

Abstract
A single nucleotide polymorphism (SNP) in codon 129 of the human prion gene, leading to a change from methionine to valine at residue 129 of prion protein (PrP), has been shown to be a determinant in the susceptibility to prion disease. However, the molecular basis of this effect remains unexplained. In the current study, we determined crystal structures of prion segments having either Met or Val at residue 129. These 6-residue segments of PrP centered on residue 129 are “steric zippers,” pairs of interacting β-sheets. Both structures of these “homozygous steric zippers” reveal direct intermolecular interactions between Met or Val in one sheet and the identical residue in the mating sheet. These two structures, plus a structure-based model of the heterozygous Met-Val steric zipper, suggest an explanation for the previously observed effects of this locus on prion disease susceptibility and progression.
Keywords: Amyloid, Neurodegeneration, Prions, Protein Folding, X-ray Crystallography, Steric Zipper
Introduction
In humans, prion diseases include neurodegenerative disorders such as Creutzfeldt-Jakob disease (CJD),2 Gerstmann–Sträussler–Scheinker syndrome, fatal familial insomnia, and kuru. All these diseases involve the aggregation and deposition of the normally cellular and monomeric prion protein (PrPC) into extracellular plaques. DNA sequencing of the human prion gene has shown that the SNP at codon 129 affects the susceptibility in all sporadic, genetic, and infectious cases of the disease (1). The frequency of this SNP is 38% in the European population, the lowest at 1% in the Japanese population, and the highest at 55% in the Fore people of Papua New Guinea (2). In Europeans, this SNP frequency translates to 39% of the population being homozygous for Met129 (both alleles of the prion gene code for methionine at codon 129), 11% being homozygous for Val129, and 50% heterozygous.
Individuals afflicted by prion disease show strikingly different allele frequencies for the SNP at codon 129 as compared with the general population. Aside from one recent exception, all known cases of new variant CJD (currently more than 200 individuals), which are believed to be acquired through ingesting beef from cows infected with bovine spongiform encephalopathy, have been Met129 homozygous (3, 4). Roughly 70% of individuals who developed CJD sporadically have been Met129 homozygous, although cases of early onset (patients <49 years of age) are equally likely to be valine or methionine homozygous (5). A prevalence of homozygosity among individuals who developed iatrogenic CJD through exposure to tainted human growth hormone derived from cadavers further underscores that propagation of prion disease occurs more efficiently when an identical codon 129 is present in both alleles (6). On the basis of this SNP frequency, it has been proposed that heterozygosity may confer a level of resistance to prion disease (2).
Although the effect of the SNP at codon 129 on disease susceptibility is pronounced, the molecular basis of this phenomenon has been difficult to explain. Structures of the globular domain of monomeric PrPC (Fig. 1) show that residues 127–132 make up a short β-strand (β1) involved in an antiparallel intramolecular β-sheet (β1-β2) (7). The exposed nature of the β1 and the modulating effect of the Met/Val129 polymorphism on susceptibility have stimulated hypotheses that this segment may serve as a nucleation site for the conformational transition of the monomeric PrPC into the aggregated pathogenic state (7). Indeed, intermolecular interactions involving β1 have been reported in crystal structures of both ovine and human PrPC showing the formation of an antiparallel β1-β1 sheet; however, these occur independently of the polymorphism (8, 9). Careful studies of the stability, dynamics, and three-dimensional structures of PrPC found no appreciable differences resulting from Met129 or Val129 residues sufficient to explain the effects on disease phenotype (10, 11). This suggests that the effects result from a property of the segment around residue 129 in the aggregated state of PrP.
FIGURE 1.
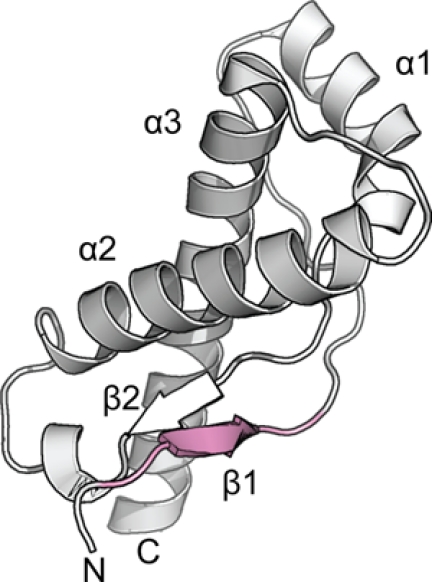
Atomic structure of the globular domain of human PrPC with Val129 (PDB ID: 3hak) shown in graphic representation (8). Segment 127–132, the focus of the present study, is shaded in pink, showing that it includes the first β-strand, β1.
Here we present two crystal structures of a segment of human PrP spanning residues 127–132, one containing Met129 and the other containing Val129. Both structures are “steric zippers,” the general class of structure we have previously found to be characteristic of aggregated amyloid and prion proteins (12–14). Steric zippers are fibril-like structures, consisting of a pair of β-sheets. In general, the interfaces between the two sheets in steric zippers are devoid of water; instead they are self-complementary in shape with the side chains of the two sheets tightly abutting or interdigitating. The polymorphic methionine or valine side chains at residue 129 are each found at the dry interface between the β-sheets. The differences we observe in these interfaces offer a hypothesis for why susceptibility to prion disease is greater in homozygous than in heterozygous individuals.
EXPERIMENTAL PROCEDURES
Crystallization
Segment peptides were purchased from CS BIO Inc. (Menlo Park, CA) in 35–50-mg batches with 99% purity as lyophilized powders. Both were dissolved in 20 mm Bis-Tris to buffer the effect of residual trifluoroacetic acid. Crystals GYMLGS (Met129) were grown within hours at peptide concentrations of 25 mg/ml by exposing the protein to 200 mm buffers with higher pH (>5) using the hanging drop method. Best diffraction quality crystals were obtained from 200 mm Tris, pH 7.0. Although GYVLGS (Val129) also readily formed crystals when exposed to higher pH buffers, they were thin and diffracted poorly. Screening using kits from Hampton Research (Aliso Viejo, CA) were used to search for better quality crystals. After optimization, diffraction quality crystals came from a 0.1 m ammonium sulfate, 0.1 m sodium acetate, pH 5.5, 17% PEG 10,000 with the Val129 segment at a concentration of 32 mg/ml.
Data Collection
Crystals of the segments grew as thin needles and required the use of microdiffraction beamlines at the Advanced Photon Source (APS) in Chicago, IL and the Swiss Light Source (SLS) in Villigen, Switzerland. Crystals were cooled to −180 °C during data collection. Data were collected using 5° wedges.
Data Processing
Indexing of diffraction images was performed using the programs DENZO (15) or XDS (16). Scaling of data was performed using the program SCALEPACK (15). The merged scaled data were imported into the CCP4 format with programs from the CCP4 program suite organized under the CCP4i interface (17).
Structure Determination and Refinement
Phases were determined using the molecular replacement method with idealized polyalanine β-strands using the program PHASER (18). The program COOT (19) was used for model building along with rounds of refinement with the program REFMAC (20). Statistics for structure determination and atomic refinement are given in Table 1.
TABLE 1.
Statistics of x-ray diffraction data collection and atomic refinement of the structures
| Crystal segment name |
||
|---|---|---|
| GYMLGS (Met129) | GYVLGS (Val129) | |
| Data Collection | ||
| Collected at | SLS X06SA | APS 24-ID-E |
| Space group | P212121 | P21212 |
| Resolution (Å) | 1.55 | 1.9 |
| Unit cell dimensions: a, b, c (Å) | 9.55, 18.15, 45.16 | 41.17, 18.95, 9.58 |
| Measured reflections | 6260 | 3616 |
| Unique reflections | 1163 | 637 |
| Overall completeness (%) | 90.9 | 91.5 |
| Last shell completeness (%) | 78.2 | 56.4 |
| Overall redundancy | 5.4 | 5.7 |
| Last shell redundancy | 5.1 | 3.2 |
| Overall Rsym | 0.19 | 0.19 |
| Last shell Rsym | 0.40 | 0.22 |
| Overall I/σ | 6.56 | 6.11 |
| Last shell I/σ | 2.54 | 8.43 |
| Last shell (Å) | 1.61-1.55 | 1.97-1.90 |
| Refinement | ||
| Rwork | 0.22 | 0.22 |
| Rfree (test set) | 0.25 (10.5%) | 0.26 (10.1%) |
| r.m.s.d. bond length (Å) | 0.013 Å | 0.015 Å |
| r.m.s.d. angle (°) | 1.54 | 1.48 |
| Number of peptide atoms | 95 | 84 |
| Number of solvent atoms | 4 | 19 |
| Average B factor of peptide (Å2) | 13.6 | 8.7 |
| Average B factor of solvent (Å2) | 39.0 | 28.0 |
| PDB code | 3NHC | 3NHD |
Creating Models of Mixed Interfaces
The program pdbset from the CCP4 program suite (17) was used to create a pair of stacked β-sheets using crystallographic symmetry operations outlined in the REMARK 350 fields found in the Protein Data Bank (PDB) files defining the biological assembly for each respective structure. The program COOT (19) was used to superimpose strands of the GYVLGS onto the biological assembly of GYMLGS and vice versa.
Features of Peptide Structures and Models
Programs from the CCP4 program suite were used to calculate structural alignments and statistics for interpreting differences and similarities between segment interfaces (17). The programs pdbset and areaimol were used to calculate the area buried in between the sheet-to-sheet interface. Similarly, the programs pdbset and sc were used to calculate the surface complementarity of the interfaces. The program lsqkab was used for structural alignments and to calculate r.m.s.d values between segments. The program hollow was used to model voids in the Met129 and Val129 mixed models (21).
Illustration of Structures
Protein structures and models were illustrated using the program PyMOL (22).
Accession Codes
The structures have been deposited in the protein data bank as 3NHC for the GYMLGS (Met129) segment and 3NHD for the GYVLGS (Val129) segment.
RESULTS
Segment 127–132 of human prion protein with methionine or valine at residue 129 forms similar steric zippers but with distinct interfaces. Segments with amino acid sequences GYMLGS (Met129) and GYVLGS (Val129) crystallized with similar space group symmetry and unit cell dimensions (Table 1). Both form antiparallel steric zippers with the adjacent β-strands in each β-sheet running in opposite directions and the pairs of β-sheets aligned face-to-face (Class 8) (13). In both structures, β-strands stack up to form nearly identical antiparallel β-sheets in which the same residues stack on each other in the main-chain hydrogen-bonding direction (fibril axis), Gly127 on Ser132, Tyr128 on Gly131,and Met/Val129 on Leu130 (Fig. 2c).
FIGURE 2.

a and b, x-ray-derived atomic structures of the homozygous steric zippers of Met129 (a) and Val129 (b) segments of human prion residues 127–132, viewed down the fibril axis. The segment backbone is shown as a ribbon with side chains represented as sticks. The steric zipper interfaces are highlighted by showing interdigitated residues in space-filling mode to emphasize the difference between the GYMLGS (Met129) and GYVLGS (Val129) segments and the efficient van der Waals packing of atoms in these interfaces. The carbon atoms in GYMLGS are colored in black, and in GYVLGS, they are in pink except when showing the side chains of Met129 and Val129, which are colored in blue. Red atoms represent oxygen, and yellow represents sulfur. c, one β-sheet of the GYMLGS viewed perpendicular to the fibril axis, which is parallel to the length of the page. Residues Gly127 and Gly131 are colored in black, residues Tyr128, Leu130, and Ser132 are in white, and residue Met129 is in blue. The antiparallel nature of the β-sheets results in the alternating stacking of blue and white as well as black and white residues up and down the fibril axis. Both faces of the β-sheets are equivalent. d, a model of one strand of GYMLGS onto the steric zipper interface of GYVLGS, viewed down the fibril axis illustrates that in such a heterozygous steric zipper, the methionine side chain would create steric clashes with the peptide backbone and the neighboring side chains. Steric clashes are represented by yellow stars. e and f, models of GYVLGS strands into the steric zipper interface of GYMLGS, viewed down the fibril axis, illustrate that the valine side chains leave large voids within the steric zipper interface. Although the voids may not be evident from the space-filling representation of the interface, f shows a surface representation (in red) of the voids created by incorporating GYVLGS strands into the GYMLGS interface. g–i, these panels show the mixed GYVLGS/GYMLGS steric zipper models viewed perpendicular to their fiber axes. This offers another perspective of the interface between the antiparallel β-sheets as well as the steric clash of a GYMLGS strand modeled onto GYVLGS interface and the voids created by GYVLGS strands modeled into the GYMLGS interface.
In contrast to the similarities of their individual β-sheets, we find key differences at the interfaces between their pairs of β-sheets. Some of these differences are immediately apparent from a view down the fibril axis (Fig. 2, a and b). The planes of the two β-sheets of GYMLGS are parallel to each other, whereas the planes of the two GYVLGS sheets form a chevron resulting in a smaller interface. In GYVLGS (Fig. 2a), the interface between β-sheets is formed by interdigitation of stacked pairs of smaller side chains, Gly127/Ser132 and Val129/Leu130, whereas in GYMLGS (Fig. 2b), the interface is dominated by interdigitation of stacked pairs of larger side chains, Tyr128/Gly131 and Met129/Leu130. In summary, both PrP segment polymorphs form steric zippers characteristic of the aggregated state of amyloids and prions, and both appear capable of lending stability to PrPSc but with different packing geometry (Table 2).
TABLE 2.
Structural features of the homozygous steric zipper interfaces at position 129
| GYMLGS (Met129) | GYVLGS (Val129) | |
|---|---|---|
| Origin of PrP sequence | Human 127-132 | Human 127-132 |
| Amino acid sequence | GYMLGS | GYVLGS |
| Surface complementarity (Sc) | 0.74 | 0.75 |
| Surface area buried in interface (Å2) | 135 ± 1 | 119 ± 4 |
| Sheet-to-sheet distance (Å) | 9.61 ± 1.24 | 9.04 ± 2.51a |
| Class of steric zipper | Class 8 (antiparallel) | Class 8 (antiparallel) |
a The large S.D. is reflective of the chevron shape of the steric zipper interface, where one end is tightly packed and close together while the other is spread farther apart. The value here is an average of the distance.
Having determined the structures of individual Met129 and Val129 steric zipper interfaces, we can model the hypothetical interface of a steric zipper made up of mixed GYMLGS and GYVLGS segments. We constructed such a model by superimposing one β-strand of GYMLGS on a β-strand of the experimentally determined steric zipper of GYVLGS. In this model, every possible rotamer of the methionine residue results in a steric clash with either the Ser132 and Val129 side chains or the main chain of the opposite β-sheet (Fig. 2, d and g). Conversely, when GYVLGS β-strands are superimposed onto β-strands of the steric zipper interface of GYMLGS, large voids are found that would not permit efficient van der Waals packing of residues in the steric zipper interface (Fig. 2, e, f, h, and i). From the hypothetical models of mixed Met129/Val129 interfaces, we infer that such “heterozygous steric zippers” are structurally disfavored for the PrP segment around residue 129, as compared with the “homozygous steric zippers” containing Met129 and Val129. In short, the heterozygous steric zipper for the PrP segment in the vicinity of residue 129 appears less likely to form than the homozygous steric zippers.
DISCUSSION
Evidence suggests that steric zippers are at the core of the pathological form of prion, PrPSc, giving rise to its appearance as unbranched rods, affinity for amyloid-specific dyes, and diagnostic cross-β diffraction pattern (23–25). In yeast prions, steric zippers are also implied in the mechanism of non-Mendelian inheritance by the obligate conversion of prion to the amyloid state (26–31). Prions in the amyloid state act as nuclei seeding the conversion to the amyloid state of the same protein from its soluble monomeric form. The amyloid state can then pass on to progeny cells. Progression and transmission of prion disease in mammals has been suggested to take place in a similar seeding of PrPC by PrPSc (32–34).
Previous work has demonstrated that the architecture of amyloid fibrils is determined by specific short segments of the fibril-forming protein that form the steric zipper spine of the fibril (12–14, 35, 36). In this sense, structures of amyloid and prion fibrils are unlike those of globular and membrane proteins in which many segments of the protein contribute to the stability of the structure. This localization of structural determinants in amyloids is why it is possible to predict protein segments that form fibrils (37), yet prediction of structures of globular and membrane proteins remains a great challenge. Furthermore, the structure-determining segments of amyloids/prions can be crystallized, and their structures can be determined. In the work reported here, we focus on segment 127–132 of PrP, which forms a steric zipper and whose structure can be studied apart from the rest of the protein.
Evidence that steric zippers, such as those reported here, are structurally related to those in full-length proteins is provided by our previous finding that these short, complementary structures can act as nuclei during the ordered aggregation of full-length proteins into amyloid (35). Both the Met129 and the Val129 steric zippers can thus represent the three-dimensional structure of an intermolecular protein-protein interface of the disease-associated PrPSc isoform found in homozygous individuals. The two structures are of interest because they are the first structural evidence for a protein-protein interface in an amyloid-like conformation involving mutual interaction of the methionine or valine side chains of the polymorphism at residue 129. The pronounced modulating effect of this polymorphism on susceptibility to prion disease suggests that this interaction must be intimately linked to the structure of PrPSc and may act as a site of nucleation.
The Met129 and Val129 steric zipper structures show how substitution of a single side chain at a nucleation site can influence the three-dimensional structure of this amyloid-like interface, highlighting the importance of primary structure (or sequence) in forming a steric zipper interface capable of efficient nucleation. Our models of heterozygous steric zippers suggest that a nucleus in individuals that are heterozygous at residue 129 would be less stable than either of the homozygous nuclei. This is analogous to the observation that the most efficient inhibitor of the growth of crystals of chicken lysozyme is the structurally identical turkey lysozyme where differences in amino acid side chains on the surfaces of the proteins make assembly into a coherent crystal less efficient (38). Simply put, forming an ordered array, such as an amyloid fibril, from two different building blocks is less probable than from just one.
The two steric zipper structures (Fig. 2, a and b) and the two models (Fig. 2, d–i) lead to a hypothesis; humans homozygous for either Met129 or Val129 are susceptible to prion disease because the PrP segment centered at either residue forms a stable homozygous steric zipper. In contrast, humans heterozygous for PrP at residue 129 are less susceptible to prion disease because a steric zipper involving one PrP molecule with Met129 and one molecule of Val129 is less stable than either of the two homozygous steric zippers. Such a hypothesis would not exclude heterozygous individuals from prion disease because heterozygotes contain many molecules of each type, and steric zippers arising from segregated pools of either polymorph are still possible but less likely due to the entropic barrier.
Our hypothesis for human susceptibility to prion disease can be extended to interpret observations of incubation time in the development of prion disease. A fundamental property of amyloid in vitro is that its formation from monomers is preceded by a lag time, which can be shortened by introduction of a nucleus (39). Similarly, the long incubation times for prion and amyloid diseases with the slow accumulation of fibrils and plaques suggest a very significant energy barrier to nucleation in vivo (12). According to our hypothesis, the presence of prion protein with both Met129 and Val129 in heterozygous individuals would lessen the probability of a stable nucleus forming at this site because of the less favorable packing and would consequently lengthen incubation times. This is indeed what is observed; case studies of heterozygous individuals with kuru have suggested that in such circumstances, the incubation period of the disease may be as long as 40–50 years (2, 40).
It is also possible that the longer incubation times in heterozygous individuals are due to nucleation of PrPSc at segments other than around residue 129, albeit less efficiently. Amyloid formation can be nucleated by more than just one segment and can involve multiple segments of a protein. For example, Wiltzius et al. (14) found that various segments of the islet amyloid polypeptide form steric zippers and serve as nucleating sites leading to different “strains” of amyloid. Similarly, PrPSc has been shown to have different strains, which have been attributed to different conformations of the protein in the aggregated state (41, 42). The crystal structures of the 6-residue 127–132 segment presented here only represent a fraction of residues known to be in the core 90–231 residues of infectious PrPSc; hence other segments must also be involved in the structure of amyloid from PrP and present possible alternative nucleation sites. Kinetic studies of amyloid formation by recombinant PrP with Met129, Val129, or a mixed population have not been consistent in recapitulating the same effects of the 129 polymorphism as seen in vivo, suggesting that in vitro, PrP amyloid may nucleate more efficiently along other interfaces (43–47). Consistent with this idea, we have found that PrP has multiple segments that form steric zippers.3
Pathogenic mutations have been documented that work synergistically with the 129 polymorphism to cause prion disease. These underscore the idea that other segments of PrP are involved in forming steric zipper interactions in the structure of PrPSc (48). Some of these mutations are distal to residue 129, such as P102L, D178N, E200K, and V210I, which have been found in multiple families affected by prion disease. These further support the involvement of intramolecular interactions between multiple steric zipper segments of PrPSc, such as those between the segments containing the 129 polymorphism and those with the pathogenic mutation. Other pathogenic mutations are proximal to the polymorphic residue 129 such as G131V, which caused Gerstmann–Sträussler–Scheinker syndrome in a patient who was homozygous for Met129 (49). In the structures of the 127–132 segments, residue Gly131 is stacked on Tyr128, which in the case of the pathogenic Val mutation would cause considerable steric clashes. The G131V pathogenic mutation would therefore cause a considerably different steric zipper packing, which as pathology suggests would nucleate amyloid assembly even more efficiently.
Clearly the transformation of PrPC to PrPSc depends on more than the segment around residue 129. However, our atomic structures, the first that show the interactions of this segment in the aggregated state, reveal a pattern of intermolecular contacts involving residues Met129 and Val129 that is consistent with previous studies of PrP disease susceptibility and progression. The structures suggest a possible mechanism for the sequence dependence of human susceptibility to prion diseases, which up to now has remained mysterious.
Acknowledgments
We thank the staff of the SLS beamline X-06-SA and APS beamline 24-ID-E for support and assistance with data collection. We thank Magdalena Ivanova and Nicole Wheatley for critical reading of the manuscript.
This work was supported, in whole or in part, by a National Institutes of Health grant (to D. E.) and Ruth L. Kirschstein National Research Service Award GM007185 (to M. I. A.). This work was also supported by grants from the National Science Foundation, Department of Defense, and Howard Hughes Medical Institute (to D. E.) and the UCLA Dissertation Year Fellowship (to M. I. A.).
The atomic coordinates and structure factors (codes 3NHC and 3NHD) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
M. I. Apostol, M. R. Sawaya, D. Cascio, and D. Eisenberg, unpublished results.
- CJD
- Creutzfeldt-Jakob disease
- PrP
- prion protein
- PrPC
- cellular prion protein
- PrPSc
- pathogenic scrapie prion
- Met129 segment
- PrP segment GYMLGS
- Val129 segment
- PrP segment GYVLGS
- r.m.s.d.
- root mean square deviation
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1.Palmer M. S., Dryden A. J., Hughes J. T., Collinge J. (1991) Nature 352, 340–342 [DOI] [PubMed] [Google Scholar]
- 2.Mead S., Stumpf M. P., Whitfield J., Beck J. A., Poulter M., Campbell T., Uphill J. B., Goldstein D., Alpers M., Fisher E. M., Collinge J. (2003) Science 300, 640–643 [DOI] [PubMed] [Google Scholar]
- 3.Kaski D., Mead S., Hyare H., Cooper S., Jampana R., Overell J., Knight R., Collinge J., Rudge P. (2009) Lancet 374, 2128. [DOI] [PubMed] [Google Scholar]
- 4.Ghani A. C., Donnelly C. A., Ferguson N. M., Anderson R. M. (2003) BMC Infect. Dis. 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alperovitch A., Zerr I., Pocchiari M., Mitrova E., de Pedro Cuesta J., Hegyi I., Collins S., Kretzschmar H., van Duijn C., Will R. G. (1999) Lancet 353, 1673–1674 [DOI] [PubMed] [Google Scholar]
- 6.Brandel J. P., Preece M., Brown P., Croes E., Laplanche J. L., Agid Y., Will R., Alpérovitch A. (2003) Lancet 362, 128–130 [DOI] [PubMed] [Google Scholar]
- 7.Riek R., Hornemann S., Wider G., Billeter M., Glockshuber R., Wüthrich K. (1996) Nature 382, 180–182 [DOI] [PubMed] [Google Scholar]
- 8.Lee S., Antony L., Hartmann R., Knaus K. J., Surewicz K., Surewicz W. K., Yee V. C. (2010) EMBO J. 29, 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haire L. F., Whyte S. M., Vasisht N., Gill A. C., Verma C., Dodson E. J., Dodson G. G., Bayley P. M. (2004) J. Mol. Biol 336, 1175–1183 [DOI] [PubMed] [Google Scholar]
- 10.Hosszu L. L., Jackson G. S., Trevitt C. R., Jones S., Batchelor M., Bhelt D., Prodromidou K., Clarke A. R., Waltho J. P., Collinge J. (2004) J. Biol. Chem. 279, 28515–28521 [DOI] [PubMed] [Google Scholar]
- 11.Riek R., Wider G., Billeter M., Hornemann S., Glockshuber R., Wüthrich K. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 11667–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson R., Sawaya M. R., Balbirnie M., Madsen A. Ø., Riekel C., Grothe R., Eisenberg D. (2005) Nature 435, 773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ø., Riekel C., Eisenberg D. (2007) Nature 447, 453–457 [DOI] [PubMed] [Google Scholar]
- 14.Wiltzius J. J., Landau M., Nelson R., Sawaya M. R., Apostol M. I., Goldschmidt L., Soriaga A. B., Cascio D., Rajashankar K., Eisenberg D. (2009) Nat. Struct. Mol. Biol. 16, 973–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 16.Kabsch W. (1993) J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 17.Collaborative Computational Project, Number 4 (1994) Acta Crystallogr. D. Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 18.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 20.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 21.Ho B. K., Gruswitz F. (2008) BMC Struct. Biol. 8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 23.Prusiner S. B., McKinley M. P., Bowman K. A., Bolton D. C., Bendheim P. E., Groth D. F., Glenner G. G. (1983) Cell 35, 349–358 [DOI] [PubMed] [Google Scholar]
- 24.Wille H., Bian W., McDonald M., Kendall A., Colby D. W., Bloch L., Ollesch J., Borovinskiy A. L., Cohen F. E., Prusiner S. B., Stubbs G. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 16990–16995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen J. T., Inouye H., Baldwin M. A., Fletterick R. J., Cohen F. E., Prusiner S. B., Kirschner D. A. (1995) J. Mol. Biol. 252, 412–422 [DOI] [PubMed] [Google Scholar]
- 26.Tanaka M., Collins S. R., Toyama B. H., Weissman J. S. (2006) Nature 442, 585–589 [DOI] [PubMed] [Google Scholar]
- 27.Glover J. R., Kowal A. S., Schirmer E. C., Patino M. M., Liu J. J., Lindquist S. (1997) Cell 89, 811–819 [DOI] [PubMed] [Google Scholar]
- 28.Wickner R. B. (1994) Science 264, 566–569 [DOI] [PubMed] [Google Scholar]
- 29.Serio T. R., Cashikar A. G., Kowal A. S., Sawicki G. J., Moslehi J. J., Serpell L., Arnsdorf M. F., Lindquist S. L. (2000) Science 289, 1317–1321 [DOI] [PubMed] [Google Scholar]
- 30.Patino M. M., Liu J. J., Glover J. R., Lindquist S. (1996) Science 273, 622–626 [DOI] [PubMed] [Google Scholar]
- 31.King C. Y., Diaz-Avalos R. (2004) Nature 428, 319–323 [DOI] [PubMed] [Google Scholar]
- 32.Jarrett J. T., Lansbury P. T., Jr. (1993) Cell 73, 1055–1058 [DOI] [PubMed] [Google Scholar]
- 33.Hall D., Edskes H. (2004) J. Mol. Biol. 336, 775–786 [DOI] [PubMed] [Google Scholar]
- 34.Griffith J. S. (1967) Nature 215, 1043–1044 [DOI] [PubMed] [Google Scholar]
- 35.Ivanova M. I., Sievers S. A., Sawaya M. R., Wall J. S., Eisenberg D. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 18990–18995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiltzius J. J., Sievers S. A., Sawaya M. R., Cascio D., Popov D., Riekel C., Eisenberg D. (2008) Protein Sci. 17, 1467–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldschmidt L., Teng P. K., Riek R., Eisenberg D. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 3487–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirschler J., Fontecilla-Camps J. C. (1996) Acta Crystallogr. D Biol. Crystallogr. 52, 806–812 [DOI] [PubMed] [Google Scholar]
- 39.Jarrett J. T., Lansbury P. T., Jr. (1992) Biochemistry 31, 12345–12352 [DOI] [PubMed] [Google Scholar]
- 40.Collinge J., Whitfield J., McKintosh E., Beck J., Mead S., Thomas D. J., Alpers M. P. (2006) Lancet 367, 2068–2074 [DOI] [PubMed] [Google Scholar]
- 41.Collinge J., Sidle K. C., Meads J., Ironside J., Hill A. F. (1996) Nature 383, 685–690 [DOI] [PubMed] [Google Scholar]
- 42.Collinge J., Clarke A. R. (2007) Science 318, 930–936 [DOI] [PubMed] [Google Scholar]
- 43.Baskakov I., Disterer P., Breydo L., Shaw M., Gill A., James W., Tahiri-Alaoui A. (2005) FEBS Lett. 579, 2589–2596 [DOI] [PubMed] [Google Scholar]
- 44.Lewis P. A., Tattum M. H., Jones S., Bhelt D., Batchelor M., Clarke A. R., Collinge J., Jackson G. S. (2006) J. Gen. Virol. 87, 2443–2449 [DOI] [PubMed] [Google Scholar]
- 45.Apetri A. C., Vanik D. L., Surewicz W. K. (2005) Biochemistry 44, 15880–15888 [DOI] [PubMed] [Google Scholar]
- 46.Jones E. M., Surewicz K., Surewicz W. K. (2006) J. Biol. Chem. 281, 8190–8196 [DOI] [PubMed] [Google Scholar]
- 47.Petchanikow C., Saborio G. P., Anderes L., Frossard M. J., Olmedo M. I., Soto C. (2001) FEBS Lett. 509, 451–456 [DOI] [PubMed] [Google Scholar]
- 48.Mead S. (2006) Eur. J. Hum Genet. 14, 273–281 [DOI] [PubMed] [Google Scholar]
- 49.Panegyres P. K., Toufexis K., Kakulas B. A., Cernevakova L., Brown P., Ghetti B., Piccardo P., Dlouhy S. R. (2001) Arch. Neurol 58, 1899–1902 [DOI] [PubMed] [Google Scholar]