versus other cell type. The investigators concluded that CCR1 partially contributed to reduction of graft accumulation of inflammatory cells, thereby attenuating chronic rejection. Notably, the authors also reported that CCR1 depletion alone did not affect the accumulation of graft inflammatory cells in the absence of anti-CD4 antibody or cyclosporine36.
Heightened expression of a large variety of chemokines accompany GAD development.14 In the acute setting, chemokines recruit leukocytes, promoting damage to the donor organ. However, during chronic allograft responses, vascular wall or leukocyte-derived chemokines may exert other important actions in GAD pathogenesis, 15, 37 specifically in the recruitment and activation of SMLC precursors. RANTES, in particular, can arise from endothelial cells, T cells, macrophages, and platelets.15, 38 Although RANTES can activate monocytes/macrophages bearing CCR1 or CCR5 when immobilized on the surfaces of endothelial cells, and promotes monocyte/macrophage migration when localized in this fashion,39 this work makes the point that mononuclear inflammatory cell recruitment into allografts does not require CCR1 expression.
Conversely, intimal SMLC recruitment does not take place in the absence of CCR1. Although several receptors can bind RANTES, including CCR1, CCR3, and CCR5, 40 SMLC express only functional CCR1. RANTES ligation of SMLC CCR1 induces their proliferation and migration. In addition, other CC chemokines not typically elaborated in rejecting allografts (including C10 and MARC) can also induce SMLC proliferation. RANTES may activate SMLC via the MAPK pathway in SMLC, as evidenced by ERK and JNK phosphorylation, and in accord with findings on other cell types41.
This study demonstrated that among the known CCR1 ligands, RANTES most potently induces intimal SMLC proliferation. Various CCR1 ligands may behave differently with regard to intimal SMLC proliferation, perhaps because of binding to other CC chemokine receptor(s)or to different binding affinity. A semi-quantitative chemokine binding assay using fluorescent chemokine ligands (Figure S5) showed that CCR1 had relatively lower binding affinity for RANTES relative to MIP-1α (IC50 was 2.6 ± 0.2 ng/mL vs. 1.3 ± 0.2 ng/mL, respectively). Other investigators have also shown different affinities of various CC-chemokine ligands (MIP-1α and RANTES) to CCR1; Gao et al found that MIP-α and RANTES induced calcium influx in human PMN with EC50 of 5 and 50 nM, respectively42, indicating that MIP-1α has higher affinity for CCR1 than RANTES. However, binding affinity does not predict signaling efficacy through this receptor43. MIP-1α exhibits both the highest binding and signaling affinity. Nevertheless, although RANTES binds with a lower affinity (KD of 460 nM), it transmits a Ca2+ signal with much greater efficacy than MIP-1α 43. Several observations suggest that RANTES might interact differently with the CCR1 than does MIP-1α; Neote et al. showed that CCR1-transfected cells clearly mobilize Ca2+ in response to as little as 1 nM RANTES, and that CCR1-transfected cells are desensitized in their ability to flux Ca2+ in response to RANTES43. The physiological significance of chemokine binding and consequent signal transduction via CCR1 receptor requires further investigation.
Human intragraft vascular cells express high levels of IP-10/CXCL10, I-TAC/CXCL11, and CXCR3; plasma levels of I-TAC elevate in patients with severe GAD.16, 17 SDF-1a/CXCR4 also appears important for EC and/or SMLC progenitor cell trafficking to and from the bone marrow and for the migration to injured peripheral organs.44, 45 Previous studies demonstrated that SMC can express CCR1 (airway smooth muscle cells),32 CCR2 (rat aortic smooth muscle cells),46 CCR3 (airway smooth muscle cells, mouse aortic SMC), CCR5, CCR8, CXCR3, and CXCR4.14, 31, 32, 46 Indeed, various chemokines, including interferon-inducible protein 10 (IP-10, rat vascular smooth muscle cells),47 SDF-1,45 monocyte chemoattractant protein-1 (MCP-1), 48, 49 CC chemokine ligand 1 (CCL1, I-309, rat aortic smooth muscle cells),46 or eotaxin (CCL11),50 promote or inhibit SMC migration and/or proliferation.14 CXCR3 chemokines in particular exert potent biological effects on vascular cells as well as their well-recognized effects on mononuclear inflammatory cells. Several studies define functional receptors as “positive induction of calcium influx, MAPK phosphorylation, or tissue factor activity” 49 Region specific chemokine receptors may explain pathogenesis of certain disease such as asthma.32 However, no previous studies have differentiated intimal SMLC in the GAD lesion and medial SMC in normal arteries. PCR analysis can detect chemokine receptor mRNA with very high sensitivity so that it may detect mRNA derived from contaminating inflammatory cells. Western blot and/or flowcytometry would be preferable methodology with certain specificity. Several investigators detected CCR3, CCR5, CXCR3, and CXCR4 in vascular SMC, receptors whose cognate ligands can promote SMC migration and proliferation in vitro. However, these findings cannot explain the lack of medial SMC proliferation or expansion in the GAD lesions, even when accompanied by increased expression of cognate ligands such as MCP-1, SDF-1, Eotaxin, or RANTES. Indeed, infusion of RANTES (a ligand for CCR1, CCR3 and CCR5) did not induce medial SMC proliferation of the normal arteries and aortas.
Clinical Implications
Graft arterial disease remains a clinically important limitation to human cardiac transplantation. One-year, 3-year, 5-year, and 10-year patient survival following adult heart transplantation is 88%, 80%, 74%, and 52%, respectively,51 with the majority of mortality attributable to GAD. Intravascular ultrasound demonstrates GAD in 75% of patients at 3 years after transplantation; 52, 53 significant vasculopathy can develop as early as 6–12 months posttransplant.
Intimal SMLC predominate in GAD lesions. Ischemia-reperfusion, allo-specific humoral and cellular effectors, donor and host antigen presenting cells, and atherogenic factors contribute to the intimal lesion formation. Chemokines indirectly affect GAD formation since chemokines contribute to ischemia reperfusion injury and acute rejection.13 However, chemokines can also contribute directly to vascular remodeling and angiogenesis beyond their role in leukocyte chemotaxis.5
Intimal SMLC likely have multiple sources, beyond simple recruitment from donor medial cells. Intimal SMLC are clonal and phenotypically distinct from medial SMC;54–57 SMLC express hematopoietic markers such as CD14, CD34, CD105, Thy-1, c-kit, flt1, and flt-3.58–60 In any event, bone marrow-derived or any other circulating precursor cell must access grafts from the circulation;5, 61 the presence of smooth muscle progenitor cells present in human peripheral blood corroborates this notion.59, 60 We hypothesized that SMLC receptors and their cognate ligands promote intimal lesion formation. Such receptors must preferentially target SMLC, compared to intrinsic arterial SMC. The current study demonstrates that RANTES can potentiate the effects of proliferative signals and implies that chemokines contribute to both recruitment and expansion of intimal cells in GAD lesions. Clinical application will require further studies evaluating the effect of CCR-1 depletion and immunosuppressive therapy, alone or in combination, on allograft rejection and GAD.
Study Limitations
We examined the function of CCR1 of the SMLC on GAD in the setting of total allo-mismatched murine aortic transplant without immunosuppression. This condition does not completely recapitulate human solid organ transplantation. We demonstrated that intimal SMLC express CCR-1 by both immunohistochemistry and by a number of in vitro molecular and cellular techniques, but the immunohistochemistry on tissue specimens cannot exclude the possibility of contamination of transdifferentiated inflammatory cells although these cells might serve as one of SMLC precursors. CCR1-depletion may affect the development and function of immune cells. The present experiments using RANTES infusion and bone marrow transplantation of normal cells into CCR-1 deficient allograft recipients provide compelling evidence for the importance of SMLC bearing CCR-1 in chronic rejection. Nonetheless, we cannot rule out an inflammatory cell chemoattracting role of CCR-1 and other chemokine receptors in the development and function of immune cells and smooth muscle cells.
Conclusion
This study demonstrated a critical function for CCR1 and RANTES in the pathogenesis of GAD beyond leukocyte recruitment. Intimal SMLC but not medial SMC express functional CCR1 that responds to RANTES by proliferation and increased migration. In the absence of host CCR1, normal allograft inflammatory cell recruitment can occur, but GAD is attenuated. These results provide new pathophysiologic insight into the mechanisms of GAD, and also suggest novel therapeutic strategies for preventing allograft arteriopathy.
Supplementary Material
Acknowledgments
We thank J. Cho, D. Cameron, W. Cho, E. Shvartz, J. N. Vaisviliene, M. Rodrigue, and E. Simon-Morrissey for their technical expertise, and J. Perry for editorial assistance.
Sources of Funding
This work was supported by an American Society of Transplantation Faculty Development grant (KS), American Heart Association Scientist Development grant (KS), a grant award from the Roche Organ Transplantation Research Foundation (KS), and a Harvard Medical School BWH Fellowship Award (KS), a Brigham & Women’s Hospital Biomedical Institute Funds to Research Excellence grant (RNM), and also by grants from the Donald W. Reynolds Foundation (PL) and the National Institutes of Health (HL-43364 to PL, RNM; GM-67049 to RNM, KS, PL; HL-67249 to KS, RNM; HL-67283 to KS; HL-34636 to PL, MM; and HL-083154 to ML).
Abbreviations
- IFN-γ
interferon gamma
- TNF-α
tumor necrosis factor-alpha
Footnotes
Disclosures
None.
References
- 1.Libby P, Salomon RN, Payne DD, Schoen FJ, Pober JS. Functions of vascular wall cells related to development of transplantation-associated coronary arteriosclerosis. Transplant Proc. 1989;21:3677–3684. [PubMed] [Google Scholar]
- 2.Libby P, Pober JS. Chronic rejection. Immunity. 2001;14:387–397. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 3.Kennedy LJ, Jr, Weissman IL. Dual origin of intimal cells in cardiac-allograft arteriosclerosis. N Engl J Med. 1971;285:884–887. doi: 10.1056/NEJM197110142851603. [DOI] [PubMed] [Google Scholar]
- 4.Shimizu K, Sugiyama S, Aikawa M, Fukumoto Y, Rabkin E, Libby P, Mitchell RN. Host bone-marrow cells are a source of donor intimal smooth- muscle-like cells in murine aortic transplant arteriopathy. Nat Med. 2001;7:738–741. doi: 10.1038/89121. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu K, Mitchell RN. Stem cell origins of intimal cells in graft arterial disease. Curr Atheroscler Rep. 2003;5:230–237. doi: 10.1007/s11883-003-0029-7. [DOI] [PubMed] [Google Scholar]
- 6.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 7.Nagai R, Suzuki T, Aizawa K, Miyamoto S, Amaki T, Kawai-Kowase K, Sekiguchi KI, Kurabayashi M. Phenotypic modulation of vascular smooth muscle cells: dissection of transcriptional regulatory mechanisms. Ann N Y Acad Sci. 2001;947:56–66. doi: 10.1111/j.1749-6632.2001.tb03930.x. discussion 66–57. [DOI] [PubMed] [Google Scholar]
- 8.Caplice NM, Bunch TJ, Stalboerger PG, Wang S, Simper D, Miller DV, Russell SJ, Litzow MR, Edwards WD. Smooth muscle cells in human coronary atherosclerosis can originate from cells administered at marrow transplantation. Proc Natl Acad Sci U S A. 2003;100:4754–4759. doi: 10.1073/pnas.0730743100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 10.Loetscher P, Moser B, Baggiolini M. Chemokines and their receptors in lymphocyte traffic and HIV infection. Adv Immunol. 2000;74:127–180. doi: 10.1016/s0065-2776(08)60910-4. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalo JA, Lloyd CM, Wen D, Albar JP, Wells TN, Proudfoot A, Martinez AC, Dorf M, Bjerke T, Coyle AJ, Gutierrez-Ramos JC. The coordinated action of CC chemokines in the lung orchestrates allergic inflammation and airway hyperresponsiveness. J Exp Med. 1998;188:157–167. doi: 10.1084/jem.188.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutierrez-Ramos JC, Lloyd C, Kapsenberg ML, Gonzalo JA, Coyle AJ. Non-redundant functional groups of chemokines operate in a coordinate manner during the inflammatory response in the lung. Immunol Rev. 2000;177:31–42. doi: 10.1034/j.1600-065x.2000.17713.x. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu K, Mitchell RN. The role of chemokines in transplant graft arterial disease. Arterioscler Thromb Vasc Biol. 2008;28:1937–1949. doi: 10.1161/ATVBAHA.107.161232. [DOI] [PubMed] [Google Scholar]
- 14.Shimizu K, Mitchell RN. Chemokine-mediated recruitment of inflammatory and smooth muscle cells in transplant-associated arteriosclerosis. Curr Opin Organ Transplant. 2003;8:55–63. [Google Scholar]
- 15.Shimizu K, Aikawa M, Takayama K, Libby P, Mitchell RN. Direct anti-inflammatory mechanisms contribute to attenuation of experimental allograft arteriosclerosis by statins. Circulation. 2003;108:2113–2120. doi: 10.1161/01.CIR.0000092949.67153.74. [DOI] [PubMed] [Google Scholar]
- 16.Kao J, Kobashigawa J, Fishbein MC, MacLellan WR, Burdick MD, Belperio JA, Strieter RM. Elevated serum levels of the CXCR3 chemokine ITAC are associated with the development of transplant coronary artery disease. Circulation. 2003;107:1958–1961. doi: 10.1161/01.CIR.0000069270.16498.75. [DOI] [PubMed] [Google Scholar]
- 17.Zhao DX, Hu Y, Miller GG, Luster AD, Mitchell RN, Libby P. Differential expression of the IFN-gamma-inducible CXCR3-binding chemokines, IFN-inducible protein 10, monokine induced by IFN, and IFN-inducible T cell alpha chemoattractant in human cardiac allografts: association with cardiac allograft vasculopathy and acute rejection. J Immunol. 2002;169:1556–1560. doi: 10.4049/jimmunol.169.3.1556. [DOI] [PubMed] [Google Scholar]
- 18.Yun JJ, Fischbein MP, Laks H, Fishbein MC, Espejo ML, Ebrahimi K, Irie Y, Berliner J, Ardehali A. Early and late chemokine production correlates with cellular recruitment in cardiac allograft vasculopathy. Transplantation. 2000;69:2515–2524. doi: 10.1097/00007890-200006270-00009. [DOI] [PubMed] [Google Scholar]
- 19.Gerard C, Frossard JL, Bhatia M, Saluja A, Gerard NP, Lu B, Steer M. Targeted disruption of the beta-chemokine receptor CCR1 protects against pancreatitis-associated lung injury. J Clin Invest. 1997;100:2022–2027. doi: 10.1172/JCI119734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huaux F, Gharaee-Kermani M, Liu T, Morel V, McGarry B, Ullenbruch M, Kunkel SL, Wang J, Xing Z, Phan SH. Role of Eotaxin-1 (CCL11) and CC chemokine receptor 3 (CCR3) in bleomycin-induced lung injury and fibrosis. Am J Pathol. 2005;167:1485–1496. doi: 10.1016/S0002-9440(10)61235-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao W, Faia KL, Csizmadia V, Smiley ST, Soler D, King JA, Danoff TM, Hancock WW. Beneficial effects of targeting CCR5 in allograft recipients. Transplantation. 2001;72:1199–1205. doi: 10.1097/00007890-200110150-00003. [DOI] [PubMed] [Google Scholar]
- 22.Manders EMM, Verbeek FJ, Aten JA. Measurement of co-localization of object in dual-colour confocal images. Journal of Microscopy. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 23.Shimizu K, Schonbeck U, Mach F, Libby P, Mitchell RN. Host CD40 ligand deficiency induces long-term allograft survival and donor-specific tolerance in mouse cardiac transplantation but does not prevent graft arteriosclerosis. J Immunol. 2000;165:3506–3518. doi: 10.4049/jimmunol.165.6.3506. [DOI] [PubMed] [Google Scholar]
- 24.Bornfeldt KE, Raines EW, Nakano T, Graves LM, Krebs EG, Ross R. Insulin-like growth factor-I and platelet-derived growth factor-BB induce directed migration of human arterial smooth muscle cells via signaling pathways that are distinct from those of proliferation. J Clin Invest. 1994;93:1266–1274. doi: 10.1172/JCI117081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogaboam CM, Gallinat CS, Taub DD, Strieter RM, Kunkel SL, Lukacs NW. Immunomodulatory role of C10 chemokine in a murine model of allergic bronchopulmonary aspergillosis. J Immunol. 1999;162:6071–6079. [PubMed] [Google Scholar]
- 26.Pardigol A, Forssmann U, Zucht HD, Loetscher P, Schulz-Knappe P, Baggiolini M, Forssmann WG, Magert HJ. HCC-2, a human chemokine: gene structure, expression pattern, and biological activity. Proc Natl Acad Sci U S A. 1998;95:6308–6313. doi: 10.1073/pnas.95.11.6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W, Bacon KB, Oldham ER, Schall TJ. Molecular cloning and functional characterization of human MIP-1 delta, a new C-C chemokine related to mouse CCF-18 and C10. J Clin Immunol. 1998;18:214–222. doi: 10.1023/a:1020535106684. [DOI] [PubMed] [Google Scholar]
- 28.Berger MS, Taub DD, Orlofsky A, Kleyman TR, Coupaye-Gerard B, Eisner D, Cohen SA. The chemokine C10: immunological and functional analysis of the sequence encoded by the novel second exon. Cytokine. 1996;8:439–447. doi: 10.1006/cyto.1996.0060. [DOI] [PubMed] [Google Scholar]
- 29.Huang CL, Takenawa T, Ives HE. Platelet-derived growth factor-mediated Ca2+ entry is blocked by antibodies to phosphatidylinositol 4,5-bisphosphate but does not involve heparin-sensitive inositol 1,4,5-trisphosphate receptors. J Biol Chem. 1991;266:4045–4048. [PubMed] [Google Scholar]
- 30.Mogami H, Kojima I. Stimulation of calcium entry is prerequisite for DNA synthesis induced by platelet-derived growth factor in vascular smooth muscle cells. Biochem Biophys Res Commun. 1993;196:650–658. doi: 10.1006/bbrc.1993.2299. [DOI] [PubMed] [Google Scholar]
- 31.Schecter AD, Berman AB, Taubman MB. Chemokine receptors in vascular smooth muscle. Microcirculation. 2003;10:265–272. doi: 10.1038/sj.mn.7800192. [DOI] [PubMed] [Google Scholar]
- 32.Joubert P, Lajoie-Kadoch S, Welman M, Dragon S, Letuvee S, Tolloczko B, Halayko AJ, Gounni AS, Maghni K, Hamid Q. Expression and regulation of CCR1 by airway smooth muscle cells in asthma. J Immunol. 2008;180:1268–1275. doi: 10.4049/jimmunol.180.2.1268. [DOI] [PubMed] [Google Scholar]
- 33.Nelson PR, Yamamura S, Mureebe L, Itoh H, Kent KC. Smooth muscle cell migration and proliferation are mediated by distinct phases of activation of the intracellular messenger mitogen-activated protein kinase. J Vasc Surg. 1998;27:117–125. doi: 10.1016/s0741-5214(98)70298-8. [DOI] [PubMed] [Google Scholar]
- 34.Imai Y, Clemmons DR. Roles of phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways in stimulation of vascular smooth muscle cell migration and deoxyriboncleic acid synthesis by insulin-like growth factor-I. Endocrinology. 1999;140:4228–4235. doi: 10.1210/endo.140.9.6980. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu K, Tanaka H, Sunamori M, Marumo F, Shichiri M. Adrenomedullin receptor antagonism by calcitonin gene-related peptide(8–37) inhibits carotid artery neointimal hyperplasia after balloon injury. Circ Res. 1999;85:1199–1205. doi: 10.1161/01.res.85.12.1199. [DOI] [PubMed] [Google Scholar]
- 36.Gao W, Topham PS, King JA, Smiley ST, Csizmadia V, Lu B, Gerard CJ, Hancock WW. Targeting of the chemokine receptor CCR1 suppresses development of acute and chronic cardiac allograft rejection. J Clin Invest. 2000;105:35–44. doi: 10.1172/JCI8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feinberg MW, Shimizu K, Lebedeva M, Haspel R, Takayama K, Chen Z, Frederick JP, Wang XF, Simon DI, Libby P, Mitchell RN, Jain MK. Essential role for Smad3 in regulating MCP-1 expression and vascular inflammation. Circ Res. 2004;94:601–608. doi: 10.1161/01.RES.0000119170.70818.4F. [DOI] [PubMed] [Google Scholar]
- 38.von Hundelshausen P, Weber K, Huo Y. Circulation. 2001;103:1772–1777. doi: 10.1161/01.cir.103.13.1772. [DOI] [PubMed] [Google Scholar]
- 39.Wiedermann C, Kowald E. Current Biology. 1993;3:735–739. doi: 10.1016/0960-9822(93)90020-o. [DOI] [PubMed] [Google Scholar]
- 40.Pakianathan DR, Kuta EG, Artis DR, Skelton NJ, Hebert CA. Distinct but overlapping epitopes for the interaction of a CC-chemokine with CCR1, CCR3 and CCR5. Biochemistry. 1997;36:9642–9648. doi: 10.1021/bi970593z. [DOI] [PubMed] [Google Scholar]
- 41.Mellado M, Rodriguez-Frade JM, Manes S, Martinez AC. Chemokine signaling and functional responses: the role of receptor dimerization and TK pathway activation. Annu Rev Immunol. 2001;19:397–421. doi: 10.1146/annurev.immunol.19.1.397. [DOI] [PubMed] [Google Scholar]
- 42.Gao JL, Kuhns DB, Tiffany HL, McDermott D, Li X, Francke U, Murphy PM. Structure and functional expression of the human macrophage inflammatory protein 1 alpha/RANTES receptor. J Exp Med. 1993;177:1421–1427. doi: 10.1084/jem.177.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neote K, DiGregorio D, Mak JY, Horuk R, Schall TJ. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell. 1993;72:415–425. doi: 10.1016/0092-8674(93)90118-a. [DOI] [PubMed] [Google Scholar]
- 44.Kaminski A, Ma N, Donndorf P, Lindenblatt N, Feldmeier G, Ong LL, Furlani D, Skrabal CA, Liebold A, Vollmar B, Steinhoff G. Endothelial NOS is required for SDF-1alpha/CXCR4-mediated peripheral endothelial adhesion of c-kit+ bone marrow stem cells. Lab Invest. 2008;88:58–69. doi: 10.1038/labinvest.3700693. [DOI] [PubMed] [Google Scholar]
- 45.Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Mopps B, Mericskay M, Gierschik P, Biessen EA, Weber C. SDF-1{alpha}/CXCR4 Axis Is Instrumental in Murine Neointimal Hyperplasia and Recruitment of Smooth Muscle Progenitor Cells. Circ Res. 2005;96:784–791. doi: 10.1161/01.RES.0000162100.52009.38. [DOI] [PubMed] [Google Scholar]
- 46.Haque NS, Fallon JT, Pan JJ, Taubman MB, Harpel PC. Chemokine receptor-8 (CCR8) mediates human vascular smooth muscle cell chemotaxis and metalloproteinase-2 secretion. Blood. 2004;103:1296–1304. doi: 10.1182/blood-2002-05-1480. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Yue TL, Ohlstein EH, Sung CP, Feuerstein GZ. Interferon-inducible protein-10 involves vascular smooth muscle cell migration, proliferation, and inflammatory response. J Biol Chem. 1996;271:24286–24293. doi: 10.1074/jbc.271.39.24286. [DOI] [PubMed] [Google Scholar]
- 48.Ikeda U, Okada K, Ishikawa S, Saito T, Kasahara T, Shimada K. Monocyte chemoattractant protein 1 inhibits growth of rat vascular smooth muscle cells. Am J Physiol. 1995;268:H1021–1026. doi: 10.1152/ajpheart.1995.268.3.H1021. [DOI] [PubMed] [Google Scholar]
- 49.Porreca E, Di Febbo C, Reale M, Castellani ML, Baccante G, Barbacane R, Conti P, Cuccurullo F, Poggi A. Monocyte chemotactic protein 1 (MCP-1) is a mitogen for cultured rat vascular smooth muscle cells. J Vasc Res. 1997;34:58–65. doi: 10.1159/000159202. [DOI] [PubMed] [Google Scholar]
- 50.Kodali RB, Kim WJ, Galaria, Miller C, Schecter AD, Lira SA, Taubman MB. CCL11 (Eotaxin) induces CCR3-dependent smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2004;24:1211–1216. doi: 10.1161/01.ATV.0000131654.90788.f5. [DOI] [PubMed] [Google Scholar]
- 51.The U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients (OPTN/ SRTR) Annual Report: Transplant Data 1997–2006. http://www.ustransplant.org/annual_reports/current/default.htm.
- 52.Pinney SP, Mancini D. Cardiac allograft vasculopathy: advances in understanding its pathophysiology, prevention, and treatment. Curr Opin Cardiol. 2004;19:170–176. doi: 10.1097/00001573-200403000-00019. [DOI] [PubMed] [Google Scholar]
- 53.Ramzy D, Rao V, Brahm J, Miriuka S, Delgado D, Ross HJ. Cardiac allograft vasculopathy: a review. Can J Surg. 2005;48:319–327. [PMC free article] [PubMed] [Google Scholar]
- 54.Cook CL, Weiser MC, Schwartz PE, Jones CL, Majack RA. Developmentally timed expression of an embryonic growth phenotype in vascular smooth muscle cells. Circ Res. 1994;74:189–196. doi: 10.1161/01.res.74.2.189. [DOI] [PubMed] [Google Scholar]
- 55.Hultgardh-Nilsson A, Krondahl U, Querol-Ferrer V, Ringertz NR. Differences in growth factor response in smooth muscle cells isolated from adult and neonatal rat arteries. Differentiation. 1991;47:99–105. doi: 10.1111/j.1432-0436.1991.tb00227.x. [DOI] [PubMed] [Google Scholar]
- 56.Majesky MW, Giachelli CM, Reidy MA, Schwartz SM. Rat carotid neointimal smooth muscle cells reexpress a developmentally regulated mRNA phenotype during repair of arterial injury. Circ Res. 1992;71:759–768. doi: 10.1161/01.res.71.4.759. [DOI] [PubMed] [Google Scholar]
- 57.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 58.Miyamoto T, Sasaguri Y, Sasaguri T, Azakami S, Yasukawa H, Kato S, Arima N, Sugama K, Morimatsu M. Expression of stem cell factor in human aortic endothelial and smooth muscle cells. Atherosclerosis. 1997;129:207–213. doi: 10.1016/s0021-9150(96)06043-1. [DOI] [PubMed] [Google Scholar]
- 59.Simper D, Stalboerger PG, Panetta CJ, Wang S, Caplice NM. Smooth muscle progenitor cells in human blood. Circulation. 2002;106:1199–1204. doi: 10.1161/01.cir.0000031525.61826.a8. [DOI] [PubMed] [Google Scholar]
- 60.Sugiyama S, Kugiyama K, Nakamura S, Kataoka K, Aikawa M, Shimizu K, Koide S, Mitchell RN, Ogawa H, Libby P. Characterization of smooth muscle-like cells in circulating human peripheral blood. Atherosclerosis. 2006;187:351–362. doi: 10.1016/j.atherosclerosis.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 61.Metharom P, Liu C, Wang S, Stalboerger P, Chen G, Doyle B, Ikeda Y, Caplice NM. Myeloid lineage of high proliferative potential human smooth muscle outgrowth cells circulating in blood and vasculogenic smooth muscle-like cells in vivo. Atherosclerosis. 2008;198:29–38. doi: 10.1016/j.atherosclerosis.2007.09.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.