

Abstract
Obligatory anaerobic bacteria are major contributors to the overall metabolism of soil and the human gut. The metabolic pathways of these bacteria remain, however, poorly understood. Using isotope tracers, mass spectrometry, and quantitative flux modeling, here we directly map the metabolic pathways of Clostridium acetobutylicum, a soil bacterium whose major fermentation products include the biofuels butanol and hydrogen. While genome annotation suggests the absence of most tricarboxylic acid (TCA) cycle enzymes, our results demonstrate that this bacterium has a complete, albeit bifurcated, TCA cycle; oxaloacetate flows to succinate both through citrate/α-ketoglutarate and via malate/fumarate. Our investigations also yielded insights into the pathways utilized for glucose catabolism and amino acid biosynthesis and revealed that the organism's one-carbon metabolism is distinct from that of model microbes, involving reversible pyruvate decarboxylation and the use of pyruvate as the one-carbon donor for biosynthetic reactions. This study represents the first in vivo characterization of the TCA cycle and central metabolism of C. acetobutylicum. Our results establish a role for the full TCA cycle in an obligatory anaerobic organism and demonstrate the importance of complementing genome annotation with isotope tracer studies for determining the metabolic pathways of diverse microbes.
In soil ecology, obligatory anaerobic bacteria are key contributors to the putrefaction of dead organic matter (18). In the human intestine, they are the dominant flora, playing a central role in metabolism, immunity, and disease (16, 24, 30). Obligatory anaerobes also encompass some of the most promising bioenergy organisms. The soil bacterium Clostridium acetobutylicum is capable of fermenting carbohydrates into hydrogen gas and solvents (acetone, butanol, and ethanol). During World War I, it was used to develop an industrial starch-based process for the production of acetone and butanol that remained the major production route for these solvents during the first half of the last century (5). Since then, and particularly during the last few decades, an active research area has developed to understand and manipulate the metabolism of this organism with the goal of improving hydrogen and solvent production (5, 15). Despite this long history, there are still key pathways of primary metabolism in C. acetobutylicum that remain unresolved. In particular, as is common for most anaerobic bacteria, the tricarboxylic acid (TCA) cycle remains ill-defined (14, 23, 28).
C. acetobutylicum is capable of growing on minimal medium (i.e., using glucose as the sole carbon source) (20), and it therefore must be able to synthesize α-ketoglutarate, the carbon skeleton of the glutamate family of amino acids. Its genome sequence, however, lacks obvious homologues of many of the enzymes of the TCA cycle, including citrate synthase, α-ketoglutarate dehydrogenase, succinyl-coenzyme A (CoA) synthetase, and fumarate reductase/succinate dehydrogenase (23). The apparent lack of these genes precludes the production of α-ketoglutarate by running the TCA cycle in either the oxidative or reductive direction.
Two recent attempts at reconstructing a genome-scale model of C. acetobutylicum metabolism have encountered this problem. In one case, it was proposed that the TCA cycle functions in the reductive (counterclockwise) direction to produce α-ketoglutarate (14). In the second, it was hypothesized that glutamate is synthesized from ornithine by running the arginine biosynthesis pathway in reverse, bypassing the need for the TCA cycle (28). With the exception of the TCA cycle, the other core metabolic pathways, e.g., of glucose catabolism and amino acid and nucleotide biosyntheses, appear based on sequence homology to be complete and analogous to those in more well-studied bacteria, such as Escherichia coli (23).
Here, we use 13C-labeled nutrients as isotopic tracers to follow the operation of C. acetobutylicum's TCA cycle and other primary metabolic pathways directly in live cells. In contrast to the previously proposed hypotheses, we find a complete, albeit bifurcated, TCA cycle. α-Ketoglutarate is produced in the oxidative direction from oxaloacetate and acetyl-CoA via citrate. Succinate can be produced in both the reductive direction via malate and fumarate and the oxidative direction via α-ketoglutarate. We also observe that C. acetobutylicum's one-carbon metabolism is distinct from that of more well-studied bacteria; the carboxyl group of pyruvate undergoes reversible exchange with free carbon dioxide, and the one-carbon units required for methionine, purine, and pyrimidine biosyntheses are derived primarily from the carboxyl group of pyruvate with minimal contribution from serine or glycine.
To obtain a quantitative understanding of the newly proposed metabolic network, we formulated an ordinary differential equation (ODE) model that allowed us to calculate the metabolic fluxes through glycolysis, the Entner-Doudoroff pathway (which was inactive), the nonoxidative pentose phosphate pathway (there is no oxidative pentose phosphate pathway), the TCA cycle, and adjacent amino acid biosynthesis pathways. Beyond providing a quantitative description of metabolic flux, this model was useful for unraveling ambiguities in the network structure that were not readily distinguished based on qualitative labeling patterns alone. This study represents the first in vivo experimental characterization of the TCA cycle and central metabolic pathways in a Clostridium species and demonstrates the importance of complementing genome annotation with isotope tracer studies in the construction of genome-scale metabolic networks.
MATERIALS AND METHODS
Media, culture conditions, and metabolite extraction.
C. acetobutylicum ATCC 824 was grown anaerobically at 37°C inside an environmental chamber (Bactron IV Shel Lab anaerobic chamber) with an atmosphere of 90% nitrogen, 5% hydrogen, and 5% carbon dioxide. The minimal medium formulation used in both liquid and filter cultures was 2 g/liter KH2PO4, 2 g/liter K2HPO4, 0.2 g/liter MgSO4·7H2O, 1.5 g/liter NH4Cl, 0.13 mg/liter biotin, 32 mg/liter FeSO4·7H2O, 0.16 mg/liter 4-aminobenzoic acid, and 10 g/liter glucose (20). In the pertinent experiments, acetate, glutamate, aspartate, or ornithine was added at a concentration of 2 g/liter. In addition to the appropriate minimal medium, the plates used in filter cultures contained 1.5% ultrapure agarose.
Detailed protocols for preparing filter cultures and extracting metabolites in Escherichia coli have been published (2, 31), and these methods were adapted for use in C. acetobutylicum. Briefly, for the preparation of filter cultures, single colonies were picked from agar-solidified reinforced clostridial medium (RCM; Difco), resuspended in liquid RCM, heat treated at 80°C for 20 min, and grown to saturation overnight. This overnight culture was then used to inoculate a liquid minimal medium culture to an initial optical density at 600 nm (OD600) of 0.03. When this liquid culture reached an OD600 of ∼0.1, 1.6-ml aliquots were taken and passed through 47-mm-diameter round hydrophilic nylon filters (HNWP04700; Millipore), which were then placed on top of agarose plates with the appropriate minimal medium. Cellular metabolism was quenched, and metabolites were extracted by submerging the filters into 0.8 ml of acetonitrile-methanol-water (40:40:20) at −20°C (25). The filters were then washed with the extraction solvent, the cellular extractions were transferred and centrifuged in Eppendorf tubes, and the supernatant was collected and stored at −20°C until analysis. To measure growth, filters from parallel cultures were washed thoroughly with 1.6 ml of fresh medium and absorbance at 600 nm was determined.
Metabolite and flux measurement.
Cell extracts were analyzed by reversed-phase ion-pairing liquid chromatography (LC) coupled with electrospray ionization (ESI) (negative mode) to a high-resolution, high-accuracy mass spectrometer (Exactive; Thermo Fisher) operated in full scan mode for the detection of targeted compounds based on their accurate masses. This analysis was complemented with liquid chromatography coupled with ESI (positive and negative modes) to Thermo TSQ Quantum triple quadrupole mass spectrometers operating in selected reaction monitoring mode (1, 19). Hydrophilic interaction chromatography was used for positive-mode ESI, and ion-pairing reversed-phase chromatography was used for negative-mode ESI. Amino acids were derivatized with benzyl chloroformate before their quantitation by negative-mode LC-ESI-tandem mass spectrometry (MS/MS) (13). Absolute intracellular metabolite concentrations were determined using an isotope ratio-based approach previously described (2). Briefly, C. acetobutylicum was grown in [U-13C]glucose medium to near-complete isotopic enrichment and then extracted with quenching solvent containing known concentrations of unlabeled internal standards. The concentrations of metabolites in the cells can then be calculated using the ratio of labeled endogenous metabolite to nonlabeled internal standard.
We used kinetic flux profiling (KFP) for measuring metabolite fluxes and elucidating the metabolic network structure (31). Filter cultures were grown on minimal medium plates to an OD600 of 0.35 and then transferred to minimal medium plates containing uniformly 13C-labeled glucose as the sole carbon source. At defined time points after the transfer (e.g., 1, 2, 4, 7, 10, 15, 30, and 60 min), metabolism was quenched and cell extracts were prepared and analyzed. The multiple isotopomers produced by the 13C labeling were monitored simultaneously using LC-MS. Metabolic fluxes were calculated based on the kinetics of the replacement of the unlabeled metabolites with the labeled ones. Similarly, the KFP experiments with uniformly 13C-labeled acetate were performed by transferring the cells to glucose minimal medium plates with added [U-13C]acetate.
For the long-term labeling experiments using [3-13C]glucose, [4-13C]glucose, [1,2-13C]glucose, [13C]glutamate, [13C]ornithine, or [13C]aspartate, the cells were extracted 2 h after they were transferred to the plates containing each labeled substrate. In all the experiments with 13C-labeled amino acids, the usual concentration of nonlabeled glucose was maintained. The 13CO2 labeling experiments were performed by adding increasing concentrations of NaH13CO3 into exponentially growing liquid cultures (OD600 = 0.35). After 1 h, the cells were quickly filtered and extracted using acetonitrile-methanol-water (40:40:20) at −20°C.
The labeling of the C1 unit pool was determined from the labeling patterns of various intermediates in nucleotide biosynthetic pathways that incorporate C1 units. We used 5′-phosphoribosyl-N-formylglycinamide and IMP, which incorporate C1 units from 10-formyl-tetrahydrofolate as well as dTMP, which incorporates a C1 unit from 5,10-methylene-trahydrofolate. In addition, methionine, which incorporates a C1 unit from 5-methyl-tetrahydrofolate, was used for corroborating data in some instances.
We used metabolic flux profiling to complement the information obtained from KFP and to determine flux ratios in various pathways. We followed the general approach described in reference 9, with the difference that instead of inferring labeling patterns from proteinogenic amino acids, we quantified them directly for most metabolites. In all experiments, the labeling data were corrected for natural abundance of 13C in nonlabeled substrates and for the 12C impurity present in 13C-labeled substrates in a fashion similar to that reported previously (2, 31).
ODE modeling and parameter identification.
We constructed an ODE model for the metabolic network shown in Fig. 4 as well as Fig. S7 in the supplemental material and then identified model parameters (fluxes and unmeasured pool sizes) that reproduce the laboratory data. The procedure was based on methods previously developed (8, 22). The ODEs describe the rates of loss of unlabeled forms of metabolites (and the creation of particular labeled forms) after feeding of [U-13C]glucose. The equations are based on flux balance of metabolites and take the form
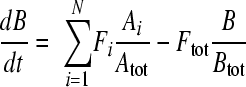 |
where metabolite B, which can be in labeled or unlabeled form, is downstream of another metabolite, Ai. The outflux, Ftot, balances the sum of N influxes, Fi, with Ai. Atot and Btot are the total pool sizes of the corresponding metabolites (sum of labeled and unlabeled forms).
FIG. 4.

Quantitation of fluxes in central metabolism. (A) Ordinary differential equation (ODE) model fitting (lines) to the [U-13C]glucose dynamic labeling data (error bars) for three representative metabolites. Complete results are in Fig. S8 in the supplemental material. (B) Metabolic fluxes identified from the ODE model. Arrow sizes indicate absolute values (in logarithmic scale) of net fluxes. The fluxes shown are median values of 1,000 sets of identified fluxes, whose distributions are plotted in Fig. S9 in the supplemental material. The flux from succinyl-CoA into succinate is a combination of the flux through succinyl-CoA synthetase (∼25% median contribution) and the fluxes through the methionine and lysine biosynthesis pathways that are coupled with the conversion of succinyl-CoA into succinate (∼75% median contribution). Hexose-P, combined pools of glucose-1-phosphate, glucose-6-phosphate, and fructose-6-phosphate; FBP, fructose-1,6-bisphosphate; DHAP, combined pools of dihydroxyacetone phosphate and glyceraldehyde-3-phosphate; 3PG, combined pools of glycerate-3-phosphate and glycerate-2-phosphate; PEP, phosphoenolpyruvate; Pentose-P, combined pools of ribose-5-phosphate, xylulose-5-phosphate, and ribulose-5-phosphate; OAA, oxaloacetate; αKG, α-ketoglutarate; SucCoA, succinyl-CoA; Asp, aspartate; Glu, glutamate; Gln, glutamine.
The unknown model parameters were identified by a genetic algorithm that minimizes a cost function (7, 8). The cost function quantifies the difference between the computational results and the laboratory measurements for the labeling dynamics, together with the additional constraints indicated in Table S1 in the supplemental material. One thousand sets of model parameters that can reproduce the laboratory data were identified, forming a distribution for each parameter (see Fig. S9 in the supplemental material). The median value and the breadth of the distribution then provide a representation of the fluxes consistent with the laboratory data. The C/C++ programs used for modeling and parameter identification are available upon request.
RESULTS
Glucose catabolism to pyruvate.
To probe metabolic flux in growing C. acetobutylicum, we monitored the dynamic (time-dependent) or long-term (steady-state) incorporation of 13C-labeled nutrients (glucose, acetate, CO2, and selected amino acids) into downstream metabolites in glycolysis, the pentose phosphate pathway, the TCA cycle, and amino acid and nucleotide biosynthetic pathways.
C. acetobutylicum can potentially metabolize glucose to trioses via three different pathways: glycolysis (the Embden-Meyerhof pathway), the Entner-Doudoroff pathway, and the pentose phosphate pathway. Homologues of enzymes of each of the above-mentioned pathways, with the exception of the oxidative pentose phosphate pathway, are present in the C. acetobutylicum genome (23). The contribution of glycolysis to pyruvate synthesis relative to that of the Entner-Doudoroff pathway can be determined from cells grown in [1,2-13C]glucose (carbons 1 and 2 are 13C labeled) or [3-13C]glucose (carbon 3 is labeled) since each pathway yields distinct positional labeled forms of pyruvate, which can be distinguished by tandem mass spectrometry (MS/MS). For C. acetobutylicum growing exponentially on glucose as the carbon source, all pyruvate appeared to be derived from glycolysis, with no detectable Entner-Doudoroff pathway flux (see Fig. S1 in the supplemental material).
The pentose phosphate pathway provides essential precursors (ribose-5P and erythrose-4P) for nucleotide and amino acid biosyntheses. Ribose-5P molecules can be produced by the oxidative pentose pathway from glucose-6P, by the nonoxidative pentose phosphate pathway via the transketolase reaction, or by the combined activity of transaldolase and transketolase. Consistent with the lack of oxidative pentose phosphate pathway enzyme homologues in the C. acetobutylicum genome, feeding of [1,2-13C]glucose resulted in no detectable production of ribose-5P containing a single 13C atom, the hallmark of oxidative pentose phosphate production. Pentoses were instead produced via transketolase (∼80%) and via transaldolase-transketolase (∼20%) (see Fig. S2 in the supplemental material).
The above-described experiments suggest normal catabolism of glucose into pyruvate via glycolysis, and consistent with this, feeding of [U-13C]glucose as the sole carbon source resulted in rapid and complete labeling of glycolysis intermediates through phosphoenolpyruvate. Pyruvate, however, appeared in roughly equimolar amounts in its fully labeled form and in an unexpected form with two 13C carbons (Fig. 1 B). Glycolysis splits glucose down the middle, converting carbon positions 1, 2, and 3 (and 6, 5, and 4) into the methyl, carbonyl, and carboxyl carbons of pyruvate, respectively. As shown in Fig. 1C, growth of C. acetobutylicum in [1,2-13C]glucose (100%) resulted, as expected, in ∼50% of phosphoenolpyruvate and pyruvate each containing two 13C carbons. In contrast, feeding of [3-13C]glucose (100%) or [4-13C]glucose (100%) resulted in 50% labeling of phosphoenolpyruvate (and upstream trioses) but only ∼25% labeling of pyruvate. This suggested that the 13C label was being lost specifically from the carboxyl carbon of pyruvate, presumably in an exchange reaction with environmental carbon dioxide (CO2), which comprises 5% of the anaerobic gaseous environment and is ∼99% nonlabeled. Consistent with exchange of the carboxyl carbon of pyruvate with carbon dioxide, growth of cells in the presence of NaH13CO2 resulted in the formation of [1-13C]pyruvate, with the fraction of labeling increasing with increasing concentrations of NaH13CO2 (Fig. 1D). There was minimal or no labeling of upstream metabolites. This confirms the rapid interchange between the carboxylic acid group in pyruvate and environmental CO2.
FIG. 1.

Glycolysis and the rapid interchange between the carboxyl group of pyruvate and CO2. (A) Overview of active and inactive pathways. Glycolysis operates normally through phosphoenolpyruvate with the Entner-Doudoroff pathway inactive (see Fig. S1 in the supplemental material). The reaction catalyzed by pyruvate ferredoxin oxidoreductase (PFOR) is partially, but not fully, reversible. GAP, glyceraldehyde-3-phosphate; DHAP, dihydroxyacetone phosphate. (B) Dynamic incorporation of uniformly 13C-labeled glucose (100%) into glycolysis intermediates. Glycolysis intermediates through phosphoenolpyruvate were labeled rapidly and completely. Pyruvate, however, appeared in roughly equimolar amounts in its fully labeled form and in an unexpected form with two 13C carbons. Environmental CO2 was ∼99% nonlabeled. The x axis represents minutes after the switch from unlabeled to [U-13C]glucose medium, and the y axis represents the fraction of the observed compound of the indicated isotopic form. (C) Steady-state labeling patterns of phosphoenolpyruvate and pyruvate obtained from cells grown in [3-13C]glucose (100%), [4-13C]glucose (100%), or [1,2-13C]glucose (100%). In [3-13C]glucose or [4-13C]glucose, about half of the phosphoenolpyruvate was labeled but only about a quarter of pyruvate was labeled. In contrast, growth in [1,2-13C]glucose resulted in identical labeling patterns for phosphoenolpyruvate and pyruvate. Environmental CO2 was ∼99% nonlabeled. These results indicate that the 13C label in pyruvate is specifically lost from the carboxyl carbon. (D) The fraction of [1-13C]pyruvate increased with increasing amounts of NaH13CO3 added to the medium. Cells were fed unlabeled glucose throughout, and labeling of upstream glycolysis intermediates was minimal or nonexistent (not shown). This experiment was performed in liquid closed-vessel cultures. The data, in conjunction with those in panels B and C, indicate exchange of the carboxyl carbon of pyruvate with carbon dioxide. (E) [U-13C]acetate was assimilated and incorporated into acetyl phosphate and acetyl-CoA. Pyruvate, however, remained unlabeled. The data suggest that the reaction catalyzed by PFOR is not fully reversible. The error bars in panels B through E show standard deviations (SD) (n = 2 to 4 independent experiments).
In anaerobic organisms, the oxidative decarboxylation of pyruvate to produce acetyl-CoA and CO2 is catalyzed by pyruvate-ferredoxin oxidoreductase (PFOR, also known as pyruvate synthase). The use of ferredoxin, whose redox potential is close to that of pyruvate, as the oxidant has the potential to make the overall reaction reversible (10, 26). When 13C-labeled acetate was added to the medium, however, there was no detectable labeling of pyruvate, even when a significant fraction of the acetyl-CoA pool was labeled (Fig. 1E). In the proposed mechanism of acetyl-CoA synthesis by PFOR, pyruvate is first decarboxylated to form the intermediate hydroxyethyl-thiamine pyrophosphate (TPP, the prosthetic group in PFOR). This intermediate then reacts with CoA (coenzyme A) to produce acetyl-CoA (26). Our data indicate that the carboxylic group in pyruvate interchanges rapidly with CO2 but that the overall PFOR reaction is essentially irreversible. We accordingly propose that the interchange results from the reversibility of the decarboxylation step of the PFOR reaction.
The interchange between the carboxyl group in pyruvate and atmospheric CO2 could also be explained by reverse flux through pyruvate dehydrogenase or pyruvate formate lyase. However, the presence of a pyruvate dehydrogenase complex has never been reported in C. acetobutylicum or in any other Clostridium species (5). Additionally, isotopic tracer experiments with aerobically and anaerobically grown E. coli strains indicate that neither pyruvate dehydrogenase nor pyruvate formate lyase is capable of producing the exchange between the carboxyl group in pyruvate and CO2 that we observe in C. acetobutylicum (see Fig. S3 in the supplemental material).
Complete bifurcated TCA cycle.
After gaining an understanding of the pathways that catabolize glucose to trioses, we examined the TCA cycle (Fig. 2). Feeding of [U-13C]glucose (100%) resulted in labeling patterns of oxaloacetate, malate, and fumarate which closely matched the labeling pattern of pyruvate, with the appearance of close to equimolar amounts of isotopomers with two or three 13C carbons (Fig. 2B). This observation is consistent with the synthesis of oxaloacetate from pyruvate and atmospheric CO2 (which is nonlabeled) and with the production of malate and fumarate from oxaloacetate by running the TCA cycle in the reductive (counterclockwise) direction. Succinate's labeling pattern, however, did not fully agree with that of pyruvate. Although succinate showed the same predominant labeled forms, their ratios were different, with nearly twice as much succinate containing three 13C carbons than two 13C carbons (Fig. 2B). This suggested that although succinate may be produced from fumarate, there must also be another source to account for the enhanced triple 13C labeling.
FIG. 2.

Complete bifurcated TCA cycle in C. acetobutylicum. (A) The diagram represents the proposed bifurcated TCA cycle in C. acetobutylicum. α-Ketoglutarate is produced from oxaloacetate and acetyl-CoA via citrate. Succinate can be produced reductively from fumarate or oxidatively from α-ketoglutarate. Gray boxes show the fate of the carbons in the incoming acetyl group from acetyl-CoA, and dotted boxes show the fate of the carbons in the carboxyl group from pyruvate. The unusual stereospecificity of citrate synthesis was confirmed by MS/MS analysis (see Fig. S5 in the supplemental material). Panels B and C show the dynamic incorporation of [U-13C]glucose (100%) and [U-13C]acetate (in the presence of nonlabeled glucose) into TCA metabolites and glutamate. The x axis represents minutes after the switch from unlabeled to 13C-labeled medium, and the y axis represents the fraction of the observed compound of the indicated isotopic form. There was no detectable labeling of oxaloacetate, malate, or fumarate in the [U-13C]acetate experiments (not shown). These results are consistent with a bifurcated TCA cycle in which oxaloacetate flows to succinate both through citrate/α-ketoglutarate and via malate/fumarate as shown in panel A. Panels D and E show the long-term labeling patterns of TCA metabolites when cells are grown in glucose minimal medium supplemented with [U-13C]aspartate or with [U-13C]glutamate. These data corroborate the results obtained for panels B and C and the existence of a bifurcated TCA cycle. AKG, α-ketoglutarate. In all experiments, the environmental CO2 comprised 5% of the anaerobic gaseous environment and was ∼99% nonlabeled. In panels B through D, the error bars show SD (n = 2 to 4 independent experiments).
The labeling pattern for α-ketoglutarate differed from that of pyruvate or succinate. If, as previously hypothesized (14, 23), α-ketoglutarate is produced from succinate and CO2 by running the TCA cycle reductively, α-ketoglutarate containing two and three 13C carbons should have appeared. However, the predominant form of α-ketoglutarate had four 13C carbons. The actual route of α-ketoglutarate production was revealed by the labeling patterns in citrate (Fig. 2B). Despite the putative lack of citrate synthase, there was a measurable intracellular pool of citrate that labeled rapidly after feeding of [U-13C]glucose. Citrate (a molecule with six carbons) was produced in two major isotopic forms, containing either four or five 13C carbons. This labeling pattern is consistent with citrate's production from oxaloacetate (with two or three 13C carbons) and acetyl-CoA (where the 2-carbon acetyl moiety is fully 13C labeled). The labeling pattern of α-ketoglutarate was then readily explained based on its production via citrate. Citrate containing either four or five 13C carbons produces α-ketoglutarate with four 13C carbons because the additional 13C carbon in citrate corresponds to the carboxyl group that is lost during oxidative decarboxylation of isocitrate to α-ketoglutarate.
The labeling of glutamate matched that of α-ketoglutarate, consistent with its formation by reductive amination of α-ketoglutarate driven by either ammonia or glutamine. To rule out the previous hypothesis that glutamate could be synthesized from ornithine by running the arginine biosynthesis pathway in reverse (28), we added [U-13C]ornithine to the medium. While arginine pathway compounds downstream of ornithine became labeled, we observed no production of labeled glutamate (see Fig. S4 in the supplemental material).
The lack of production of succinate containing four 13C carbons initially suggested that there was no production of succinate via α-ketoglutarate. However, experiments with additional 13C-labeled nutrients proved that this does occur. When cells were grown in unlabeled glucose plus [U-13C]acetate, 13C was assimilated into acetyl-CoA. Consistent with turning of the TCA cycle in the oxidative direction, citrate, α-ketoglutarate, and glutamate with two 13C carbons were produced, but the cycle was incomplete; there was no detectable labeling in oxaloacetate, malate, or fumarate. Interestingly, however, we observed the production of succinate with one 13C carbon (Fig. 2C).
This labeling of succinate is consistent with its production from α-ketoglutarate, but the stereospecificity of citrate synthase was the opposite of that found in common bacterial model organisms and eukaryotes. This unusual Re-stereospecificity of citrate synthase was confirmed by examining the positions of 13C carbons within glutamate and proline by MS/MS analysis (see Fig. S5 in the supplemental material). Production of succinate from α-ketoglutarate explains the succinate labeling patterns in the [U-13C]glucose labeling experiments; succinate was synthesized both via fumarate (producing succinate containing two and three 13C carbons) and via α-ketoglutarate (producing succinate containing three 13C carbons). With glucose as the sole carbon source, the relative contributions from each route to succinate production are ∼60% and 40%, respectively.
Aspartate and glutamate can be deaminated to oxaloacetate and α-ketoglutarate, respectively, to enter the TCA cycle. When [U-13C]aspartate is added to the medium (in the presence of unlabeled glucose), a large fraction (>80%) of the malate, fumarate, succinate, and citrate pools becomes quadrupoly 13C labeled. α-Ketoglutarate and glutamate become triply 13C labeled (Fig. 2D). When cells are grown in the presence of [U-13C]glutamate plus unlabeled glucose, both α-ketoglutarate and succinate become fully 13C labeled. In this case, oxaloacetate, malate, and fumarate are not 13C labeled (Fig. 2E). These observations corroborate the existence of a complete bifurcated TCA cycle in C. acetobutylicum.
Amino acid biosynthetic pathways and C1 metabolism.
By analyzing the labeling patterns of amino acids and key biosynthetic intermediates, we were able to resolve most of the amino acid biosynthesis pathways in C. acetobutylicum. The observed labeling patterns were consistent with those expected based on canonical amino acid biosynthesis pathways, with the exception of isoleucine and glycine production (see Table S2 in the supplemental material). The labeling patterns in isoleucine indicated that it is not synthesized by the canonical pathway via threonine but are instead consistent with its production from acetyl-CoA and pyruvate via the citramalate pathway (see Table S2).
For glycine, there are two alternative pathways (Fig. 3 A). The more common pathway involves synthesis of glycine from serine by the enzyme serine hydroxymethyltransferase, which transfers the methanol group from serine to tetrahydrofolate (THF). The resulting methyl-folate species provide C1 units for the biosynthesis of purines, thymidine, and methionine. Alternatively, in Saccharomyces cerevisiae and some bacteria, glycine can be synthesized by degradation of threonine, e.g., into acetaldehyde and glycine (12, 21). When cells were grown on [U-13C]glucose, serine (synthesized via 3-phosphoglycerate) became fully labeled, whereas threonine (synthesized via pyruvate) was ∼50% triply 13C labeled and ∼50% doubly 13C labeled. Consistent with its predominant formation from threonine but not from serine, glycine was ∼50% fully labeled and ∼50% singly 13C labeled (Fig. 3B). The synthesis of glycine from threonine was further corroborated by growing cells in [U-13C]glucose plus nonlabeled aspartate and observing that both the threonine and glycine pools are mostly nonlabeled while serine was largely fully labeled (Fig. 3C).
FIG. 3.

One-carbon metabolism in C. acetobutylicum. (A) Proposed network of one-carbon metabolism in C. acetobutylicum. Blue arrows highlight the major production routes for glycine, serine, and one-carbon units (C1 folates). The fate of the carbons originating from pyruvate is highlighted by gray and dotted boxes. (B) Dynamic incorporation of [U-13C]glucose (100%) into the amino acids serine, glycine, and threonine. The labeling patterns observed in glycine indicate that its primary route of production is via threonine and not serine. (C) The synthesis of glycine from threonine was confirmed by growing cells on [U-13C]glucose plus nonlabeled aspartate and observing that both the threonine and glycine pools are largely nonlabeled while the serine pool remains largely labeled. (D) Cells grown in [1,2-13C]glucose (100%) showed less than a 5% label in C1 units, even though the precursor carbon in glycine (methylene group highlighted in gray in panel A) is ∼50% labeled. (E) Correlation between the labeled fractions of the carboxylic acid carbon of pyruvate and the labeled fractions of C1 units across diverse labeling experiments. The addition of unlabeled aspartate to cells growing in [U-13C]glucose (100%), which results in the production of unlabeled glycine (C), does not affect labeling of C1 units. (F) Cells grown with increasing concentrations of NaH13CO3 showed increasing labeling of C1 units that closely followed the labeling in the carboxyl group of pyruvate but not the labeling of CO2 present in the medium. The fraction of labeled CO2 medium was determined based on labeling of CO2 assimilated into pyrimidines. In panels B through E, the environmental CO2 comprised 5% of the anaerobic gaseous environment and was ∼99% nonlabeled. In panel F, the experiments were performed in liquid closed-vessel cultures to minimize the interchange between atmospheric 12CO2 and NaH13CO3.
Since glycine is synthesized primarily from threonine, C1 units must be obtained from a precursor other than serine. Glycine is also commonly used as a precursor of C1 units, but we also found that this route is nearly inactive in C. acetobutylicum. When cells were grown in [1,2-13C]glucose, less than 5% of C1 units were 13C labeled, even though the methylene group in glycine (the precursor of C1 units) was ∼50% labeled (Fig. 3D). Conversely, when cells were grown in either [3-13C]glucose or [4-13C]glucose, the methylene group in glycine was nonlabeled but ∼25% of the C1 unit pool was 13C labeled (Fig. 3E). The possibility that C1 units could be synthesized from the carboxyl group of glycine via some noncanonical pathway was ruled out by the observation that the percentage of 13C-labeled C1 units is essentially unchanged between cells grown in [U-13C]glucose and cells grown in [U-13C]glucose plus nonlabeled aspartate (Fig. 3E), even though the 13C label in the carboxyl group of glycine decreases from ∼50% to ∼10% (Fig. 3B and C). These experiments show that there is minimal production of C1 units via serine or glycine. We found, however, a strong correlation between the labeled fraction of the carboxylic group of pyruvate and the labeled fraction of C1 units across all these labeling experiments (Fig. 3E). In addition, when NaH13CO2 was added to the medium, 13CO2 was incorporated into C1 units. The fraction of labeled C1 units did not correspond directly to the fraction of labeled CO2 but did correspond to the fraction of labeled CO2 that was incorporated into pyruvate (Fig. 3F). Our data therefore indicate that in C. acetobutylicum, C1 units are derived primarily (>90%) from the carboxylic group of pyruvate, likely through the combined action of pyruvate formate lyase and formate-tetrahydrofolate ligase.
Metabolic flux quantitation.
Among the most important characteristics of a biochemical network are the in vivo reaction rates. To achieve a quantitative understanding of the fluxes in the newly proposed metabolic network, we developed an ordinary differential equation (ODE) model that describes the isotope labeling kinetics of metabolites following the addition of universally labeled [13C]glucose (see Materials and Methods and Fig. S7 in the supplemental material). Given the model equations, we employed a nonlinear global search algorithm to identify the fluxes that can quantitatively reproduce the laboratory data (8). In addition to the labeling kinetics, inputs to the model included intracellular concentrations of glycolysis and TCA cycle intermediates and amino acids (see Table S3 in the supplemental material), nutrient uptake rates, excretion rates (see Fig. S6 in the supplemental material), and specific flux branch point data obtained from the steady-state labeling experiments discussed previously. The details of the cost function used for model fitting are presented in Table S1 in the supplemental material. To avoid overfitting the data, any simulations which fell within the 95% confidence limits of the laboratory data were considered acceptable; only more severe misfits were penalized during the search. A total of 1,000 well-fitting sets of fluxes were identified and used to estimate flux confidence intervals.
Figure 4 shows representative results for the ODE model fitting and a map of the identified median flux values in central metabolism. The complete results are presented in Fig. S8 and S9 in the supplemental material. The ODE model fits all of the observed data. Most of the identified fluxes, with the exception of several exchange fluxes, were tightly constrained, indicating that they are reliably defined by the available laboratory data. The results show that glycolytic flux predominates and is directed primarily toward acid production. Other significant fluxes include aspartate production via pyruvate/oxaloacetate, fatty acid production from dihydroxyacetone phosphate, and ribose-phosphate production from glycolytic intermediates. Within the TCA cycle, the flux through the oxidative branch is slightly larger than through the reductive branch. The production of succinate from succinyl-CoA can occur via succinyl-CoA synthetase but is also expected to occur via the canonical methionine and lysine pathways. The computational results indicate that the median contribution of the succinyl-CoA synthetase flux to the total succinyl-CoA flux into succinate is about 25%, while the methionine and lysine pathways combined contribute to ∼75% of the total flux (see Fig. S9).
In addition to providing quantitative flux values, the ODE model also helped to resolve an ambiguity in the network structure that was not adequately addressed by qualitative analysis of the isotope labeling patterns alone. Malate and fumarate production can occur directly via the reductive TCA cycle or alternatively from passage of carbon from aspartate to fumarate, which would then be oxidized to malate (see the alternative pathway in Fig. S7 in the supplemental material). Both pathways result in qualitatively indistinguishable labeling patterns. To distinguish them, we constructed ODE models for the two alternative pathways and performed flux identification using the procedure described above. Both models were able to describe the quantitative dynamic data following [U-13C]glucose labeling. However, in the second model, because fumarate is partly used for the production of malate, the contribution of fumarate to succinate production is smaller than that in the first model. Quantitatively, the percentage of succinate produced from fumarate is ∼54% for the first model and ∼6% for the second model. Compared with the experimentally measured value of ∼60%, the computational results indicate that the second model is inaccurate and the first one is correct, meaning that malate is produced primarily reductively from oxaloacetate rather than oxidatively from fumarate.
DISCUSSION
Comparative genome sequence analysis has become the predominant tool for genome-scale reconstruction of the metabolic network of microorganisms. Frequently, however, due to incomplete annotation or undocumented functional genes, there are gaps and uncertainties within the metabolic network that need to be resolved experimentally. These limitations get in the way of a comprehensive understanding of their metabolism and interfere with the creation of quantitative genome-scale models of metabolism. This hinders the ability to rationally modulate metabolism for biotechnological or medical purposes.
Here, we used 13C-labeled tracer experiments to elucidate the in vivo function of the TCA cycle and other primary metabolic pathways in C. acetobutylicum. In contrast to the prevailing hypothesis, we found that this organism has a complete, albeit bifurcated, TCA cycle; oxaloacetate flows to succinate both through citrate/α-ketoglutarate and via malate/fumarate. Although there is currently no gene annotated as citrate synthase in C. acetobutylicum, our data revealed the presence of a citrate synthase with Re-stereospecificity. An Re-citrate synthase has recently been identified in Clostridium kluyveri as the product of a gene predicted to encode isopropylmalate synthase (17). The corresponding protein in C. acetobutylicum, CAC0970, has a 64% amino acid sequence identity and is one candidate for the Re-citrate synthase in this organism. While aconitase and isocitrate dehydrogenase were not annotated when the genome sequence of C. acetobutylicum was first released (23), the genes CAC0971 and CAC0971 are now annotated as such in the Kyoto Encyclopedia of Genes and Genomes (KEGG). The products of these genes, however, have not yet been characterized in C. acetobutylicum or in any other clostridia. The α-ketoglutarate dehydrogenase complex is missing in the C. acetobutylicum genome, but it has been hypothesized that a putative 2-oxoacid ferredoxin oxidoreductase (CAC2458) could catalyze succinyl-CoA formation from α-ketoglutarate (23). There are still no candidate genes encoding fumarate reductase/succinate dehydrogenase or succinyl-CoA synthetase.
Initially defined by a set of broad phenotypic characteristics such as rod-like morphology, Gram-positive cell walls, endospore formation, and strict anaerobic metabolism, Clostridium is one of the most heterogeneous bacterial genera (5). In a sequence-based species tree, there are a number of independent and deeply branching sublines within the Clostridium subdivision, which also includes many nonclostridial species (4). Among the clostridia, C. kluyveri shows a unique metabolism; it grows anaerobically on ethanol and acetate as sole energy sources (27). Only about half of the genes in C. kluyveri show more than 60% similarity in C. acetobutylicum (3). The similarities that we observe between C. acetobutylicum and C. kluyveri regarding the oxidative production of α-ketoglutarate and one-carbon metabolism (as discussed further below) are therefore noteworthy.
In both the initial genome sequencing and a subsequent genome-scale reconstruction of the C. acetobutylicum metabolic network, it was proposed that α-ketoglutarate is synthesized from oxaloacetate by running the TCA cycle reductively. It was argued that a reductive TCA cycle would be favored given the low redox potential of the internal anaerobic environment of C. acetobutylicum. It is therefore intriguing that C. acetobutylicum synthesizes α-ketoglutarate exclusively oxidatively. The reasons for this remain unclear, but the conversion of α-ketoglutarate into succinyl-CoA appears to be irreversible in this organism; although succinate is readily synthesized via α-ketoglutarate, there is no back-flux from succinate to α-ketoglutarate, even under conditions in which there is ample production of succinate by the reductive TCA cycle (as when cells are grown in the presence of aspartate). The irreversibility is expected if this reaction is catalyzed by a yet-to-be-identified α-ketoglutarate dehydrogenase but not if it is catalyzed, as previously proposed, by a reversible 2-oxoacid ferredoxin oxidoreductase.
Given that α-ketoglutarate is synthesized solely via citrate, succinate becomes a metabolite of limited biosynthetic value. The benefit of maintaining two different routes for its production is therefore unclear. A possibility is that a bifurcated TCA cycle ending in succinate plays a role in cellular redox balance. However, the rate of succinate excretion (∼4 μmol/h/g cells [dry weight]) is very low compared to that of other fermentation products such as acetate and butyrate (∼4 mmol/h/g cells [dry weight]) (see Fig. S6 in the supplemental material). Another possibility is that this particular arrangement of the TCA cycle facilitates the utilization of certain amino acids as nitrogen sources. For example, when C. acetobutylicum is grown in glutamate or aspartate as the sole nitrogen source, large amounts of α-ketoglutarate or oxaloacetate are produced during deamination. While a fraction of these carbon skeletons may be used for biosynthetic purposes, most must be discarded. Their conversion to succinate, and subsequent excretion, provides a short and rapid route. These hypotheses are consistent with the data obtained from our experiments with [13C]glutamate and [13C]aspartate.
In most organisms, glycine is synthesized from serine, producing a C1 unit during the process. Glycine, in turn, can also be used to produce a C1 unit. In contrast, in C. acetobutylicum, the major route (∼90%) for the production of glycine is via threonine. This necessitates C1 unit production from a precursor other than serine, and we found that C1 units are derived predominantly (∼90%) from the carboxyl group of pyruvate. A related situation has been observed in C. kluyveri, in which 67% of glycine is formed from threonine and 33% from serine, and about 25% of C1 units are synthesized from serine and 75% from CO2 (11). The production of C1 units from the carboxyl group of pyruvate (oxidation state, +3) can be viewed as a reductive pathway while their production from the methylene group of serine or glycine (oxidation state, −1) can be considered an oxidative pathway. For example, using serine as the source for C1 units, the production of 10-formyl-tetrahydrofolate (used in purine biosynthesis) is accompanied by the production of one NADH; using glycine, two NADHs are produced. However, no NADH is produced when pyruvate is used as the source of C1 units for the production of 10-formyl-tetrahydrofolate. Therefore, for an anaerobic bacterium such as C. acetobutylicum, it makes sense to derive C1 units from the carboxyl group of pyruvate. Also, the capacity to produce C1 units both reductively and oxidatively suggests that the relative utilization of these pathways may be yet another way to control cellular redox balance.
Our observations strengthen the notion that pyruvate constitutes a pivotal metabolic crossroads in C. acetobutylicum, linking the TCA cycle, amino acid biosynthesis pathways, one-carbon metabolism, and acid/solvent-producing pathways. It therefore represents a control point that could be exploited to improve biofuel production. For example, decreasing the activity of pyruvate carboxylase should decrease the flux of pyruvate into the TCA cycle and associated amino acid biosynthesis pathways and increase pyruvate flux into acetyl-CoA and solvent production.
The dynamic isotope labeling approach (kinetic flux profiling) that we use here is different from the steady-state isotopic approach (metabolic flux analysis) recently used in similar contexts (6, 29). One major advantage of our approach is that it provides absolute fluxes throughout the network instead of just ratios of fluxes at branch points. Additional advantages include easy data deconvolution and short labeling time. The quantitative modeling technique used in this study is generally applicable for the identification of metabolic fluxes from dynamic isotope tracer experiments (22). In addition to providing a quantitative understanding of the target metabolic networks, we have shown its ability to discriminate among competing network structures that produce qualitatively indistinguishable labeling patterns. Moreover, given the appropriate input data, the general nonlinear identification strategy can also be employed for the construction of dynamic models that reflect the regulation of metabolic fluxes (8, 32). Such dynamic models can enable a more comprehensive understanding and rational engineering of metabolic networks. In the case of C. acetobutylicum, for example, a model of dynamic regulation could be used to design genetic and nutrient perturbations that enhance solvent and/or biohydrogen production.
This study represents the first in vivo experimental characterization of the TCA cycle and central metabolism in C. acetobutylicum and exemplifies the potential of dynamic isotope tracer studies and quantitative flux modeling in complementing genome-based metabolic network reconstruction.
Supplementary Material
Footnotes
Published ahead of print on 9 July 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Bajad, S. U., W. Lu, E. H. Kimball, J. Yuan, C. Peterson, and J. D. Rabinowitz. 2006. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J. Chromatogr. A 1125:76-88. [DOI] [PubMed] [Google Scholar]
- 2.Bennett, B. D., J. Yuan, E. H. Kimball, and J. D. Rabinowitz. 2008. Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat. Protoc. 3:1299-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brinkac, L. M., T. Davidsen, E. Beck, A. Ganapathy, E. Caler, R. J. Dodson, A. S. Durkin, D. M. Harkins, H. Lorenzi, R. Madupu, Y. Sebastian, S. Shrivastava, M. Thiagarajan, J. Orvis, J. P. Sundaram, J. Crabtree, K. Galens, Y. Zhao, J. M. Inman, R. Montgomery, S. Schobel, K. Galinsky, D. M. Tanenbaum, A. Resnick, N. Zafar, O. White, and G. Sutton. 2010. Pathema: a clade-specific bioinformatics resource center for pathogen research. Nucleic Acids Res. 38:D408-D414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dehal, P. S., M. P. Joachimiak, M. N. Price, J. T. Bates, J. K. Baumohl, D. Chivian, G. D. Friedland, K. H. Huang, K. Keller, P. S. Novichkov, I. L. Dubchak, E. J. Alm, and A. P. Arkin. 2010. MicrobesOnline: an integrated portal for comparative and functional genomics. Nucleic Acids Res. 38:D396-D400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dürre, P. 2005. Handbook on clostridia. Taylor & Francis, Boca Raton, FL.
- 6.Feng, X., H. Mouttaki, L. Lin, R. Huang, B. Wu, C. L. Hemme, Z. He, B. Zhang, L. M. Hicks, J. Xu, J. Zhou, and Y. J. Tang. 2009. Characterization of the central metabolic pathways in Thermoanaerobacter sp. strain X514 via isotopomer-assisted metabolite analysis. Appl. Environ. Microbiol. 75:5001-5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng, X. J., S. Hooshangi, D. Chen, G. Li, R. Weiss, and H. Rabitz. 2004. Optimizing genetic circuits by global sensitivity analysis. Biophys. J. 87:2195-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng, X. J., and H. Rabitz. 2004. Optimal identification of biochemical reaction networks. Biophys. J. 86:1270-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer, E., and U. Sauer. 2003. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem. 270:880-891. [DOI] [PubMed] [Google Scholar]
- 10.Furdui, C., and S. W. Ragsdale. 2000. The role of pyruvate ferredoxin oxidoreductase in pyruvate synthesis during autotrophic growth by the Wood-Ljungdahl pathway. J. Biol. Chem. 275:28494-28499. [DOI] [PubMed] [Google Scholar]
- 11.Jungermann, K. A., W. Schmidt, F. H. Kirchniawy, E. H. Rupprecht, and R. K. Thauer. 1970. Glycine formation via threonine and serine aldolase. Its interrelation with the pyruvate formate lyase pathway of one-carbon unit synthesis in Clostridium kluyveri. Eur. J. Biochem. 16:424-429. [DOI] [PubMed] [Google Scholar]
- 12.Kataoka, M., M. Ikemi, T. Morikawa, T. Miyoshi, K. Nishi, M. Wada, H. Yamada, and S. Shimizu. 1997. Isolation and characterization of d-threonine aldolase, a pyridoxal-5′-phosphate-dependent enzyme from Arthrobacter sp. DK-38. Eur. J. Biochem. 248:385-393. [DOI] [PubMed] [Google Scholar]
- 13.Kraml, C. M., D. Zhou, N. Byrne, and O. McConnell. 2005. Enhanced chromatographic resolution of amine enantiomers as carbobenzyloxy derivatives in high-performance liquid chromatography and supercritical fluid chromatography. J. Chromatogr. A 1100:108-115. [DOI] [PubMed] [Google Scholar]
- 14.Lee, J., H. Yun, A. M. Feist, B. O. Palsson, and S. Y. Lee. 2008. Genome-scale reconstruction and in silico analysis of the Clostridium acetobutylicum ATCC 824 metabolic network. Appl. Microbiol. Biotechnol. 80:849-862. [DOI] [PubMed] [Google Scholar]
- 15.Lee, S. Y., J. H. Park, S. H. Jang, L. K. Nielsen, J. Kim, and K. S. Jung. 2008. Fermentative butanol production by clostridia. Biotechnol. Bioeng. 101:209-228. [DOI] [PubMed] [Google Scholar]
- 16.Ley, R. E., M. Hamady, C. Lozupone, P. J. Turnbaugh, R. R. Ramey, J. S. Bircher, M. L. Schlegel, T. A. Tucker, M. D. Schrenzel, R. Knight, and J. I. Gordon. 2008. Evolution of mammals and their gut microbes. Science 320:1647-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li, F., C. H. Hagemeier, H. Seedorf, G. Gottschalk, and R. K. Thauer. 2007. Re-citrate synthase from Clostridium kluyveri is phylogenetically related to homocitrate synthase and isopropylmalate synthase rather than to Si-citrate synthase. J. Bacteriol. 189:4299-4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ljungdahl, L. G. 2003. Biochemistry and physiology of anaerobic bacteria. Springer, New York. NY.
- 19.Lu, W., B. D. Bennett, and J. D. Rabinowitz. 2008. Analytical strategies for LC-MS-based targeted metabolomics. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 871:236-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monot, F., J. R. Martin, H. Petitdemange, and R. Gay. 1982. Acetone and butanol production by Clostridium acetobutylicum in a synthetic medium. Appl. Environ. Microbiol. 44:1318-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monschau, N., K. P. Stahmann, H. Sahm, J. B. McNeil, and A. L. Bognar. 1997. Identification of Saccharomyces cerevisiae GLY1 as a threonine aldolase: a key enzyme in glycine biosynthesis. FEMS Microbiol. Lett. 150:55-60. [DOI] [PubMed] [Google Scholar]
- 22.Munger, J., B. D. Bennett, A. Parikh, X. J. Feng, J. McArdle, H. A. Rabitz, T. Shenk, and J. D. Rabinowitz. 2008. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26:1179-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nolling, J., G. Breton, M. V. Omelchenko, K. S. Makarova, Q. Zeng, R. Gibson, H. M. Lee, J. Dubois, D. Qiu, J. Hitti, Y. I. Wolf, R. L. Tatusov, F. Sabathe, L. Doucette-Stamm, P. Soucaille, M. J. Daly, G. N. Bennett, E. V. Koonin, and D. R. Smith. 2001. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J. Bacteriol. 183:4823-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin, J., R. Li, J. Raes, M. Arumugam, K. S. Burgdorf, C. Manichanh, T. Nielsen, N. Pons, F. Levenez, T. Yamada, D. R. Mende, J. Li, J. Xu, S. Li, D. Li, J. Cao, B. Wang, H. Liang, H. Zheng, Y. Xie, J. Tap, P. Lepage, M. Bertalan, J. M. Batto, T. Hansen, D. Le Paslier, A. Linneberg, H. B. Nielsen, E. Pelletier, P. Renault, T. Sicheritz-Ponten, K. Turner, H. Zhu, C. Yu, M. Jian, Y. Zhou, Y. Li, X. Zhang, N. Qin, H. Yang, J. Wang, S. Brunak, J. Dore, F. Guarner, K. Kristiansen, O. Pedersen, J. Parkhill, J. Weissenbach, P. Bork, and S. D. Ehrlich. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rabinowitz, J. D., and E. Kimball. 2007. Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal. Chem. 79:6167-6173. [DOI] [PubMed] [Google Scholar]
- 26.Ragsdale, S. W. 2003. Pyruvate ferredoxin oxidoreductase and its radical intermediate. Chem. Rev. 103:2333-2346. [DOI] [PubMed] [Google Scholar]
- 27.Seedorf, H., W. F. Fricke, B. Veith, H. Bruggemann, H. Liesegang, A. Strittmatter, M. Miethke, W. Buckel, J. Hinderberger, F. Li, C. Hagemeier, R. K. Thauer, and G. Gottschalk. 2008. The genome of Clostridium kluyveri, a strict anaerobe with unique metabolic features. Proc. Natl. Acad. Sci. U. S. A. 105:2128-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senger, R. S., and E. T. Papoutsakis. 2008. Genome-scale model for Clostridium acetobutylicum. I. Metabolic network resolution and analysis. Biotechnol. Bioeng. 101:1036-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang, Y. J., S. Yi, W.-Q. Zhuang, S. H. Zinder, J. D. Keasling, and L. Alvarez-Cohen. 2009. Investigation of carbon metabolism in “Dehalococcoides ethenogenes” strain 195 by use of isotopomer and transcriptomic analyses. J. Bacteriol. 191:5224-5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turnbaugh, P. J., and J. I. Gordon. 2009. The core gut microbiome, energy balance and obesity. J. Physiol. 587:4153-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan, J., B. D. Bennett, and J. D. Rabinowitz. 2008. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat. Protoc. 3:1328-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan, J., C. D. Doucette, W. U. Fowler, X. J. Feng, M. Piazza, H. A. Rabitz, N. S. Wingreen, and J. D. Rabinowitz. 2009. Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol. Syst. Biol. 5:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.