

Abstract
Although it is well established that changes in endothelial intracellular [Ca2+] regulate endothelium-dependent vasodilatory pathways, the molecular identities of the ion channels responsible for Ca2+ influx in these cells are not clearly defined. The sole member of the ankyrin (A) transient receptor potential (TRP) subfamily, TRPA1, is a Ca2+-permeable non-selective cation channel activated by electrophilic compounds such as acrolien (tear gas), allicin (garlic) and allyl isothiocyanate (AITC) (mustard oil). The current study examines the hypothesis that Ca2+ influx via TRPA1 causes endothelium-dependent vasodilation. The effects of TRPA1 activity on vascular tone were examined using isolated, pressurized cerebral arteries. AITC induced concentration-dependent dilation of pressurized vessels with myogenic tone that was accompanied by a corresponding decrease in smooth muscle intracellular [Ca2+]. AITC-induced dilation was attenuated by disruption of the endothelium and when the TRPA1 channel blocker HC-030031 was present in the arterial lumen. TRPA1 channels were found to be present in native endothelial cells, localized to endothelial cell membrane projections proximal to vascular smooth muscle cells. AITC-induced dilation was insensitive to nitric oxide synthase or cyclooxygenase inhibition, but was blocked by luminal administration of the small and intermediate conductance Ca2+-activated K+ channel blockers apamin and TRAM34. BaCl2, a blocker of inwardly rectifying K+ (KIR) channels, also inhibited AITC-induced dilation. AITC-induced smooth muscle cell hyperpolarization was blocked by apamin and TRAM34. We conclude that Ca2+ influx via endothelial TRPA1 channels elicits vasodilation of cerebral arteries by a mechanism involving endothelial cell Ca2+- activated K+ channels and KIR channels in arterial myocytes.
Keywords: TRP channels, EDHF, AITC, inwardly-rectifying K+ channels
INTRODUCTION
Changes in endothelial intracellular [Ca2+] regulate endothelium-dependent vasodilation through diverse signaling pathways. Production of potent vasoactive substances such as nitric oxide (NO)1 and prostacyclin (PGI2)2 are stimulated by increases in intracellular [Ca2+]. In addition, small (KCa2.3) and intermediate (KCa3.1) conductance Ca2+-activated K+ channels hyperpolarize the endothelial cell plasma membrane and influence smooth muscle excitability, vascular tone, and arterial blood pressure by direct electrical communication via myoendothelial gap junctions3, 4 or release of K+ ions5. Despite the impact of endothelial cell Ca2+ mobilization and dynamics on critical aspects of vascular function, the molecular identities and regulation of Ca2+ entry channels present in these cells is poorly understood. Further characterization of the ion channels responsible for endothelial cell Ca2+ influx is expected to provide critical insight into the nature of endothelium-dependent vasodilation.
Ca2+-permeable ion channels belonging to the Transient Receptor Potential (TRP) superfamily6 are present in vascular endothelial cells and likely play a major role in Ca2+-dependent signaling processes7–9. The sole member of the ankyrin (A) TRP subfamily, TRPA110, is a Ca2+-permeable non-selective cation channel that is activated by electrophilic compounds such as acrolien (an active component of tear gas), allicin (found in garlic), and allyl isothiocyanate (AITC, derived from mustard oil)11, 12. Unsaturated aldehydes produced endogenously in response to oxidative stress, such as 4-hydroxy-2-nonenal (4-HNE)13, 14, 4-oxo-nonenal (4-ONE)13, and 4-hydroxyhexenal (4-HHE)13, also activate TRPA1. TRPA1 is expressed by a subset of nociceptive sensory neurons and mediates inflammatory pain in response to stimuli such as topical administration of chemical irritants15 and inhalation of cigarette smoke16. TRPA1 is also present in non-neuronal tissues such as basal urothelial cells17 and prostate epithelial cells18, although the functional significance of the channel in these tissues is not known.
The effects of TRPA1 activity on endothelium-dependent dilation of cerebral resistance arteries was investigated. We find that endothelial cell TRPA1 channels mediate vasodilation by a novel pathway involving Ca2+-activated K+ channels in endothelial cells and KIR channels in arterial myocytes.
MATERIAL AND METHODS
Cerebral and cerebellar arteries used for these studies were isolated from male Sprague-Dawley rats (250–350 g; Harlan, Indianapolis, Indiana, USA). All animal use procedures were in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committeeof Colorado State University.
For isolated vessel experiments, arteries were cannulated, pressurized with physiological saline solution (PSS), and superfused with aerated PSS at 37°C. To monitor changes in vessel wall [Ca2+] (representative of smooth muscle intracellular [Ca2+]), arteries were loaded with the ratiometric Ca2+ indicator dye fura-2AM from the abluminal surface. Inner diameter was continuously monitored using video microscopy and edge-detection software (Ionoptix). Pressurized vessels were intermittently excited with UV light at 340 and 380 nm (10 Hz) and deep red wavelength emissions were recorded using a photomultiplier tube and expressed as the ratio of emissions during 340 nm excitation vs. 380 nm excitation (340/380 ratio). For some experiments, endothelial cell function was disrupted by passage of air and distilled water through the vessel lumen. AITC-induced dilation was found to be very reproducible (Online Figure I), allowing all experiments examining the effects of endothelial disruption, NOS and COX inhibition, or block of K+ channels on vasodilation to be performed and analyzed using a paired design. Smooth muscle cell membrane potential was recorded in pressurized (70 mmHg) cerebral arteries with intracellular microelectrodes using previously described methods19, 20.
To assess TRPA1 mRNA expression in vascular tissue, enzymatically-dispersed native endothelial cells were visually identified using phase contrast microscopy and were collected using a micromanipulator controlled pipette. Total RNA was isolated from these cells and RT-PCR was performed using TRPA1-specific primers yielding a product of 500 bp. RT-PCR was performed using RNA isolated from three animals.
Immunostaining for TRPA1 was performed using intact cerebral arteries with the endothelium exposed by cutting the vessel lengthwise and pinning the tissue to a Silgard block. Fixed tissue was probed with anti-TRPA1 (Santa Cruz, 1:1000)21, anti- KCa3.1 and/or anti-KCa2.3 (both Alomone, 1:1000) overnight at 4°C. Arteries were probed with fluorescent secondary antibodies (Texas Red, Santa Cruz, 1:500 or Alexa 633, Molecular Probes, 1:500) and immunofluorescence was detected using a Zeiss LSM 510 Meta laser scanning confocal microscope. Immunofluorescence was not detected in tissues probed with secondary antibodies alone.
An Expanded Materials and Methods section can be found in the Online Data Supplement available at http://www.circresaha.org.
RESULTS
TRPA1 Agonists Dilate Cerebral Arteries by an Endothelium-Dependent Mechanism
To investigate the effects of TRPA1 channel activity on vasomotor tone of cerebral arteries, changes in luminal diameter and vessel wall [Ca2+] were recorded when the TRPA1 agonist AITC was present in the bathing solution. For these experiments, fura-2AM was administered to the abluminal surface, preferentially loading vascular smooth muscle cells22. Arteries were pressurized to 70 mmHg and spontaneous myogenic tone was allowed to develop. AITC administration resulted in robust, persistent vasodilation of preconstricted arteries that was accompanied by a corresponding decrease in vessel wall [Ca2+] (Figure 1A). Arteries contracted to their original diameter when AITC was removed from the bathing solution (Online Figure I), and subsequent AITC administration elicited equivalent dilator responses (Online Figure I), demonstrating that TRPA1-mediated vasodilation is reversible and reproducible. AITC elicited statistically significant vasodilation and reduction in vessel wall [Ca2+] at concentrations as low as 3 μM, and induced maximal changes in luminal diameter at a concentration of 30 to 100 μM (Figure 1B and C). The half-maximal effective concentration (EC50) for vasodilation was 16.4 μM (Figure 1D). These findings show that activation of TRPA1 channels causes dilation of preconstricted cerebral arteries by reducing smooth muscle intracellular [Ca2+]. During myogenic constriction, voltage-dependent Ca2+ channels are the primary Ca2+ influx pathway in cerebral artery myocytes22. Thus, these findings suggest that TRPA1 activity elicits dilation by hyperpolarizing the membrane potential of vascular smooth muscle cells.
Figure 1. The TRPA1 Agonist AITC Elicits Vasodilation of Cerebral Arteries.
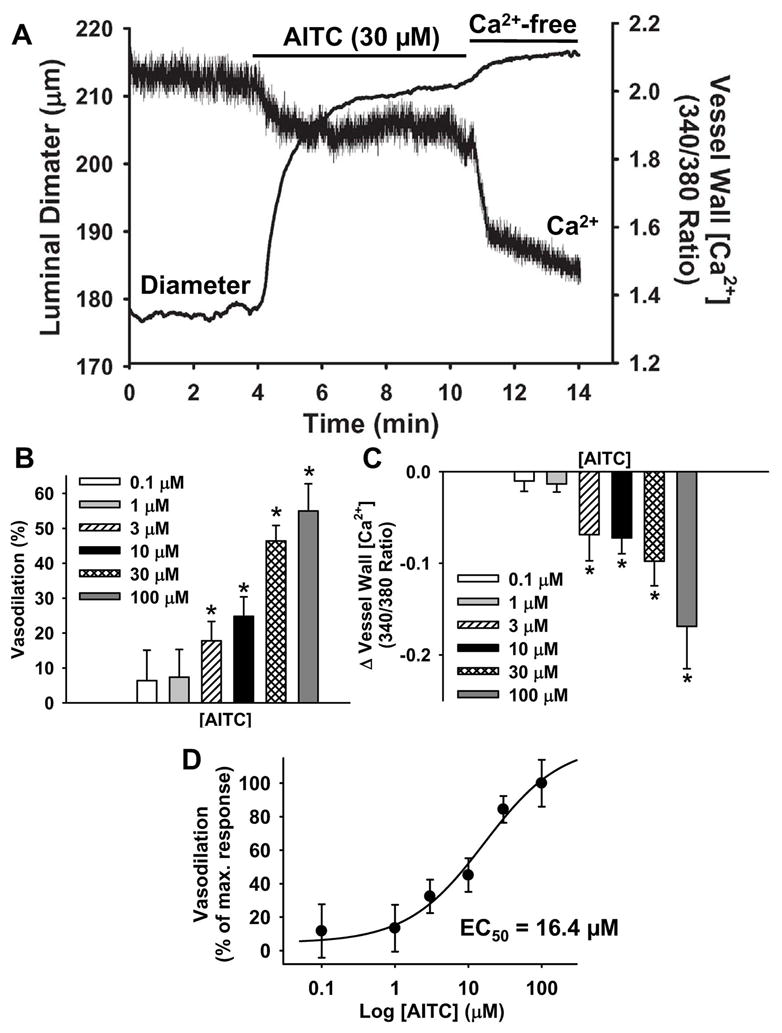
A: Representative recording of AITC (30 μM)-induced dilation and decrease in vessel wall [Ca2+] of an isolated cerebral artery with myogenic tone (70 mmHg). B: Vasodilation (normalized to passive diameter recorded under Ca2+-free conditions) in response to increasing concentrations of AITC. n=5–11 per concentration. C: Decrease in vessel wall [Ca2+] (expressed as change in 340/380 ratio) in response to increasing concentrations of AITC. n=6–11 per concentration. D: Concentration response data normalized to maximum vasodilation. EC50 = 16.4 μM.
Activation of TRPA1 channels in any of the cell types within the vascular wall, including smooth muscle, endothelial cells, or perivascular nerve terminals could be responsible for AITC-induced vasodilation. To determine if the endothelium contributes to TRPA1-dependent vasodilation, endothelial cell function was disrupted by briefly perfusing isolated cerebral arteries with distilled water followed by air. Vasodilation and changes in vessel wall [Ca2+] in response to AITC were recorded before and after endothelial cell disruption. Damage to the endothelium resulted in impaired AITC-induced vasodilation (Figure 2A and B) demonstrating that endothelial cells are involved in arterial dilation associated with TRPA1 channel activation. In addition, AITC administration did not cause significant changes in vessel wall [Ca2+] in endothelium-disrupted arteries, suggesting that this agonist has no direct effect on vascular smooth muscle cells.
Figure 2. Endothelial Cell TRPA1 Channels Mediate AITC-Induced Vasodilation.

A: Vasodilation in response to AITC (100 μM) before and after disruption of endothelial cell function. B: Summary data of the effects of endothelium disruption on AITC-induced vasodilation. n=3. *P≤0.05 vs. Control. C: Vasodilation in response to AITC (100 μM) before and after luminal administration of the TRPA1 blocker HC-030031 (3 μM). D: Summary data for the effects of luminal HC-030031 on AITC-induced vasodilation. n=5. *P≤0.05 vs. Control.
A recently described blocker of TRPA1 channels, HC-03003123, was used to further probe the role of TRPA1 channels in endothelium-dependent vasodilation. The half-maximal inhibitory concentration (IC50) of HC-030031 for AITC-induced TRPA1 currents in patch clamp studies is reportedly 0.7±0.1 μM23. HC-030031 is selective for TRPA1 channels and does not inhibit TRPV1, TRPV3, TRPV4, hERG, or NaV1.2 channel activity in the concentration range used for the current study (IC50 for these channels >10–20 μM)23. To block endothelial cell TRPA1 channels, HC-030031 (3 μM) was administered to the lumen of isolated cerebral arteries. In paired experiments, AITC-induced dilation was impaired in the presence of luminal HC-030031 (Figure 2C and D), demonstrating that activation of TRPA1 channels in vascular endothelial cells causes vasodilation of cerebral arteries. Interestingly, luminal administration of HC-030031 also caused a small, yet statistically significant increase in myogenic tone (Online Figure II). These data suggest that endothelial TRPA1 channels support a tonic vasodilatory influence that is independent of exogenous TRPA1 activators.
TRPA1 Channels Are Present in Cerebral Artery Endothelial Cells
Endothelial cell damage (Figure 2B) and luminal administration of the TRPA1 blocker HC-030031 inhibits AITC-induced vasodilation (Figure 2D), suggesting that TRPA1 channels present in vascular endothelial cells mediate this response. Consistent with these results, message encoding TRPA1 is present in total RNA extracted from native cerebral artery endothelial cells (Figure 3A).
Figure 3. TRPA1 is Present in Endothelial Cells Isolated from Rat Cerebral Arteries.

A: RT-PCR for TRPA1 using total RNA from freshly-isolated rat cerebral artery endothelial cells (EC). NT = No template control, −RT = no reverse transcriptase control. Data are representative of RNA isolated from three rats. B–H: Localization of TRPA1 and KCa3.1 channels in cerebral artery endothelial cell membrane projections. Images show immunostaining for TRPA1 (red) (C–E) and KCa3.1 (red) (F–H). The level of the internal elastic lamina (IEL) is shown in green. Black holes (arrow) in the IEL indicate endothelial cell membrane projections in the direction of vascular smooth muscle cells (C and F). Superimposed images demonstrate that TRPA1 channels and KCa3.1 channels are abundant in the holes in the IEL (E, H). Bar = 20 μm. I: Z-stack image showing projection of TRPA1 immunostaining though the IEL. Bar =10 μm. I–L: Co-immunostaining for TRPA1 and KCa3.1 in cerebral artery endothelial cell membrane projections. The level of the internal elastic lamina (IEL) is shown in green (I). Black holes (arrow) in the IEL indicate endothelial cell membrane projections in the direction of vascular smooth muscle cells. J: Immunostaining for TRPA1 (red) K: Immunostaining for KCa3.1 (blue). L: Superimposed images demonstrate that TRPA1 channels and KCa3.1 channels co-localize in black holes in the IEL. Bar = 25 μm. All immunostaing data are representative of arteries isolated from at least three animals.
To assess the localization of TRPA1 channels in cerebral arteries, vessels were cut longitudinally, pinned to Silgard blocks, fixed, and immunostained using an anti-TRPA1 antibody (Santa Cruz)21. To localize the endothelium in this preparation, the internal elastic lamina (IEL) was identified based upon its characteristic green autofluorescence24, 25 (Figures 3B and E). Trans-IEL endothelial cell membrane projections24, 25 are clearly visible in these images (arrow). Immunostaining for TRPA1 (red, Figure 3C) is abundant in these membrane depressions (Figure 3D, arrow) demonstrating that the channel is expressed at high levels in these structures. Diffuse immunostaining for TRPA1 channels in the endothelium was also present outside of endothelial cell membrane projections (Figure 3C). In agreement with recent findings reported by Sandow et al24. and Ledoux et al.25, KCa3.1 channels are also highly enriched in these membrane structures (Figures 3E–G). Dual-label experiments confirm co-localization of TRPA1 and KCa3.1 in black holes (Figure 3I–L). Consistent with an earlier study24, immunostaining for the SKCa channel KCa2.3 was present in cerebral arteries but did not localize to myoendothelial membrane projections (Online Figure III). A reconstructed series of images taken vertically through the depth of the tissue (“z-stack”) demonstrates that TRPA1 immunostaining projects though the IEL (Figure 3H). TRPA1 immunostaining was not present following mechanical disruption of the endothelium (Online Figure IV). These findings demonstrate that TRPA1 channels are present in the endothelium and are abundant in trans-IEL membrane domains with elevated levels of KCa3.1 channels.
TRPA1-Mediated Vasodilation Requires Endothelial KCa and Smooth Muscle KIR Channels
Stimulation of TRPA1 activity causes endothelium-dependent vasodilation. Pharmacological inhibition of the NOS and COX pathways was used to examine their contribution to TRPA1-induced vasodilation. Superfusion of arteries with the NOS inhibitor N-nitro-L-arginine (L-NNA) (300 μM, 20 minutes) or a combination of L-NNA and the COX inhibitor indomethacin (10 μM, 15 minutes) caused an increase in basal myogenic tone (myogenic tone = 19.4 ± 4.4% under control condition, 25.0 ± 7.6% in the presence of L-NNA, and 27.4±10.0% in the presence of L-NNA+ indomethacin, n=5). AITC-induced vasodilation was not diminished by NOS or COX inhibition (Online Figure V), demonstrating that activation of TRPA1 channels in the endothelium causes vasodilation by a pathway that is independent of NO and PGI2 production. These data do not directly address the effects of AITC on NOS and COX activity.
Ca2+-activated K+ channels are present in vascular endothelial cells and activation of these channels can cause vasodilation3, 4, 26. To determine if TRPA1- dependent Ca2+ influx elicits dilation by activating these channels, the KCa3.1 inhibitor TRAM34 (1 μM) or a combination of TRAM34 and the SKCa blocker apamin (1 μM) were administered to the lumen of cerebral arteries that had been allowed to develop spontaneous myogenic tone (70 mmHg). TRAM34 significantly decreased AITC-induced vasodilation (Figure 4B), whereas the combination of apamin and TRAM34 abolished sustained AITC-induced vasodilation (Figure 4A and C), indicating that SKCa and KCa3.1 channel activity contribute to TRPA1-mediated vasodilatory responses. Arteries treated with apamin and TRAM34 dilated to nearly maximal diameters (99.3 ± 1.4% of Ca2+-free diameter, n=3) in response to the ATP-sensitive K+ (KATP) channel opener pinacidil (10 μM) (Figure 4A), demonstrating that KCa channel inhibition did not block vasodilation in response to a stimulus that hyperpolarizes vascular smooth muscle cells. These data show that Ca2+ influx via endothelial TRPA1 stimulates SKCa/KCa3.1 channels, causing K+ efflux and vasodilation. Selective activation of SKCa/KCa3.1 vs. NOS and COX pathways in response to AITC is in agreement with the hypothesis that stimulation of TRPA1 channels does not globally increase intracellular [Ca2+] but instead results in localized elevation of intracellular [Ca2+] in subcellular microdomains containing KCa channels. Consistent with this possibility, TRPA1 and KCa3.1 channels are co-localized in endothelial cell membrane domains spanning the IEL (Figure 3I–L).
Figure 4. TRPA1-Dependent Vasodilation Requires KCa and KIR Channels.

A: Representative recordings of vasodilation in response to AITC (10 μM) before and after luminal administration of the SKCa blocker apamin (1 μM) and the KCa3.1 blocker TRAM34 (1 μM). B: Summary of the effects of KCa3.1 blockade on AITC-induced dilation. n=5. C: Summary of the effects of SKCa/KCa3.1 blockade on AITC-induced vasodilation. n=5. *P≤0.05 vs. Control. D: Representative recording of vasodilation in response to AITC (10 μM) in the presence of the KIR blocker BaCl2 (30 μM). E: Summary of the effects BaCl2 on AITC-induced vasodilation. n=5. *P≤0.05 vs. Control.
Activation of endothelial SKCa/KCa3.1 channels hyperpolarizes the endothelial cell plasma membrane3, 4, 26. Myoendothelial gap junctions conduct changes in endothelial cell membrane potential to vascular smooth muscle, resulting in myocyte hyperpolarization and vasodilation3, 4, 26. In cerebral arteries, endothelium-dependent smooth muscle hyperpolarization can be amplified by the negative slope conductance of inwardly rectifying K+ (KIR) channels27–29. Alternatively, K+ released from endothelial cell KCa channels could locally elevate [K+] in the interstial space proximal to vascular smooth muscle cells to levels (8–12 mM) sufficient to activate KIR channels present in cerebral artery myocytes5. Activation of KIR channels in smooth muscle cells at physiologically relevant membrane potentials −50 mV to −35 mV) causes K+ efflux which hyperpolarizes the sarcolemma, resulting in myocyte relaxation and vasodilation30. Low (μM) concentrations of Ba2+ ions are effective at selectively blocking KIR channels in smooth muscle cells30. Extracellular BaCl2 (30 μM) essentially abolished AITC-induced vasodilation (Figure 4D and E), demonstrating that KIR channel activity is required for cerebral artery dilation in response to TRPA1 activation. These experiments also show that pinacidil-induced vasodilation was not blocked by BaCl2 (Figure 4D, 96.5 ±1% of Ca2+-free diameter, n=4), indicating that inhibition of KIR channels does not impair KATP-induced myocyte hyperpolarization and arterial dilation.
TRPA1 Channel Activation Hyperpolarizes Cerebral Artery Myocytes
The effects of TRPA1 channel activation on smooth muscle cell membrane potential were directly assessed using intracellular microelectrodes. The resting membrane potential of arterial myocytes in cerebral arteries pressurized to 70 mmHg under control conditions is −37.0 ± 1.0 mV (n=7) (Figure 5A and D), whereas in the presence of AITC (30 μM) smooth muscle membrane potential is −44.6 ± 2.1 mV (n=7) (Figure 5B and D). These data demonstrate that TRPA1 channel activity hyperpolarizes the sarcolemma. To investigate a potential role for SKCa/KCa3.1 channels in AITC-induced hyperpolarization, experiments were performed when apamin and TRAM34 (1 μM each) were present in the arterial lumen. This treatment had no significant effect on membrane potential or myogenic tone under control conditions. In addition, smooth muscle cell membrane potential did not differ when recordings were obtained under control conditions (37.3 ± 1.8 mV, n=7), following luminal administration of apamin and TRAM34 (33.6 ± 2.6 mV, n=5), and when apamin and TRAM34-treated arteries were superfused with AITC (30 μM) (−36.7 ± 1.8 mV, n=7, Figure 5C and E). These findings show that activation of TRPA1 in the endothelium hyperpolarizes smooth muscle cells by a mechanism that requires SKCa and KCa3.1 channel activity (Figure 6).
Figure 5. TRPA1 Channel Activation Hyperpolarizes Cerebral Artery Myocytes.

AC: Representative membrane potential recordings of smooth muscle cells in pressurized (70 mmHg) cerebral arteries under control conditions (A), in the presence of AITC (30 μM) (B), and in the presence of AITC (30 μM) following luminal administration apamin (1 μM) and TRAM34 (1 μM) (C). D: Summary data for the effects of AITC (30 μM) on smooth muscle cell membrane potential. Data are from tissue isolated from three animals. n= 7 cells for each group. *P≤0.05 vs. Control. E: Summary data for the effects of AITC (30 μM) on smooth muscle cell membrane potential when apamin (1 μM) and TRAM34 (1 μM) was present in the arterial lumen. Data are from tissue isolated from three animals. n=5–7 cells per group. There were no significant differences.
Figure 6. Proposed Signaling Pathway for TRPA1-Mediated Vasodilation of Cerebral Arteries.

EC, endothelial cell; VSMC, vascular smooth muscle cell; IEL, internal elastic lamina; MEGJ, myoendothelial gap junction; AITC, Ally isothiocyanate; KCa3.1, intermediate conductance Ca2+-activated K+ channel; KCa2.3, small conductance Ca2+-activated K+ channel; KIR, inwardly rectifying K+ channel.
DISCUSSION
This study examined the functional consequences of TRPA1-mediated endothelial cell Ca2+ influx on the vasomotor activity of cerebral arteries. The major findings are: 1) The TRPA1 agonist AITC causes concentration-dependent dilation of cerebral arteries that is associated with a decrease in smooth muscle intracellular [Ca2+]; 2) TRPA1 agonist-dependent dilation is impaired by disruption of the endothelium and by luminal administration of a TRPA1 blocking compound; 3) TRPA1 channels are present in the vascular endothelium and are localized to membrane projections that approach underlying smooth muscle cells; 4) Vasodilation resulting from stimulation of TRPA1 activity is independent of NOS and COX activity but is attenuated by blockade of SKCa and KCa3.1 channels; 5) Inhibition of KIR channel activity abolishes TRPA1-dependent vasodilation; and 6) Activation of TRPA1 causes smooth muscle cell hyperpolarization by a mechanism that requires the activity of SKCa and KCa3.1 channels. The findings of this study demonstrate that endothelial TRPA1 channels are part of a vasodilatory signaling complex that includes KCa channels in endothelial cells and KIR channels in cerebral artery myocytes (Figure 6).
Endothelium-dependent dilation of cerebral arteries following stimulation of TRPA1 channels is not altered by blockade of NOS and COX, but is sensitive to inhibition of small- and intermediate-conductance Ca2+-activated K+ channels. In addition, activation of TRPA1 results in smooth muscle cell hyperpolarization that is sensitive to KCa channel blockade. These properties are hallmarks of “endothelium-derived hyperpolarizing factor” (EDHF)-induced dilation31. Despite considerable effort, the identity of the underlying molecular nature of EDHF remains elusive. Compelling evidence in support of a number of mechanisms, including cytochrome P450 epoxygenase products32, K+ ions5, hydrogen peroxide33, and gap junctional communication34, 35 has been reported. The current findings demonstrate that endothelial TRPA1 channels promote EDHF-type vasodilation in cerebral arteries. These observations are in agreement with those of earlier studies demonstrating that dilation of pulmonary36 and mesenteric arteries37 in response to the TRPA1 agonist allicin is independent of NOS and COX activity. However, in contrast to findings presented here demonstrating a critical role for the vascular endothelium, Bautista et al. showed that allicin-induced relaxation of mesenteric arteries was attenuated by pre-treatment with an calcitonin gene-related peptide (CGRP) antagonist37. Bautista and coworkers conclude that activation of TRPA1 channels in perivascular sensory nerve endings causes vasodilation of mesenteric arteries by stimulating release of CGRP37. A potential role for the endothelium in mesenteric artery relaxation in response to TRPA1 stimulation was not investigated 37. The current findings show that disruption of the endothelium or luminal administration of a TRPA1 blocking compound eliminates approximately 75% of AITC-induced cerebral artery dilation (Figure 2B and D), clearly demonstrating that endothelial cells mediate changes in vascular tone in response to TRPA1 activity in this vascular bed. Transient vasodilation in response to AITC following disruption of the endothelium (Figure 2A) or in the presence of apamin and TRAM34 (Figure 4A) may result from release of CGRP or other endothelium-independent mechanisms. Although it is possible that stimulation of TRPA1 channels present in perivascular nerve endings mediates a component of the response37, the endothelium appears to play a major role in steady-state TRPA1-dependent vasodilation in the cerebral vasculature.
The findings of this study provide new insight into the relationship between endothelial cell Ca2+ dynamics and endothelium-dependent vasodilation. The TRPA1 vasodilatory response is attenuated by blockade of Ca2+-activated K+ channels (Figure 4A–C), but not by inhibition of the NOS/COX pathways. Since all of these vasodilatory mechanisms are activated by increases in endothelial cell [Ca2+], the current findings suggest that spatially distinct Ca2+ signals regulate particular endothelium-dependent vasodilatory pathways. This conclusion is consistent with recent work by Ledoux et al., demonstrating that localized Ca2+ release events, (“Ca2+ pulsars”) exist in native endothelial cells and are dependent on Ca2+ release from inositol trisphoshphate (IP3) receptors located on the endoplasmic reticulum (ER)25. These dynamic Ca2+ events occur in structures that project from endothelial cells through the internal elastic lamina to underlying vascular smooth muscle cells25. Ledoux and colleagues also report that ER elements, IP3 receptors, and KCa3.1 channels colocalize with myoendothelial membrane projections in mesenteric arteries25. The current study demonstrates that TRPA1 channels and KCa3.1 channels are associated with these endothelial cell membrane projections in cerebral vessels. These findings are consistent with the hypothesis that TRPA1 channels and KCa3.1 channels form a vasodilatory signaling complex in endothelial cell membrane projections proximal to arterial myocytes. In this proposed pathway, localized TRPA1 Ca2+ signals can either directly stimulate KCa channel activity, or can be amplified by causing a Ca2+-dependent increase in the open probability of IP3 receptors located in myoendothelial membrane projections25, which serves to elevate the frequency of Ca2+ pulsars and activate KCa channels25. A combination of the SKCa channel blocker apamin and the KCa3.1 channel blocker TRAM34 appears to be more effective in blocking TRPA1-induced vasodilation compared with TRAM34 alone. KCa2.3, the most abundant SKCa channel in cerebral artery smooth muscle, is critical for endothelium-dependent vasodilation and regulation of endothelial cell membrane potential4. This channel is absent from myoendothelial membrane projections that span the IEL24 (Online Figure III). It is not clear from the current studies if SKCa channels are directly involved in TRPA1-dependent responses, or if the effects of apamin reflect a tonic hyperpolarizing effect of SKCa channel activity on the endothelial cell membrane4. KCa channel activity hyperpolarizes the endothelial cell plasma membrane, and serves to hyperpolarize underlying smooth muscle by charge transfer mediated by myoendothelial gap junctions present at the tip of the endothelial cell membrane projection spanning the internal elastic lamina (Figure 6).
AITC-induced dilation was blocked by Ba2+, demonstrating that KIR channel activity is required for vasodilation in response to activation of TRPA1 channels. KIR channels are prominent in smooth muscle cells and may be present in the endothelium of some vascular beds. Although the current study does not rule out the possibility that endothelial KIR channels contribute to TRPA1-mediated vasodilation, prior reports provide a strong conceptual framework supporting an important role for smooth muscle KIR activity in vasodilatory responses. For example, release of K+ from KCa channels into the interstitial space between the endothelium and underlying vascular smooth muscle cells could increase [K+] from a nominal 3 mM to 8–12 mM, sufficient to activate smooth muscle KIR channels and account for TRPA1-dependent vasodilation30. Biophysical properties of KIR channels suggest another mechanism that could explain involvement of this channel in TRPA1-mediated vasodilation. KIR channels composed of KIR2.1 and 2.2 subunits have an unusual current-voltage relationship known as “negative slope conductance”27, 28. Over a limited voltage range, outward K+ currents increase as membrane potential becomes more hyperpolarized. This effect is apparent for membrane potentials that are physiologically relevant for vascular smooth muscle cells. Message encoding KIR2.1 and 2.2 is present in cerebral artery smooth muscle cells and KIR channels appear to facilitate endothelium-dependent vasodilation in these vessels29. Furthermore, using both computational models and empirical studies, Smith et al. demonstrate that the negative slope conductance of KIR channels serves to amplify endothelium-derived hyperpolarizing stimuli29. KIR-dependent amplification of gap junction-mediated change transfer could also enhance smooth muscle hyperpolarization and vasodilation resulting from TRPA1 activity. This proposed signaling pathway highlights the importance of spatial and temporal relationships of ion channels and Ca2+ signals in endothelial and smooth muscle cells.
Although the electrophilic agonist AITC is commonly used to stimulate TRPA1 activity, recent reports suggest that TRPA1 channels may also be also be activated by endogenously occurring substances. 4-HNE, 4-ONE, and 4-HHE are lipid peroxidation products that potently activate TRPA1 channels expressed by HEK cells and cultured sensory neurons13, 14. The cyclopentanone prostaglandin 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) also stimulates TRPA1 activity13. All of these compounds are produced in response to oxidative stress, consistent with a possible role for endothelial TRPA1 channels in vascular responses to pathophysiological conditions that increase generation of reactive oxygen species, such as ischemia/reperfusion injury, hypoxia, and inflammation13, 14, 38. Inhibition of TRPA1 activity in isolated cerebral arteries results in a small, yet statistically significant, increase in myogenic tone, providing evidence that TRPA1 channels mediate tonic relaxation of cerebral arteries in the absence of exogenous agonists. Thus, in addition to eliciting vasodilation in response to environmental factors, TRPA1 channels may play an important role in vasomotor tone regulation under normal conditions and during pathophysiological situations that result in the generation of reactive oxygen species.
Pungent compounds found in garlic and mustard oil stimulate TRPA1, and activation of TRPA1 channels in the endothelium may contribute to improvements in vascular function attributed to these foods. Additionally, TRPA1 channels are activated by compounds produced in response to oxidative stress and this mechanism could mediate vascular responses to certain pathological conditions. The findings of this study suggest that endothelial cell TRPA1 channels present an interesting and novel target for anti-cardiovascular disease drug development efforts.
Supplementary Material
Acknowledgments
We thank Alainna McPhaul for technical assistance and Drs. Kevin S. Thorneloe, Gregory C. Amberg, and Michael M. Tamkun for critical comments on the manuscript.
SOURCES OF FUNDING This work was supported by NIH RO1HL091905, American Heart Association Grant AHA0535226N (SE), and NIH F31HL094145 (ALG).
Footnotes
DISCLOSURES None.
Literature Cited
- 1.Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brotherton AF, Hoak JC. Role of Ca2+ and cyclic AMP in the regulation of the production of prostacyclin by the vascular endothelium. Proc Natl Acad Sci U S A. 1982;79:495–499. doi: 10.1073/pnas.79.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coleman HA, Tare M, Parkington HC. K+ currents underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig, rat and human blood vessels. J Physiol. 2001;531:359–373. doi: 10.1111/j.1469-7793.2001.0359i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP, Nelson MT. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–131. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- 5.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 6.Venkatachalam K, Montell C. TRP channels. Annual review of biochemistry. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwan HY, Huang Y, Yao X. TRP channels in endothelial function and dysfunction. Biochimica et biophysica acta. 2007;1772:907–914. doi: 10.1016/j.bbadis.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Yao X, Garland CJ. Recent developments in vascular endothelial cell transient receptor potential channels. Circulation research. 2005;97:853–863. doi: 10.1161/01.RES.0000187473.85419.3e. [DOI] [PubMed] [Google Scholar]
- 9.Ahmmed GU, Malik AB. Functional role of TRPC channels in the regulation of endothelial permeability. Pflugers Arch. 2005;451:131–142. doi: 10.1007/s00424-005-1461-z. [DOI] [PubMed] [Google Scholar]
- 10.Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112:819–829. doi: 10.1016/s0092-8674(03)00158-2. [DOI] [PubMed] [Google Scholar]
- 11.Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Patapoutian A. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron. 2004;41:849–857. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- 12.Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- 13.Andersson DA, Gentry C, Moss S, Bevan S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci. 2008;28:2485–2494. doi: 10.1523/JNEUROSCI.5369-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, Imamachi N, Andre E, Patacchini R, Cottrell GS, Gatti R, Basbaum AI, Bunnett NW, Julius D, Geppetti P. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A. 2007;104:13519–13524. doi: 10.1073/pnas.0705923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 16.Andre E, Campi B, Materazzi S, Trevisani M, Amadesi S, Massi D, Creminon C, Vaksman N, Nassini R, Civelli M, Baraldi PG, Poole DP, Bunnett NW, Geppetti P, Patacchini R. Cigarette smoke-induced neurogenic inflammation is mediated by alpha, beta-unsaturated aldehydes and the TRPA1 receptor in rodents. The Journal of clinical investigation. 2008;118:2574–2582. doi: 10.1172/JCI34886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gratzke C, Streng T, Waldkirch E, Sigl K, Stief C, Andersson KE, Hedlund P. Transient Receptor Potential A1 (TRPA1) Activity in the Human Urethra-Evidence for a Functional Role for TRPA1 in the Outflow Region. European urology. 2008 doi: 10.1016/j.eururo.2008.04.042. [DOI] [PubMed] [Google Scholar]
- 18.Du S, Araki I, Kobayashi H, Zakoji H, Sawada N, Takeda M. Differential expression profile of cold (TRPA1) and cool (TRPM8) receptors in human urogenital organs. Urology. 2008;72:450–455. doi: 10.1016/j.urology.2007.11.127. [DOI] [PubMed] [Google Scholar]
- 19.Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res. 2004;95:922–929. doi: 10.1161/01.RES.0000147311.54833.03. [DOI] [PubMed] [Google Scholar]
- 20.Earley S, Straub SV, Brayden JE. Protein kinase C regulates vascular myogenic tone through activation of TRPM4. Am J Physiol Heart Circ Physiol. 2007;292:H2613–2622. doi: 10.1152/ajpheart.01286.2006. [DOI] [PubMed] [Google Scholar]
- 21.Corey DP, Garcia-Anoveros J, Holt JR, Kwan KY, Lin SY, Vollrath MA, Amalfitano A, Cheung EL, Derfler BH, Duggan A, Geleoc GS, Gray PA, Hoffman MP, Rehm HL, Tamasauskas D, Zhang DS. TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature. 2004;432:723–730. doi: 10.1038/nature03066. [DOI] [PubMed] [Google Scholar]
- 22.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM. TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci U S A. 2007;104:13525–13530. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandow SL, Neylon CB, Chen MX, Garland CJ. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (K(Ca)) and connexins: possible relationship to vasodilator function? Journal of anatomy. 2006;209:689–698. doi: 10.1111/j.1469-7580.2006.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI, Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105:9627–9632. doi: 10.1073/pnas.0801963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coleman HA, Tare M, Parkington HC. EDHF is not K+ but may be due to spread of current from the endothelium in guinea pig arterioles. Am J Physiol Heart Circ Physiol. 2001;280:H2478–2483. doi: 10.1152/ajpheart.2001.280.6.H2478. [DOI] [PubMed] [Google Scholar]
- 27.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. The American journal of physiology. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 28.Nelson MT, Huang Y, Brayden JE, Hescheler J, Standen NB. Arterial dilations in response to calcitonin gene-related peptide involve activation of K+ channels. Nature. 1990;344:770–773. doi: 10.1038/344770a0. [DOI] [PubMed] [Google Scholar]
- 29.Smith PD, Brett SE, Luykenaar KD, Sandow SL, Marrelli SP, Vigmond EJ, Welsh DG. KIR channels function as electrical amplifiers in rat vascular smooth muscle. J Physiol. 2008;586:1147–1160. doi: 10.1113/jphysiol.2007.145474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol. 1996;492:419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends in pharmacological sciences. 2002;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- 32.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 33.Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. The Journal of clinical investigation. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium- derived hyperpolarizing factor-mediated responses. Circ Res. 2000;86:341–346. doi: 10.1161/01.res.86.3.341. [DOI] [PubMed] [Google Scholar]
- 35.Fukuta H, Koshita M, Yamamoto Y, Suzuki H. Inhibition of the endothelium-dependent relaxation by 18beta-glycyrrhetinic acid in the guinea-pig aorta. Jpn J Physiol. 1999;49:267–274. doi: 10.2170/jjphysiol.49.267. [DOI] [PubMed] [Google Scholar]
- 36.Kaye AD, De Witt BJ, Anwar M, Smith DE, Feng CJ, Kadowitz PJ, Nossaman BD. Analysis of responses of garlic derivatives in the pulmonary vascular bed of the rat. J Appl Physiol. 2000;89:353–358. doi: 10.1152/jappl.2000.89.1.353. [DOI] [PubMed] [Google Scholar]
- 37.Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Hogestatt ED, Julius D, Jordt SE, Zygmunt PM. Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci U S A. 2005;102:12248–12252. doi: 10.1073/pnas.0505356102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, Jordt SE. TRPA1 is a major oxidant sensor in murine airway sensory neurons. The Journal of clinical investigation. 2008;118:1899–1910. doi: 10.1172/JCI34192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.