


Abstract
The accurate mapping of reads that span splice junctions is a critical component of all analytic techniques that work with RNA-seq data. We introduce a second generation splice detection algorithm, MapSplice, whose focus is high sensitivity and specificity in the detection of splices as well as CPU and memory efficiency. MapSplice can be applied to both short (<75 bp) and long reads (≥75 bp). MapSplice is not dependent on splice site features or intron length, consequently it can detect novel canonical as well as non-canonical splices. MapSplice leverages the quality and diversity of read alignments of a given splice to increase accuracy. We demonstrate that MapSplice achieves higher sensitivity and specificity than TopHat and SpliceMap on a set of simulated RNA-seq data. Experimental studies also support the accuracy of the algorithm. Splice junctions derived from eight breast cancer RNA-seq datasets recapitulated the extensiveness of alternative splicing on a global level as well as the differences between molecular subtypes of breast cancer. These combined results indicate that MapSplice is a highly accurate algorithm for the alignment of RNA-seq reads to splice junctions. Software download URL: http://www.netlab.uky.edu/p/bioinfo/MapSplice.
INTRODUCTION
Alternative splicing is a fundamental mechanism that generates transcript diversity. Particular combinations of cis-acting sequences, trans-acting splicing regulators and histone modifications contribute to differential exon usage among diverse cell types (1,2). Through shuffling of exons, splice sites and untranslated regions can drastically alter the cellular function of proteins (3,4). Notably, SNPs have been linked to changes in transcript isoform proportions among different individuals (5). In some cases, rare mutations that alter splicing patterns have been linked to disease (6–9). Thus, transcriptome profiling should comprise a comprehensive survey of alternative splicing.
Microarrays were the first technology to enable global assessment of alternative splicing (10–13). Oligonucleotides designed to span two adjacent exons can be used to measure the abundance of splice junctions. However, these splice junction probes only interrogate a predefined set of transcript isoforms. Due to the large number of hypothetical exon–exon combinations, microarrays are not efficient at discovering novel transcript isoforms.
Deep transcriptome sequencing provides sufficient read counts to measure relative proportions of transcript isoforms, as well as to discover new isoforms (1,14–17). Several high-throughput technologies currently sample short sequence tags, typically <200 bp in size. The accurate mapping of sequence tags that span splice junctions is the foundation for transcript isoform reconstruction (18,19). One approach relies on existing transcript annotations to create a database of potential splice junction sequences. Similar to the above limitation with microarrays, the construction of predefined alignment databases limits the set of possible splice junctions interrogated.
Recently methods have been developed to find novel splice junctions from short sequence tags. The pioneering QPALMA algorithm adopted a machine learning algorithm to predict splice junctions from a training set of positive controls (20). The TopHat algorithm constructs candidate splice junctions by pairing candidate exons and evaluating the alignment of reads to such candidates (21). SpliceMap is another method that uses splice site flanking bases in locating potential splice sites (22).
We introduce the MapSplice algorithm to detect splice junctions without any dependence on splice site features. This enables MapSplice to discover non-canonical junctions and other novel splicing events, in additional to the more common canonical junctions. MapSplice can be generally applied to both short and long RNA-seq reads. In addition, MapSplice leverages the quality and diversity of read alignments that include a given splice to increase specificity in junction discovery. As a result, MapSplice demonstrates high specificity and sensitivity. Performance results are established using synthetic data sets and validated experimentally.
We have used MapSplice to investigate significant differences in alternative splicing between a set of basal and luminal breast cancer tissues. Experimental validation of 20 exon skipping events by quantitative RT–PCR (qRT–PCR) correctly identified isoform proportions that are highly correlated (Pearson's correlation = 0.86) with their estimates based on splice junctions. Splice junctions also recapitulated the difference between molecular subtypes of breast cancer. On a global level, the proportion of splice junctions in various categories of alternative splicing was concordant with a previous RNA-seq study.
MATERIALS AND METHODS
The goal of MapSplice is to find the exon splice junctions present in the sampled mRNA transcriptome, and to determine the most likely alignment of each mRNA sequence tag to a reference genome. Each tag corresponds to a number of consecutive nucleotides read from an mRNA transcript, where the length of the tag is determined by the protocol and the sequencing technology. For example, the Illumina Genome Analyzer IIx generates over 20M tags of size up to 100 bp per sequencing lane.
MapSplice operates in two phases to achieve its goal. In the ‘tag alignment' phase, candidate alignments of the mRNA tags to the reference genome G are determined. Tags with a contiguous alignment fall within an exon and can be mapped directly to 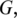 but tags that include one or more splice junctions require a gapped alignment, with each gap corresponding to an intron spliced out during transcription. Since multiple possible alignments may be found, the result of this phase is, in general, a set of candidate alignments for each tag.
but tags that include one or more splice junctions require a gapped alignment, with each gap corresponding to an intron spliced out during transcription. Since multiple possible alignments may be found, the result of this phase is, in general, a set of candidate alignments for each tag.
In the ‘splice inference phase,’ splice junctions that appear in the alignments of one or more tags are analyzed to determine a splice significance score based on the quality and diversity of alignments that include the splice. The purpose of this phase is to reject spurious splices and to provide a basis for choosing the most likely alignment for each tag based on a combination of alignment quality and splice significance. An overview of the algorithm can be found in Figure 1. The two phases are described in the following two sections.
Figure 1.

An overview of the MapSplice pipeline. The algorithm contains two phases: tag alignment (Step 1–Step 4) and splice inference (Step 5–Step 6). In the ‘tag alignment' phase, candidate alignments of the mRNA tags to the reference genome 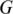 are determined. In the ‘splice inference' phase, splice junctions that appear in one or more tag alignments are analyzed to determine a splice significance score based on the quality and diversity of alignments that include the splice. Ambiguous candidate alignments are resolved by selecting the alignment with the overall highest quality match and highest confidence splice junctions.
are determined. In the ‘splice inference' phase, splice junctions that appear in one or more tag alignments are analyzed to determine a splice significance score based on the quality and diversity of alignments that include the splice. Ambiguous candidate alignments are resolved by selecting the alignment with the overall highest quality match and highest confidence splice junctions.
Tag alignment
Let Θ be the set of tags and let m be the tag length. A tag 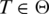 has an ‘exonic alignment' if it can be aligned in its entirety to a consecutive sequence of nucleotides in G. T has a ‘spliced alignment' if its alignment to G requires one or more gaps.
has an ‘exonic alignment' if it can be aligned in its entirety to a consecutive sequence of nucleotides in G. T has a ‘spliced alignment' if its alignment to G requires one or more gaps.
MapSplice identifies candidate tag alignments in three steps. First, tags are partitioned into consecutive short segments and an exonic alignment to G is attempted for each segment. In the second step, segments that do not have an exonic alignment are considered for spliced alignment using a splice junction search technique that starts from neighboring segments already aligned. In the final step, segment alignments for a tag are merged to find candidate overall alignments for each tag. The details of the steps follow next.
Step 1: partition tags into segments
Tags in Θ of length m are partitioned into n consecutive segments of length k where 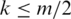 . Typically k is 20–25 for tags of length 50 or greater. As k is decreased, the chance that a segment contains one or more splice junctions decreases correspondingly, but the chance for multiple spurious alignments of the segment increases. The segments making up a tag T are labeled
. Typically k is 20–25 for tags of length 50 or greater. As k is decreased, the chance that a segment contains one or more splice junctions decreases correspondingly, but the chance for multiple spurious alignments of the segment increases. The segments making up a tag T are labeled  where the number of segments
where the number of segments 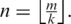
Step 2: exonic alignment of segments
Exonic alignment of segments can be performed using fast approximate aligners such as Bowtie (23), and BWA (24), or aligners with more general error tolerance models such as SOAP2 (25), BFAST (26) and MAQ (27). For each segment 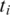 of tag T, let
of tag T, let 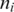 be the number of possible exonic alignments of
be the number of possible exonic alignments of 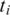 to the genome, determined using one of the algorithms mentioned above with an error tolerance of
to the genome, determined using one of the algorithms mentioned above with an error tolerance of 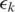 mismatches. When
mismatches. When 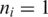 the segment has a unique exonic alignment. When
the segment has a unique exonic alignment. When 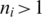 the segment has multiple alignments, each of which is considered in the subsequent steps.
the segment has multiple alignments, each of which is considered in the subsequent steps.
Step 3: spliced alignment of segments
If 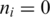 , segment
, segment 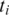 does not have an exonic alignment. One possible reason is that it may have a gapped alignment crossing a splice junction. In general, if the minimum exon length is at least 2k, then for every pair of consecutive segments in T at least one segment should have an exonic alignment. Therefore, the alignment of segment
does not have an exonic alignment. One possible reason is that it may have a gapped alignment crossing a splice junction. In general, if the minimum exon length is at least 2k, then for every pair of consecutive segments in T at least one segment should have an exonic alignment. Therefore, the alignment of segment 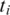 is localized to the alignments of its neighbors. The following two techniques are used to find a spliced alignment for segments, and are illustrated in Figure 2.
is localized to the alignments of its neighbors. The following two techniques are used to find a spliced alignment for segments, and are illustrated in Figure 2.
Figure 2.

A portion of an mRNA transcript sampled by tag 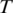 consists of the 3′ end of exon 1, all of exon 2 and the 5′ end of exon 3.
consists of the 3′ end of exon 1, all of exon 2 and the 5′ end of exon 3. 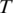 is split into segments t1,…, tn each of length
is split into segments t1,…, tn each of length 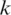 to identify the alignment of
to identify the alignment of 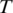 to the genome. Provided no exon has a length less than
to the genome. Provided no exon has a length less than 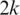 nucleotides, at least one of every two consecutive segments must have an exonic alignment. In this example with
nucleotides, at least one of every two consecutive segments must have an exonic alignment. In this example with 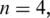 segments t1 and t3 have exonic alignment. Segment t2 has spliced alignment; the splice junction
segments t1 and t3 have exonic alignment. Segment t2 has spliced alignment; the splice junction 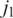 can be easily discovered using the double-anchor search method starting from t1 and t3. The spliced alignment for t4 is discovered by searching downstream in the genome for an occurrence of the suffix
can be easily discovered using the double-anchor search method starting from t1 and t3. The spliced alignment for t4 is discovered by searching downstream in the genome for an occurrence of the suffix 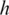 -mer of t4. When such an occurrence is found, the double-anchor search method is used to evaluate a possible splice junction
-mer of t4. When such an occurrence is found, the double-anchor search method is used to evaluate a possible splice junction 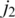 between
between 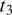 and the
and the 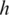 -mer occurrence.
-mer occurrence.
If 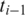 and
and 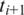 both have exonic alignments, then we perform a ‘double-anchored' spliced alignment of
both have exonic alignments, then we perform a ‘double-anchored' spliced alignment of 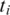 for all combinations of exonic alignments of
for all combinations of exonic alignments of 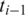 and
and 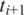 . If only one neighboring segment
. If only one neighboring segment 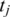 has an exonic alignment, then we perform a ‘single-anchored' alignment starting from the
has an exonic alignment, then we perform a ‘single-anchored' alignment starting from the 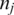 possible alignments of
possible alignments of 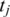 .
.
-
Double-anchored spliced alignment: the spliced alignment of
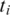 to the genomic interval between anchors
to the genomic interval between anchors 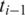 and
and 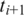 need only consider the
need only consider the 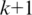 possible positions of the splice junction x within
possible positions of the splice junction x within 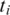 and minimize alignment mismatch.Formally, the ‘Hamming distance'
and minimize alignment mismatch.Formally, the ‘Hamming distance'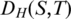 between two equal length sequences S and T is defined as the number of corresponding positions with mismatching bases. We define the spliced alignment between the segment
between two equal length sequences S and T is defined as the number of corresponding positions with mismatching bases. We define the spliced alignment between the segment 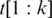 and the genomic interval
and the genomic interval 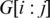 as
as
which yields the optimal position x of the splice junction in t that gives the best spliced alignment to the given genomic interval. The splice junction x defines the intron as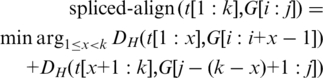
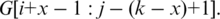
To find the spliced alignment for
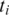 between two aligned segments, let
between two aligned segments, let 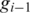 and
and 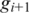 be the leftmost genomic coordinates in the alignment of
be the leftmost genomic coordinates in the alignment of 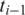 and
and 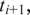 respectively, and compute spliced–align
respectively, and compute spliced–align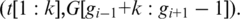
In case the alignment cost for the splice junction exceeds the error tolerance threshold
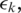 the alignment for
the alignment for 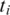 fails. If there exists more than one splice junction position with minimum cost for
fails. If there exists more than one splice junction position with minimum cost for 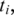 multiple alignments are recorded for
multiple alignments are recorded for 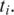
-
Single-anchored spliced alignment: in the case of a single anchor
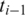 upstream of the unaligned
upstream of the unaligned 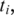 we conduct a search for
we conduct a search for 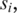 the h-base suffix of
the h-base suffix of 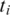 in the genomic region downstream from
in the genomic region downstream from 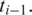 Similarly, in the case of a single anchor
Similarly, in the case of a single anchor 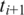 downstream from
downstream from 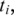 the search is for the h-base prefix
the search is for the h-base prefix 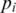 of
of 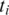 in the region upstream from
in the region upstream from 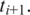 In either case this search is limited in range by a parameter D, the maximum intron size for single anchor search, typically set to 50 000 bp.
In either case this search is limited in range by a parameter D, the maximum intron size for single anchor search, typically set to 50 000 bp.All single anchored alignments can be resolved with a single traversal of (the expressed portion of) the genome using a sliding window of size D. An h-mer index is maintained during this traversal, mapping occurrences of an h-mer
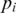 to the downstream anchor
to the downstream anchor 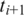 within distance D and occurrences of an h-mer
within distance D and occurrences of an h-mer 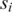 to the upstream anchor
to the upstream anchor 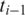 within distance D. As the window moves, new entries are added as anchors come within range, and old entries are deleted as anchors fall out of range.
within distance D. As the window moves, new entries are added as anchors come within range, and old entries are deleted as anchors fall out of range.When the h-mer at the current coordinate c in the genome scan is mapped to a downstream segment
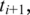 spliced-align
spliced-align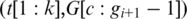 gives the best spliced alignment which is recorded if it is within the segment error threshold
gives the best spliced alignment which is recorded if it is within the segment error threshold 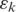 . Similarly, when the h-mer is mapped to an upstream segment
. Similarly, when the h-mer is mapped to an upstream segment 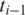 , we record spliced-align
, we record spliced-align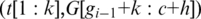 if it is within the segment error threshold
if it is within the segment error threshold 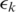 .
. -
Spliced alignment in the presence of small exons: if an exon shorter than
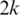 is included in a transcript, it is possible that two adjacent segments
is included in a transcript, it is possible that two adjacent segments 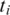 and
and 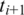 both include a splice junction so that neither can be aligned continuously within an exonic region. If the exon is shorter than k, even a single segment might include more than one splice junction. The following approach allows us to detect exons with size less than
both include a splice junction so that neither can be aligned continuously within an exonic region. If the exon is shorter than k, even a single segment might include more than one splice junction. The following approach allows us to detect exons with size less than 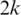 .
.Assume S is a sequence of one or two missed segments between two anchors
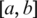 that cannot be successfully aligned in the previous steps, potentially due to short exons. We divide S into a sequential set of h-mers and index S with these h-mers. By extending the sequential scan of the genome used in single-anchored spliced alignment, h-mers on the reference genome in
that cannot be successfully aligned in the previous steps, potentially due to short exons. We divide S into a sequential set of h-mers and index S with these h-mers. By extending the sequential scan of the genome used in single-anchored spliced alignment, h-mers on the reference genome in 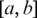 can all be searched simultaneously. When a match exists, two double-anchored spliced alignments will be performed: one is between a and the 5′-site of the h-mer alignment; and the other is between the 3′-site of h-mer alignment and b.
can all be searched simultaneously. When a match exists, two double-anchored spliced alignments will be performed: one is between a and the 5′-site of the h-mer alignment; and the other is between the 3′-site of h-mer alignment and b.According to the pigeon-hole principle, if the exon is no shorter than
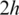 , one of the h-mers in the unaligned segments will fall within an exon and thus trigger the subsequent spliced alignments. Therefore, this method is guaranteed to detect small exons longer than
, one of the h-mers in the unaligned segments will fall within an exon and thus trigger the subsequent spliced alignments. Therefore, this method is guaranteed to detect small exons longer than 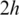 and possibly detect shorter exons. The typical h-mer size is 6–8 bps. When exons are shorter than
and possibly detect shorter exons. The typical h-mer size is 6–8 bps. When exons are shorter than 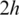 , the chances of finding a spliced alignment decrease. Reducing h will lead to an increasing number of spurious matches that will be difficult to filter out.
, the chances of finding a spliced alignment decrease. Reducing h will lead to an increasing number of spurious matches that will be difficult to filter out.
Step 4: merging segment alignments
The assembly of a complete tag alignment from individual alignments of its segments is straightforward if each segment is aligned uniquely and connects to its neighboring segments without a gap. However, a given segment 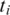 may be aligned at multiple locations. In this case, the possible combinations of alignments must be searched to find the best overall alignment for the tag.
may be aligned at multiple locations. In this case, the possible combinations of alignments must be searched to find the best overall alignment for the tag.
Let 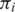 be the set of alignments for segment
be the set of alignments for segment 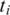 and when
and when 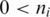 , let
, let 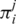 be the
be the 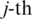 alignment of
alignment of 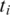 , where
, where 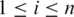 and
and 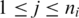 . In principle there exist
. In principle there exist 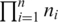 different combinations of alignments, but most can be ruled out by a simple coherence test based on contiguity of consecutive segment alignments.
different combinations of alignments, but most can be ruled out by a simple coherence test based on contiguity of consecutive segment alignments.
Two adjacent segments 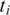 and
and 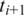 with exonic alignment that are not contiguous on the genome are checked for a splice junction between the two segments using the double-anchored spliced alignment method. This procedure also corrects inaccurate splice points due to the error tolerance in the alignment of
with exonic alignment that are not contiguous on the genome are checked for a splice junction between the two segments using the double-anchored spliced alignment method. This procedure also corrects inaccurate splice points due to the error tolerance in the alignment of 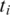 and
and 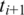 .
.
For each assembly of segments that yields a candidate alignment for T, we compute its ‘mismatch score', a modified Hamming distance between T and its alignment to the genome 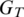 .
.
The mismatch score takes into account base call qualities when available. A poor quality base call can improve the score when associated with a mismatched base, but can also decrease the score when associated with a matched base (28). The base call quality for a given base x in the overall alignment of a tag T can be converted to a probability p that x was called incorrectly, and so the expected mismatch 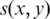 in the alignment of base x to base y in the genome is given by
in the alignment of base x to base y in the genome is given by
| (1) |
where 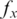 is the probability of base x in the background distribution of nucleotides. We assume a uniform distribution for nucleotides hence
is the probability of base x in the background distribution of nucleotides. We assume a uniform distribution for nucleotides hence 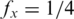 for all x. Thus, given a proposed alignment of
for all x. Thus, given a proposed alignment of 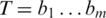 and
and 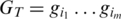 and
and 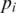 the probability that nucleotide
the probability that nucleotide 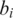 was called incorrectly, the expected mismatch is
was called incorrectly, the expected mismatch is 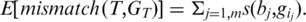
The candidate alignment is retained if 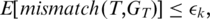 otherwise it is discarded. Note that while each segment was aligned allowing
otherwise it is discarded. Note that while each segment was aligned allowing 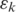 mismatches, the overall tag alignment only allows a total of
mismatches, the overall tag alignment only allows a total of 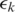 expected mismatches. We define the quality of the alignment to be
expected mismatches. We define the quality of the alignment to be 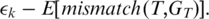
Splice junction inference
Splice junction alignments introduce a multiplicity of ways in which a tag may be split into pieces, each of which may be separately aligned to the genome. For a given tag, at most one of these is the true alignment. Splice inference leverages the extensive sampling of splice junctions by tags to compute a junction quality that can be used to distinguish true splice junctions from spurious splice junctions and to determine the best alignment among the remaining candidate alignments of a tag.
Step 5: splice junction quality
For a given splice 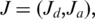 where
where 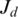 is the last coordinate of the donor exon and
is the last coordinate of the donor exon and 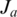 is the first coordinate of the acceptor exon, we consider the set
is the first coordinate of the acceptor exon, we consider the set 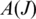 of tags that include a splice junction for J in a candidate alignment. We define two statistical measures on
of tags that include a splice junction for J in a candidate alignment. We define two statistical measures on 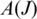 : the ‘anchor significance'
: the ‘anchor significance' 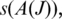 determined by the alignment in
determined by the alignment in 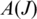 that maximizes significance as a result of long anchors on each side of the splice junction, and the ‘entropy'
that maximizes significance as a result of long anchors on each side of the splice junction, and the ‘entropy' 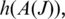 measured by the diversity of splice junction positions in
measured by the diversity of splice junction positions in 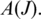
-
Anchor significance of a splice junction: a tag that includes a splice junction has some number of contiguous bases aligned on either side of the splice site. An alignment with a short anchor on one side has low confidence, since we expect it to be easy to find other occurrences of the sequence of nucleotides found in the short anchor each of which might equally well be the correct target. We define the anchor significance of a splice J in a tag
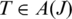 as follows. Let
as follows. Let 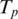 be the maximal contiguous sequence of bases in T with exonic alignment ending at coordinate
be the maximal contiguous sequence of bases in T with exonic alignment ending at coordinate 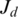 in the genome, and let
in the genome, and let 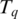 be the maximal contiguous sequence of bases in T with exonic alignment starting from coordinate
be the maximal contiguous sequence of bases in T with exonic alignment starting from coordinate 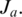 These are the two anchors, each of which has at least one alignment (the one in T). The expected number of alignments in the genome for an anchor
These are the two anchors, each of which has at least one alignment (the one in T). The expected number of alignments in the genome for an anchor 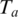 is therefore given by
is therefore given by
Here, we model the genome as a sequence of independent random variables with uniform distribution over A, C, T, G, so that the chance that a length n sequence aligns at a given coordinate is simply 4−n. For double-anchor alignments, the search space is effectively the entire genome of length N. For single anchor alignments, we only consider occurrences within distance D.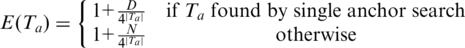
Since we assume that only one of the potential alignments is correct and the rest are spurious, the chance of a spurious alignment of anchor
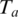 is
is 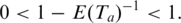 Thus the log-transformed significance of anchor
Thus the log-transformed significance of anchor 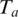 is
is 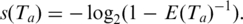 The anchor alignment of junction J in T is only as significant as the anchor with least confidence, hence is
The anchor alignment of junction J in T is only as significant as the anchor with least confidence, hence is 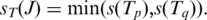 The anchor significance of junction J over all occurrences in
The anchor significance of junction J over all occurrences in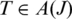 is the occurrence with greatest anchor significance:
is the occurrence with greatest anchor significance:
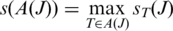
-
Entropy: in principle, the RNA-seq protocol samples each transcript uniformly, so that the position of a true splice junction J within
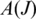 is expected to be uniformly distributed on
is expected to be uniformly distributed on 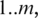 provided the sampling is sufficiently deep and the splice junction is not too close to the end of the transcript. To measure the uniformity of the sampling, we apply Shannon maximum entropy to the distribution of splice junction positions in
provided the sampling is sufficiently deep and the splice junction is not too close to the end of the transcript. To measure the uniformity of the sampling, we apply Shannon maximum entropy to the distribution of splice junction positions in 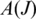 for splice J. Let
for splice J. Let 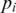 with
with 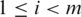 be the frequency of occurrence of a splice junction J at position i within
be the frequency of occurrence of a splice junction J at position i within 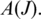 The Shannon entropy can be measured as
The Shannon entropy can be measured as
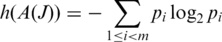
The higher the Shannon entropy, the closer the distribution is to uniform, and therefore the higher the chance that the junction is part of some transcript that was sampled uniformly.
- Combined Metric: the combined metric
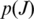 is the posterior probability that junction J is a true junction determined using Bayesian regression. The observed data of
is the posterior probability that junction J is a true junction determined using Bayesian regression. The observed data of 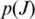 are the entropy and anchor significance of J within
are the entropy and anchor significance of J within 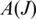 and the average quality of read alignments that include J.
and the average quality of read alignments that include J.
We apply linear regression to obtain the best configuration of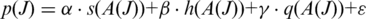
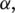
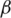 and
and 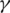 that achieves the maximum sensitivity and specificity in junction classification.
that achieves the maximum sensitivity and specificity in junction classification.
Step 6: best alignment for tags
For each tag T, we select the candidate alignment 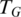 that achieves the highest score when combining alignment quality from Step 4 and junction quality from Step 5.
that achieves the highest score when combining alignment quality from Step 4 and junction quality from Step 5.
Synthetic data generation for validation
To evaluate the sensitivity and specificity of MapSplice, we generated synthetic data sets of tags derived from transcripts cataloged in the Alternative Splicing and Transcript Diversity (ASTD) database (29).
This database collects full-length transcripts illustrating alternative splicing events in genes from human, mouse and rat. A synthetic `transcriptome' is generated by randomly selecting genes and expression levels according to an empirical distribution of tags per gene observed in Ref. (1). Within a gene, transcripts are selected at random following various submodels that determine expression level of individual transcripts relative to the overall gene. The synthetic transcriptome characterized in this fashion is then sampled to yield two synthetic RNA-seq data sets. The noise-free data set samples the transcripts exactly and the resultant tags align to the reference genome exactly to model single-nucleotide variations in the data base transcripts. The noisy data set introduces mutations into base calls following empirical Illumina base call quality profiles. The resulting data sets mimic the observed distribution of errors in tags in Ref. (30).
Experimental validation by qRT–PCR
Total RNA isolated from MCF-7 and SUM-102 cells was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit with RNase inhibitor (Applied Biosystems, Foster City, CA, USA) as per manufacturer’s instructions. Relative expression levels of the transcripts of interest were determined by qRT–PCR on the Applied Biosystems 7300 Real Time PCR System with premade or custom TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) containing primers flanking the splice sites of interest and FAM/MGB-labeled oligonucleotide probes. PCR reactions were carried out as per manufacturer’s instructions. cDNA equivalent to 100 ng of total RNA was amplified with 1 μl of TaqMan assay in Gene Expression master mix in a total volume of 20 μl. Each assay was performed in triplicate. Thermal Cycling conditions were as follows: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min. Ct values were determined in the manufacturer’s software, data was further analyzed in Excel utilizing comparative Ct method. For comparisons of relative expression levels between the two cell lines, Ct values for the transcripts of interest were first normalized to those of HPRT1.
RESULTS
Junction inference
We constructed a synthetic noise-free RNA-seq data set with 20M 100 bp tags sampling 46 311 distinct transcripts from the ASTD. The tags were aligned to the reference genome (hg18) using the MapSplice algorithm steps 1–4 with 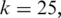
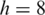 and
and 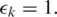 To establish the training data set containing both true and false junctions, no restrictions on splice site flanking sequences or maximum intron size were enforced.
To establish the training data set containing both true and false junctions, no restrictions on splice site flanking sequences or maximum intron size were enforced.
We randomly selected 10K true junctions and 10K false junctions as a training set and analyzed the three different junction classification metrics utilized in Step 5 of MapSplice: alignment quality entropy and anchor significance, as well the combined metric obtained by linear regression of the first three metrics. Five-fold cross-validation was applied to avoid sample bias in training. The ROC curve that illustrates the sensitivity and specificity of each metric is shown in Figure 3. The combined metric (solid green curve) offers better classification results than individual metrics simply because individual metrics only capture one property of a junction. At the best point, the combined metric achieves a true-positive rate of 96.3% and false-positive rate of 8%.
Figure 3.

ROC curves for junction classification. A synthetic data set of 20M 100 bp tags was generated from transcripts selected from the ASTD database. 10K true-positive junctions and 10K false-positive junctions were selected as training data sets. Five different metrics were evaluated. They include (i) alignment quality; (ii) anchor significance; (iii) entropy; (iv) coverage; and (v) combination of metrics (i–iii). The red cross in each curve marks the point with best balance of sensitivity and specificity.
We also compared the results with one of the most commonly used metrics: junction coverage (the number of tags aligned to a junction). In many studies, a junction is considered to be true if at least three tags are aligned to the junction. However, as shown in Figure 3, coverage (solid red curve) is the least reliable metric and yields the worst performance in terms of junction classification.
The specific parameters 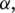
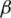 and
and 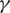 in the combined metric obtained from this synthetic data set were used for all data sets processed by MapSplice in this article. Slightly improved sensitivity can be obtained by using parameters obtained by logistic regression that are specialized for data sets with specific tag lengths. Experiments on the robustness of the parameters and their sensitivity to tag length and sampling depth are included in the Supplementary Data.
in the combined metric obtained from this synthetic data set were used for all data sets processed by MapSplice in this article. Slightly improved sensitivity can be obtained by using parameters obtained by logistic regression that are specialized for data sets with specific tag lengths. Experiments on the robustness of the parameters and their sensitivity to tag length and sampling depth are included in the Supplementary Data.
Sensitivity and specificity of splice inference
Three programs that map splice junctions using RNA-seq data were compared, MapSplice, TopHat (1.0.12) and SpliceMap (C++, v3.0, 15 April 2010). We applied all three algorithms to two representative synthetic data sets. One was a data set with 20M tags of length 50 bp. The other was a data set with 20M 100 bp tags. For both MapSplice and TopHat, we set 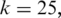
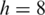 and
and 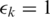 . For SpliceMap, the only configurable parameter is the mismatch in the segment (seeds), which was set to 1 as well. In comparison (Table 1), both TopHat and MapSplice were more memory efficient and much faster than SpliceMap. The filtering criteria adopted by SpliceMap including minimum anchor (extension) of 10 bp and no multiple alignments within a 400 kb region improved its specificity with some tradeoff in sensitivity in 100 bp tags. MapSplice performed best in both categories by detecting more true-positive junctions and fewer false-positive junctions.
. For SpliceMap, the only configurable parameter is the mismatch in the segment (seeds), which was set to 1 as well. In comparison (Table 1), both TopHat and MapSplice were more memory efficient and much faster than SpliceMap. The filtering criteria adopted by SpliceMap including minimum anchor (extension) of 10 bp and no multiple alignments within a 400 kb region improved its specificity with some tradeoff in sensitivity in 100 bp tags. MapSplice performed best in both categories by detecting more true-positive junctions and fewer false-positive junctions.
Table 1.
Comparison of TopHat (21), SpliceMap (22) and MapSplice on two synthetic data sets with tags of length 50 and 100 bp, respectively
| Data set | Method | Performance |
Junction discovery |
|||
|---|---|---|---|---|---|---|
| Time | Peak Mem. | Total | True | False | ||
| 50 bp | TopHat (1.0.12) | 50 min | <4 GB | 85 356 | 76 486 | 8870 |
| SpliceMap (C++3.0) | 13 h | 9.3 GB | 88 807 | 87 205 | 1602 | |
| MapSplice | 25 min | <4 GB | 88 180 | 87 330 | 750 | |
| 100 bp | TopHat (1.0.12) | 3 h 40 min | <4 GB | 100 012 | 90 720 | 9292 |
| SpliceMap (C++3.0) | 41 h | 12 GB | 91 259 | 89 991 | 1268 | |
| MapSplice | 1 h 50 min | <4 GB | 94 112 | 92 849 | 1263 | |
Both data sets have 20 million tags.
The best values in each comparison are shown in bold.
Due to the incompleteness of SpliceMap's output (tag alignments were not generated), we limit the more comprehensive comparison to TopHat and MapSplice. We investigated the sensitivity and specificity in splice inference as a function of tag length and sampling depth. We generated synthetic data sets to study the effect of these variations on junction discovery. In the synthetic data set, we have ground truth junctions and know their actual coverage, i.e. the number of tags spanning each junction. Two measures were used to evaluate the algorithms. The ‘sensitivity' is the ratio of the total number of true junctions discovered to the total number of junctions sampled in the synthetic data. The ‘specificity' is the ratio of the total number of true junctions discovered over the total number of discovered junctions. Since coverage of a junction is essential for the junction to be discovered, we plot sensitivity and specificity at coverage x as the sensitivity and specificity for all junctions with coverage x or greater, as shown in Figure 4. We also show the recovered ‘coverage ratio' for junctions identified as true in Figure 5.
Figure 4.

Sensitivity and specificity of splice inference in synthetic data sets with different characteristics. The sensitivity is the fraction of true junctions discovered among the true junctions sampled in the synthetic data. The specificity is the fraction of true junctions within the reported junctions. Since the depth of sampling is essential for the junction to be discovered, we plot the sensitivity and specificity as a function of the coverage threshold. (A) and (B) The sensitivity and specificity for perfect tags and tags seeded with sequencing errors. (C) and (D) Sensitivity and specificity compared at different tag lengths (50 bp, 75 bp and 100 bp). (E) and (F) Sensitivity and specificity compared at two different depths of sampling (10M and 20M tags, respectively).
Figure 5.

Fraction of tags containing a true junction recovered (i.e. aligned to include the junction) as a function of junction coverage (defined by exponential bins). (A) TopHat recovers about 63% tags while (B) MapSplice recovers an average of 84% of the tags at each junction. The whiskers in the box plot with a recovery ratio >1 at very low coverage are due to false positives or repeats in rare cases.
Effect of noise
In the first experiment, we constructed the error-free and noisy versions of a 100 bp synthetic RNA-seq data sets of 20M tags as described above. MapSplice and TopHat were run on both data sets and given the same error tolerance of 4% (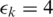 ). Figure 5A and B shows that performance was only impaired at low coverage. When coverage is high, the sensitivity is similar despite the presence of errors. Specificity is more affected, but also converges to similar performance when coverage is high. With low coverage, more spurious junctions were discovered in the data set with error than the one without. Comparing MapSplice with TopHat, MapSplice has higher sensitivity and specificity in identifying junctions in both data sets. Specificity is substantially higher even at low coverage.
). Figure 5A and B shows that performance was only impaired at low coverage. When coverage is high, the sensitivity is similar despite the presence of errors. Specificity is more affected, but also converges to similar performance when coverage is high. With low coverage, more spurious junctions were discovered in the data set with error than the one without. Comparing MapSplice with TopHat, MapSplice has higher sensitivity and specificity in identifying junctions in both data sets. Specificity is substantially higher even at low coverage.
Effect of tag length
In the second experiment, we generated a synthetic data set of 20M 100 bp tags and created two additional data sets by selecting a 50 and 75 bp random subsequences of the 100 bp tags, respectively. Both MapSplice and TopHat were applied to these data sets with maximum percentage of mismatches as 4% of the tag length. The result is shown in Figure 5C and D. In general, for both TopHat and MapSplice, longer tags not only improve the sensitivity but also improve the specificity of the junction discovery. In comparison, MapSplice has higher sensitivity for all three tag lengths. The difference in sensitivity is more pronounced in junctions with low coverage, where junction discovery is most difficult.
Effect of sampling depth
In the final experiment, we generated two 100 bp data sets with a different number of tags: 10M and 20M, respectively. Doubling the sampling depth does not double the specificity of junctions, but it does improve the sensitivity. Doubling the depth has a negative effect on specificity, especially in the low coverage areas. This is mostly because increasing the number of tags sampled from a fixed number of transcripts increases the chances for a repeated tag, especially one with a high error rate, to be incorrectly aligned on the genome.
Breast cancer transcriptomes
We performed cDNA sequencing to obtain about 25 million tags of length 75 bp for four primary breast tumors and replicate samples for two breast cancer cell lines. In total, four samples correspond to the basal subtype of breast cancer, and four samples correspond to the luminal subtype. We applied both MapSplice and TopHat to detect splice junctions using the same parameter settings employed with the synthetic data sets. The mapping result is shown in Table 2. In summary, between 10% and 16% of the tags in each sample include splice junctions in their alignment. Over 77% of these canonical junctions were confirmed by known transcripts in GenBank, which represented between 6% and 11% more confirmed junctions than TopHat. MapSplice identified 2421 ∼ 3173 semi-canonical junctions, much less than the number reported by TopHat. But for both sets, very similar sets of junctions are known, which suggests that MapSplice has a higher specificity for non-canonical splice junctions.
Table 2.
Tag mapping and splice junction detection results on eight breast cancer samples: two basal (BAS) primary tumors, two SUM-102 (SUM) cell lines, two luminal (LUM) primary tumors and two MCF-7 (MCF) cell lines
| Sample | Tag mapping |
Canonical junctionsa |
Semi-canonical junctionsb |
Non-canonical junctionsc |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total tags | MS spliced (%) | TH spliced (%) | MS |
TH |
MS |
TH |
MS |
TH |
|||||||
| Total | Knownd | Total | Knownd | Total | Knownd | Total | Knownd | Total | Knownd | Total | Knownd | ||||
| BAS | 23.9M | 12.7 | 10.7 | 168.6K | 131.4K | 140.3K | 114.5K | 2914 | 970 | 8441 | 958 | 1967 | 96 | 4874 | 0 |
| BAS | 25.9M | 12.7 | 10.7 | 178.1K | 138.1K | 150.3K | 122.7K | 3173 | 1036 | 8276 | 1027 | 1691 | 109 | 4994 | 0 |
| SUM | 25.4M | 15.8 | 13.7 | 149.7K | 119.8K | 132.6K | 109.3K | 2691 | 910 | 7828 | 940 | 1665 | 91 | 2468 | 0 |
| SUM | 25.5M | 15.8 | 13.7 | 149.8K | 119.9K | 132.5K | 109.4K | 2683 | 909 | 7989 | 921 | 1685 | 99 | 2138 | 0 |
| LUM | 25.8M | 10.3 | 8.6 | 175.2K | 137.3K | 145.2K | 119.4K | 2447 | 1011 | 8230 | 995 | 1365 | 93 | 4308 | 0 |
| LUM | 25.0M | 11.1 | 9.4 | 173.7K | 137.6K | 144.6K | 118.8K | 2507 | 1014 | 9467 | 989 | 1591 | 93 | 5944 | 0 |
| MCF | 24.6M | 15.4 | 13.3 | 154.7K | 120.2K | 135.5K | 110.5K | 2421 | 937 | 6387 | 951 | 1240 | 92 | 1129 | 0 |
| MCF | 23.1M | 15.4 | 13.2 | 152.3K | 119.4K | 133.4K | 109.5K | 2287 | 935 | 6222 | 946 | 1157 | 91 | 1139 | 0 |
MapSplice (MS) detected 177 875 splice junctions occurring in at least two tags in any of the breast tumors or cell lines. Of the tags, 10–16% in each sample contained splice junctions. MapSplice detected 149.7K–178.1K canonical junctions, among which about 109.3K–122.7K are confirmed by known transcripts in GenBank. In general, MapSplice detected 10K–18K more canonical junctions than TopHat (TH). MapSplice identified 2421–3173 semi-canonical junctions, far fewer than the number reported by TopHat. But in both sets, a very similar subset of junctions are known. There are 91–99 non-canonical junctions known out of the 1157–1967 non-canonical junctions reported by MapSplice. While TopHat did report up to 5944 non-canonical junctions, none of them are confirmed.
aFlanked by GT-AG.
bFlanked by AT-AC or GC-AG.
cOther flanking dinucleotide.
dA junction is known if it is included in at least one transcript in GenBank.
MapSplice reported between 1157 and 1967 non-canonical splice junctions, of which 5–8% were confirmed in known GenBank transcripts. While TopHat reported up to 5944 non-canonical junctions, none of them were confirmed in GenBank transcripts. Since the TopHat program does not search for non-canonical junctions, this result might be an artifact. We found 9205 genes that showed evidence of alternative splicing, ranging from 7371 to 8942 genes per tumor. There were 420 to 430 canonical junctions identified by MapSplice within 2 bp of a known semi-canonical or non-canonical junction. For almost all of them, the tags aligned to the canonical junction have fewer mismatches than if they were aligned to the nearby non-canonical or semi-canonical junction. Such findings suggest that there exist errors in the current database and RNA-seq data might be able to correct these errors.
MapSplice detected the expected proportion of alternative splicing categories, even though it did not rely on a database of transcript annotations. We investigated how many alternative splicing events could be detected at different minimum thresholds for the minor transcript isoforms (Table 3). For instance, at a cutoff of two or more tags per splice junction, MapSplice detected between 7535 and 8270 alternative splicing events in each tumor. These events comprised: 34.5% skipped exons; 30.3% alternative 5′-sites; 33.8% alternative 3′-sites; and 1.4% mutually exclusive exons. A previous RNA-seq study across 10 different tissues and 10 different cell lines (1) reported similar values: 35% skipped exons; 28% alternative 5′-sites and first exons; 31% alternative 3′-sites, last exons and UTRs; and 4% mutually exclusive exons. The high concordance between these two studies further suggested that the MapSplice alignments were highly accurate.
Table 3.
A survey of alternative exon splicing events identified with MapSplice junctions
| Coverage | Alternative exon events |
Mutual Excl. | |||
|---|---|---|---|---|---|
| Sample | Skipped Exon | Alt. Start | Alt. End | ||
 1 1 |
BAS | 6880 | 6700 | 7474 | 442 |
| BAS | 7365 | 7611 | 8005 | 454 | |
| SUM | 5574 | 5690 | 6359 | 353 | |
| SUM | 5491 | 5701 | 6451 | 337 | |
| LUM | 6523 | 7326 | 7777 | 387 | |
| LUM | 6321 | 6928 | 7625 | 355 | |
| MCF | 6776 | 6338 | 7350 | 472 | |
| MCF | 6352 | 6063 | 7083 | 444 | |
 2 2 |
BAS | 2726 | 2144 | 2564 | 101 |
| BAS | 2941 | 2529 | 2689 | 111 | |
| SUM | 2271 | 2098 | 2347 | 103 | |
| SUM | 2277 | 2096 | 2359 | 95 | |
| LUM | 2599 | 2542 | 2574 | 95 | |
| LUM | 2333 | 2031 | 2387 | 86 | |
| MCF | 2949 | 2410 | 2778 | 129 | |
| MCF | 2669 | 2331 | 2588 | 109 | |
 5 5 |
BAS | 651 | 476 | 614 | 26 |
| BAS | 718 | 522 | 641 | 23 | |
| SUM | 644 | 538 | 643 | 25 | |
| SUM | 623 | 528 | 656 | 25 | |
| LUM | 618 | 538 | 582 | 22 | |
| LUM | 503 | 386 | 528 | 21 | |
| MCF | 815 | 686 | 780 | 30 | |
| MCF | 757 | 656 | 735 | 22 | |
Four different types of alternative splicing events involving at least two splice junctions were identified in each sample. They are exon skipping events, alternative 3′-end, alternative 5′ start and mutually exclusive exons. Alternative splicing events were examined at different expression levels, only junctions with coverage larger than the given threshold (1, 2, 5) were considered. In general, there are about 35% exon skipping events, 30% alternative 5′ start events, 34% alternative 3′-end events and 1.3% mutually exclusive exon events.
We randomly selected skipped exon events for the experimental validation of MapSplice alignments to splice junctions. We calculated the proportion of splice junction tags aligning to the skipped exon isoform, and then compared this to the total number of splice junction tags aligning to either the skipped exon isoform or the included exon isoform (Figure 6). We compared these calculations with the splicing ratio determined by qRT–PCR in the MCF-7 and SUM-102 cell lines. With a Pearson's correlation of 0.84 across these 20 events, MapSplice achieved very high accuracy for splice junction counting.
Figure 6.

Correlation of exon skipping ratio detected by MapSplice and Taqman. Each point represents the exon skipping ratio measured in either the MCF-7 (black) or SUM-102 (blue) cell lines.
We identified 12 exon skipping events with significant differences between the basal and luminal subtypes. For instance, NUMB is an adaptor protein in the Notch and Hedgehog pathways with a potential skipped exon in an N-terminal PTB domain, as well as another skipped exon in a C-terminal proline-rich region (31). While all breast cancer samples had similar skipping ratios for the PTB domain exon, we detected significant differences for the skipped exon in the proline-rich region. This longer isoform had exon inclusion ratios ranging 45–78% in the luminal samples, compared with 16–22% of the basal samples (Figure 7). We expect that as more samples are sequenced, we will have more statistical power to identify alternative splicing events that can distinguish between cancer subtypes.
Figure 7.

Examples of alternative exon skipping events. The second exon in NUMB shows differential alternative splicing between two cancer subtypes. The exon skipping ratios in basal samples are ∼70% while in luminal samples they are <50%.
We investigated whether molecular subtypes of tumors may have different patterns of alternative splicing regardless of their gene expression levels. We selected 129 single exon skipping events that were detected by at least three tags in each tumor. The matrix of splicing ratios was then hierarchically clustered, with each row representing a distinct splicing event and each column representing a single tumor (Figure 8). Notably, the two primary breast tumors from the luminal subtype clustered together, as did the two primary breast tumors from the basal subtype. The breast cancer cell lines clustered in between the primary tumors, which indicates that these cell lines resemble their primary tumors of origin, but also share some major differences in splicing. Principal components analysis on these splicing ratios reached similar conclusions: the first principal component distinguished cell lines from primary tumors, while the second principal component segregated luminal versus basal subtypes (Figure 8B and D).
Figure 8.

Clustering of tumor subtypes with skipping ratios of alternative exon skipping events. One hundred twenty-nine alternative exon skipping events with minimum junction support of at least three for each sample were selected. (A) Heatmap (red to blue scale) of skipping ratios, where each row corresponds to one distinct exon skipping event and each column represents a single sample. We performed hierarchical clustering on both the rows and columns. The dendrograms are shown on the left and top of the heatmap, respectively. (B) We applied principal component analysis (PCA) on the correlation matrix of the eight samples. The scatter plot shows the relative position of the eight samples in the 2D space formed by the first principal component and the second principal component. The plot shows good separation between two cancer subtypes along the second principal component. (C) We applied an ANOVA test on the skipping ratio matrix in (A). We selected 12 events that significantly differentiate between the two tumor subtypes with a  0.001. The matrix of their skipping ratios are shown in the heatmap. Both rows and columns were clustered. (D) A scatter plot of the eight samples along the first and second principal components generated from the PCA of the correlation distance matrix of the eight samples based on the 11 selected events.
0.001. The matrix of their skipping ratios are shown in the heatmap. Both rows and columns were clustered. (D) A scatter plot of the eight samples along the first and second principal components generated from the PCA of the correlation distance matrix of the eight samples based on the 11 selected events.
DISCUSSION
Accurate identification and quantification of transcript isoforms is crucial to characterize alternative splicing among different cell types. In addition, sequence variants found within splice sites or splicing enhancer sequences may have functional consequences on alternative splicing. Thus, methods to accurately detect alternative splicing events will be necessary to determine whether these sequence variants affect the transcript isoform proportions. Since certain splice junctions can unambiguously distinguish transcript isoforms, we have focused on increasing the accuracy of aligning splice junctions de novo. For this task, we developed a new splice discovery algorithm, MapSplice, that meets three goals.
First, MapSplice performs a sensitive, complete and unbiased search to find splice junctions using approximate sequence similarity that is not dependent on features or locations of the splice sites. As a result, the algorithm can be applied equally to RNA-seq data from well-studied model organisms and also data from organisms with sparse transcripts annotations. The algorithm is capable of finding short-range as well as long-range and inter-chromosomal splices such as that might arise in gene fusion and other chimeric splicing events that result from damage to the DNA.
Second, MapSplice utilizes efficient approximate sequence alignment methods combined with a local search to create a fast and memory-efficient algorithm. Its alignment strategy can be readily generalized to reads >100 bp. With a processing power of 10 million reads (100 bp) per hour and peak memory usage below 4 GB, MapSplice can run on both desktop and servers with high efficiency.
Third, MapSplice incorporates a rigorous approach to increase specificity of the splice search, necessitated by the multiple ways in which some RNA-seq tags can find spliced alignments to the genome. By leveraging the deep sampling of the transcriptome in RNA-seq data sets, spurious splices can be discriminated from true splices. High specificity is critical as a typical RNA-seq data set can contain some evidence for hundreds of thousands of splices.
In this article, we have made a rigorous measurement of sensitivity and specificity of splice finding algorithms using realistic synthetic data sets. The performances are further assessed by experimental validation of results obtained from breast cancer samples. Using synthetic data sets, we determined that read lengths of 75 or 100 bp yield significantly better sensitivity and specificity for splice detection than 50 bp data sets. We determined that splices can be found despite the presence of errors. Finally, we used synthetic data to calibrate several filtering criteria to achieve over 98% specificity and 96% sensitivity in the detection of splice junctions in the simulated data. These filtering criteria provided superior accuracy in our comparisons to the TopHat (21) and SpliceMap (22) algorithms.
Several experimental lines of evidence also confirmed a high accuracy of the MapSplice algorithm's splice junction alignments. First, the distribution of splice junctions in various categories of alternative splicing are highly concordant with previous studies (Table 3). Second, experimental validation of 10 predictions by qRT-PCR correctly identified isoform proportions that are highly correlated (Pearson's correlation = 0.86) with their estimates based on splice junctions. Third, hierarchical clustering of splicing ratios recapitulated known molecular subtypes of four breast tumors and two breast cancer cell lines. As sample size increases, we will achieve more power to identify candidate genes with significant differences in splicing isoform proportions between molecular subtypes of cancer.
This deep sequencing study represents the first survey of alternative splicing differences between cancer subtypes. At a sequencing depth of approximately 20 million reads with a length of 75 bp, we identified between 149 722 and 178 107 canonical splice junctions, as well as 3661 to 4884 semi-canonical and non-canonical splice junctions. Notably, we discovered that 19–22% of these splice junctions have not been previously observed in full-length transcripts in GenBank. Among these junctions, ∼15% connected two known exons, suggesting novel isoforms with exon skipping events.
We anticipate that tests between sample groups will be crucial to interpret data from large-scale transcriptome sequencing projects, such as the Cancer Genome Atlas. Future research efforts will be needed to distinguish splicing patterns that are enriched in a (potentially heterogeneous) disease state, compared to the natural variation in alternative splicing within populations (5).
The reconstruction of full-length transcripts from short sequence reads is a challenging task, especially for low abundance transcripts. Splice junctions constitute the building blocks for these algorithms (19,32–35). We anticipate that further advances in sequencing technologies, such as higher read depths and longer reads, will continue to improve these methods. Recent studies have combined both splice junction reads and exon reads to provide an integrated partitioning of alignments (36).
FUNDING
National Science Foundation (grant number 0850237 to J.L., J.N.M. and J.F.P.); National Institutes of Health (grant number CA143848 to C.M.P. and grant number P20RR016481 to J.L.); Alfred P. Sloan Foundation (to D.Y.C.). Funding for open access charge: National Institutes of Health (grant number CA143848).
Conflict of interest statement. None declared.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We wish to thank Zefeng Wang, Ben Berman, Corbin Jones, Oleg Evgrafov and the anonymous reviewers for their critical comments on the manuscript.
REFERENCES
- 1.Wang ET, Sandberg R, Luo SJ, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersen LB, Ballester R, Marchuk DA, Chang E, Gutmann DH, Saulino AM, Camonis J, Wigler M, Collins FS. A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol. Cell. Biol. 1993;13:487–495. doi: 10.1128/mcb.13.1.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Screaton GR, Bell MV, Jackson DG, Cornelis FB, Gerth U, Bell JI. Genomic structure of DNA encoding the lymphocyte homing receptor CD44 reveals at least 12 alternatively spliced exons. Proc. Natl Acad. Sci. USA. 1992;89:12160–12164. doi: 10.1073/pnas.89.24.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwan T, Benovoy D, Dias C, Gurd S, Provencher C, Beaulieu P, Hudson TJ, Sladek R, Majewski J. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008;40:225–231. doi: 10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- 6.Meyers GA, Day D, Goldberg R, Daentl DL, Przylepa KA, Abrams LJ, Graham JM, Jr, Feingold M, Moeschler JB, Rawnsley E, et al. FGFR2 exon IIIa and IIIc mutations in Crouzon, Jackson-Weiss, and Pfeiffer syndromes: evidence for missense changes, insertions, and a deletion due to alternative RNA splicing. Am. J. Hum. Genet. 1996;58:491–498. [PMC free article] [PubMed] [Google Scholar]
- 7.Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H, Mohammadi M, Futreal PA, Stratton MR, Trent JM, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene. 2007;26:7158–7162. doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 9.Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, Nicoletti R, Winckler W, Grewal R, Hanna M, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc. Natl Acad. Sci. USA. 2008;105:8713–8717. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 11.Pan Q, Shai O, Misquitta C, Zhang W, Saltzman AL, Mohammad N, Babak T, Siu H, Hughes TR, Morris QD, et al. Revealing global regulatory features of mammalian alternative splicing using a quantitative microarray platform. Mol. Cell. 2004;16:929–941. doi: 10.1016/j.molcel.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, et al. Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 2005;37:844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 13.Castle JC, Zhang C, Shah JK, Kulkarni AV, Kalsotra A, Cooper TA, Johnson JM. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008;40:1416–1425. doi: 10.1038/ng.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan Q, Shai O, Lee LJ, Frey J, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 15.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 16.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 17.Mereau A, Anquetil V, Cibois M, Noiret M, Primot A, Vallee A, Paillard L. Analysis of splicing patterns by pyrosequencing. Nucleic Acids Res. 2009;37:e126. doi: 10.1093/nar/gkp626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xing Y, Yu T, Wu YN, Roy M, Kim J, Lee C. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res. 2006;34:3150–3160. doi: 10.1093/nar/gkl396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang H, Wong WH. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009;25:1026–1032. doi: 10.1093/bioinformatics/btp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Bona F, Ossowski S, Schneeberg K, Ratsch G. Optimal spliced alignments of short sequence reads. Bioinformatics. 2008;24:i174–i180. doi: 10.1093/bioinformatics/btn300. [DOI] [PubMed] [Google Scholar]
- 21.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Au K, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;2010 doi: 10.1093/nar/gkq211. doi:10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 26.Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS ONE. 2009;4:e7767. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malde K. The effect of sequence quality on sequence alignment. Bioinformatics. 2008;24:897–900. doi: 10.1093/bioinformatics/btn052. [DOI] [PubMed] [Google Scholar]
- 29.Koscielny G, Le Texier V, Gopalakrishnan C, Kumanduri V, Riethoven JJ, Nardone F, Stanley E, Fallsehr C, Hofmann O, Kull M, et al. ASTD: the Alternative Splicing and Transcript Diversity database. Genomics. 2009;93:213–220. doi: 10.1016/j.ygeno.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 30.Kircher M, Stenzel U, Kelso J. Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome Biol. 2009;10:R83. doi: 10.1186/gb-2009-10-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gulino A, Di Marcotullio L, Screpanti I. The multiple functions of Numb. Exp. Cell Res. 2010;316:900–906. doi: 10.1016/j.yexcr.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 32.Heber S, Alekseyev M, Sze SH, Tang H, Pevzner PA. Splicing graphs and EST assembly problem. Bioinformatics. 2002;18(Suppl. 1):S181–S188. doi: 10.1093/bioinformatics/18.suppl_1.s181. [DOI] [PubMed] [Google Scholar]
- 33.Xing Y, Lee C. Reconstruction of full-length isoforms from splice graphs. Methods Mol. Biol. 2008;452:199–205. doi: 10.1007/978-1-60327-159-2_10. [DOI] [PubMed] [Google Scholar]
- 34.Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, Stazyk G, Morin RD, Zhao Y, Hirst M, Schein JE, et al. De novo transcriptome assembly with ABySS. Bioinformatics. 2009;25:2872–2877. doi: 10.1093/bioinformatics/btp367. [DOI] [PubMed] [Google Scholar]
- 35.Zheng S, Chen L. A hierarchical Bayesian model for comparing transcriptomes at the individual transcript isoform level. Nucleic Acids Res. 2009;37:e75. doi: 10.1093/nar/gkp282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richard H, Schulz MH, Sultan M, Nurnberger A, Schrinner S, Balzereit D, Dagand E, Rasche A, Lehrach H, Vingron M, et al. Prediction of alternative isoforms from exon expression levels in RNA-Seq experiments. Nucleic Acids Res. 2010;8:e112. doi: 10.1093/nar/gkq041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.