

Abstract
Gene expression is regulated in part by protein transcription factors (TFs) that bind target regulatory DNA sequences. Predicting DNA binding sites and affinities from transcription factor sequence or structure is difficult; therefore, experimental data are required to link TFs to target sequences. We present a microfluidics-based approach for de novo discovery and quantitative biophysical characterization of DNA target sequences. We validated our technique by measuring sequence preferences for 28 S. cerevisiae TFs with a variety of DNA binding domains, including several that have proven difficult to study via other techniques. For each TF, we measured relative binding affinities to oligonucleotides covering all possible 8-bp DNA sequences to create a comprehensive map of sequence preferences; for 4 TFs, we also determined absolute affinities. We anticipate that these data and future use of this technique will provide information essential for understanding TF specificity, improving identification of regulatory sites, and reconstructing regulatory interactions.
Recent evidence suggests that knowledge of both strongly-and weakly-bound sequences and their interaction affinities is required for an accurate understanding of transcriptional regulation. Weak-affinity sites are evolutionarily conserved, make significant contributions to overall transcription1,2, and may allow closely related TFs to mediate different transcriptional responses3. In addition, quantitative models require both strongly-and weakly-bound sequences and their binding affinities to recapitulate transcriptional responses4-7.
Unfortunately, quantitative data detailing TF binding are often lacking, even for model organisms. In vivo immunoprecipitation-based methods (e.g. ChIP-chip8 and ChIP-SEQ9 provide genome-wide information about promoter occupancy. However, these techniques require knowledge of physiological states under which TFs are bound to promoters, cannot distinguish whether a TF contacts DNA directly or is tethered via another DNA-binding protein, and do not measure affinities.
In vitro methods complement in vivo data by measuring binding affinities, distinguishing whether TFs directly bind DNA, and allowing manipulation of post-translational modifications and buffer conditions. Furthermore, in vitro methods can be used without knowledge of conditions under which TFs are active. However, current in vitro methods cannot simultaneously discover both high-and low-affinity target sequences and measure their affinities. Electromobility shift assays (EMSAs)10 DNAse footprinting11 and surface plasmon resonance12 require prior knowledge of potential binding sites, precluding motif discovery. Conversely, selection techniques (i.e. SELEX) and one-hybrid systems13 discover motifs from a large sequence space, but recover only the most strongly bound sequences, without affinity information. Protein binding microarrays (PBMs)3,14-18 can discover both strongly-and weakly-bound sequences but cannot measure reactions at equilibrium, preventing affinity measurements. PBMs also suffer from reduced sensitivity: a recent study using PBMs to probe TF binding in S. cerevisiae failed to recover consensus motifs for 49 of 101 TFs with previous evidence of direct DNA binding15. Embedding immobilized DNA in hydrogels19 extends the PBM technique to allow affinity and kinetic measurements, but limits available DNA sequences to ∼ 100.
An alternative approach is Mechanically-Induced Trapping of Molecular Interactions (MITOMI), a technique that uses a microfluidic device to measure binding interactions at equilibrium, allowing construction of detailed maps of binding energy landscapes. The first-generation MITOMI device measured 640 parallel interactions and required TF-specific DNA libraries20.
Here, we report a second-generation MITOMI device (MITOMI 2.0) capable of measuring 4,160 parallel interactions. Devices were fabricated in polydimethylsiloxane (PDMS) using multilayer soft lithography; each device had 4,160 unit cells and approximately 12,555 valves to control fluid flow (Fig. 1a). Each unit cell contained a DNA chamber and a protein chamber, controlled by micromechanical valves: a ‘neck’ valve, ‘sandwich’ valves, and a ‘button’ valve (Fig. 1a, Supplementary Fig. 1). Unit cells were programmed with particular DNA sequences by aligning and bonding the device with a non-covalently spotted DNA microarray containing a library of 1457 double-stranded Cy5-labeled oligonucleotides. To accommodate all 65,536 DNA 8-mers, each 70-bp oligonucleotide contained 45 overlapping, related 8-mer de Bruijn sequences21 (Fig. 1b). Each oligonucleotide sequence appeared in at least 2 unit cells.
Figure 1.
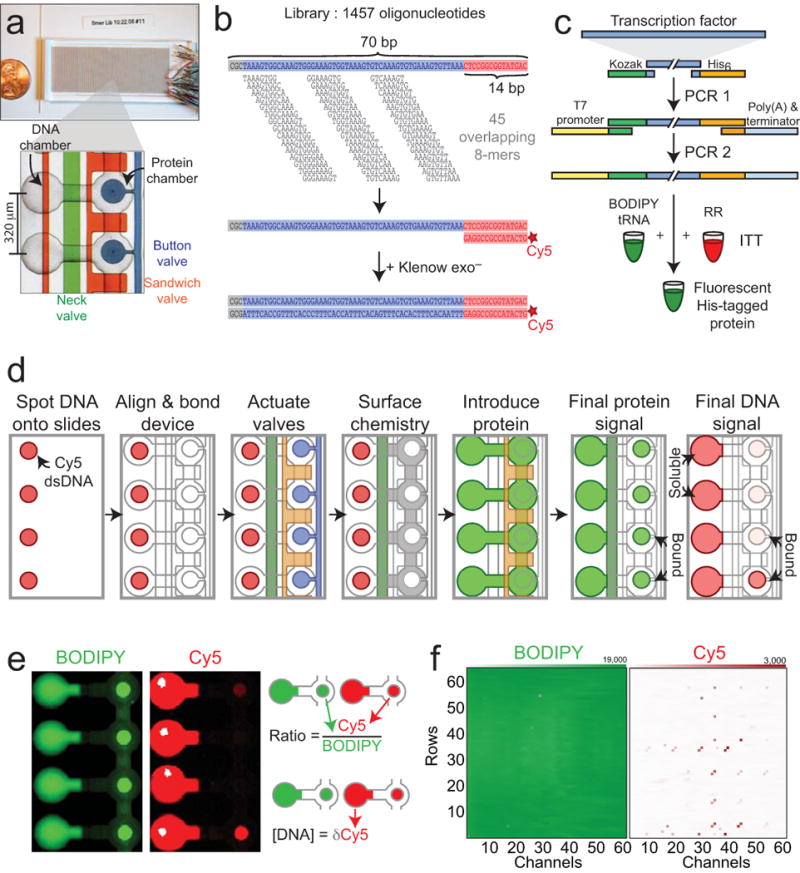
Overall experimental design and procedure. (a) Microfluidic device hybridized to glass slide. Unit cells contain two chambers (a ‘DNA chamber’ and a ‘protein’ chamber) controlled by three valves: a ‘neck’ valve (green) to separate the two chambers, a ‘sandwich’ valve (orange) to isolate unit cells, and a ‘button’ valve (blue) to protect molecular interactions. (b) DNA 8mer library design. Each 70 bp oligonucleotide contains 45 overlapping 8mers, a 3 bp GC-clamp at the 5′ end, and an identical 14-bp sequence at the 3′ end for Cy5 labeling and primer extension. (c) PCR generation of linear templates for protein expression. In PCR1, template-specific primers attach a Kozak sequence, 6× His tag, and universal overhangs. In PCR2, universal primers add a T7 promoter, poly-A tail, and T7 terminator. In vitro transcription/translation (ITT) of this template in rabbit reticulocyte lysate (RR) with BODIPY-labeled lysine charged tRNA produces labeled, His-tagged protein. (d) Overview of experimental procedure. Devices are manually aligned to a spotted microarray. Neck valves are closed to protect DNA within chambers, and slide surfaces are derivatized with anti-pentaHis antibodies below the button (white) and passivated elsewhere (grey). Lysate containing fluorescently labeled His-tagged TFs is introduced and neck valves are opened to allow interaction between transcription factors and DNA; sandwich valves are closed to isolate each unit cell. Following an incubation, button valves are pressurized to protect protein:DNA interactions, unbound DNA and proteins are washed out, and the device is scanned. (e) Scanned picture showing final protein (BODIPY, left) and DNA (Cy5, right) intensities in the chamber and under the button. (f) Arrays showing example protein intensities (left) and DNA intensities (right) under the button for each unit cell within a device.
To evaluate the performance of this technique, we measured DNA binding for 28 S. cerevisiae TFs from 10 different families (Supplementary Table 1). Of these, 26 TFs had prior evidence of direct, sequence-specific DNA binding and 2 TFs had no previously annotated literature motifs, despite multiple previous attempts14,15,22.
All TF protein was produced by in vitro transcription/translation. PCR-generated linear expression templates were added directly to rabbit reticulocyte lysate off-chip in the presence of a small fraction of BODIPY-labeled lysine charged tRNA to produce BODIPY-labeled, His-tagged TFs (Fig. 1c, Supplementary Fig. 2). In each experiment, ∼ 50 μL of extract (∼ 100 ng of protein) was loaded into the device.
Following alignment to DNA microarrays, slide surfaces within the protein chamber were derivatized with anti-pentaHis antibodies beneath the button valve and passivated elsewhere (Fig. 1d). Introduction of His-tagged TFs into both chambers solubilized spotted DNA, allowing TFs and DNA to interact. TF-DNA complexes were captured on the surface beneath the button valve during a ∼ 1 hour incubation; rapid closure of the button valve trapped interactions at equilibrium concentrations prior to a final wash to remove unbound material before imaging20.
BODIPY intensities under the button valve reflect the number of surface-bound protein molecules; Cy5 intensities under the button valve reflect the number of DNA molecules bound by surface-immobilized protein (Fig. 1d,e,f). Therefore, the ratio of Cy5 to BODIPY fluorescence is linearly proportional to the number of protein molecules with bound DNA, or protein fractional occupancy. Cy5 intensities within the DNA chamber reflect the amount of soluble DNA available for binding.
All 28 TFs showed oligonucleotide-specific variations in bound Cy5 intensities, demonstrating marked preferences for individual oligonucleotides (Fig. 2a, Supplementary Fig. 3). By contrast, the distribution of intensities for rabbit reticulocyte extract alone was well-fit by a Gaussian (reduced χ2 = 1.0, p = 0.47), establishing that binding is due to expressed TFs and not components of the in vitro transcription/translation system (Fig. 2a).
Figure 2.
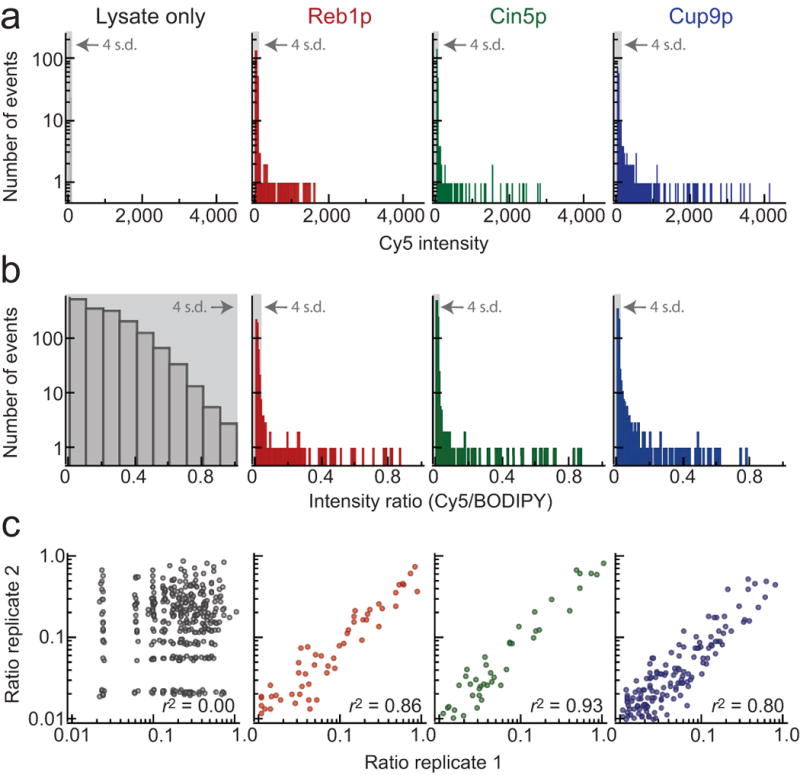
Detailed analysis of measured Cy5 intensities and fluorescence intensity ratios (Cy5/BODIPY-FL) for rabbit reticulocyte lysate alone, Reb1p, Cin5p, and Cup9p. (a) Distribution of measured Cy5 intensities for all oligonucleotides. Light grey box indicates measurements within 4 standard deviations of the mean (as determined by a Gaussian fit). Measured Cy5 intensities for rabbit reticulocyte lysate alone are well-fit by a Gaussian (reduced χ2 = 1.0, p = 0.47). For all TFs, measured Cy5 intensities deviate significantly from a Gaussian distribution, with measured events many standard deviations above the mean. (b)Distribution of measured intensity ratios for all oligonucleotides. Light grey box indicates measurements within 4 standard deviations of the mean (as determined by a Gaussian fit). Measured intensity ratios in the presence of TFs deviate significantly from a normal distribution (Supplementary Table 2). (c) Correlation between ratios measured for the same oligonucleotide at two separate locations within the device.
Variations in fluid flow between channels can lead to differences in the number of protein molecules beneath each button valve. To account for these differences and generate a quantity proportional to fractional occupancy, Cy5 intensities were normalized by BODIPY intensities to yield a dimensionless intensity ratio (Cy5 intensity/BODIPY intensity) (Fig. 1e). Intensity ratios also showed strong preferences for individual oligonucleotide sequences, with no clear preference detected for rabbit reticulocyte lysate alone (Fig. 2b; Supplementary Fig. 4, Supplementary Table 2). Intensity ratios were well correlated both between measurements of the same 70-mer oligonucleotide at different locations within a given device (Fig. 2c; Supplementary Table 3) and between experiments (Supplementary Fig. 5).
Binding affinity can be described by a single-site binding model relating intensity ratio (r) to DNA concentration ([D]); Kd, the DNA concentration at which measured intensities reach half their maximum value (rmax) provides a quantitative measure of binding affinity (Eqn. 1):
| (1) |
At low DNA concentrations, measured intensity ratios are approximately inversely proportional to Kd. Calibrated measurements of DNA chamber intensities in our experiments establish that soluble DNA concentrations are indeed low (150 ± 25 nM, mean ± s.e.m.) (Supplementary Fig. 6), suggesting it might be possible to accurately estimate interaction affinities from intensity ratios measured at a single, low DNA concentration.
To test this hypothesis, we first measured concentration-dependent binding for 4 TFs from 2 different families (Cbf1p, Cin5p, Pho4p, and Yap1p), each interacting with 10 oligonucleotides from the 8-mer DNA library. We then globally fit Eqn. 1 over all oligonucleotides at all concentrations to get accurate Kd measurements (Fig. 3a,b; Supplementary Fig. 7, Supplementary Fig. 8, Supplementary Fig. 9).
Figure 3.
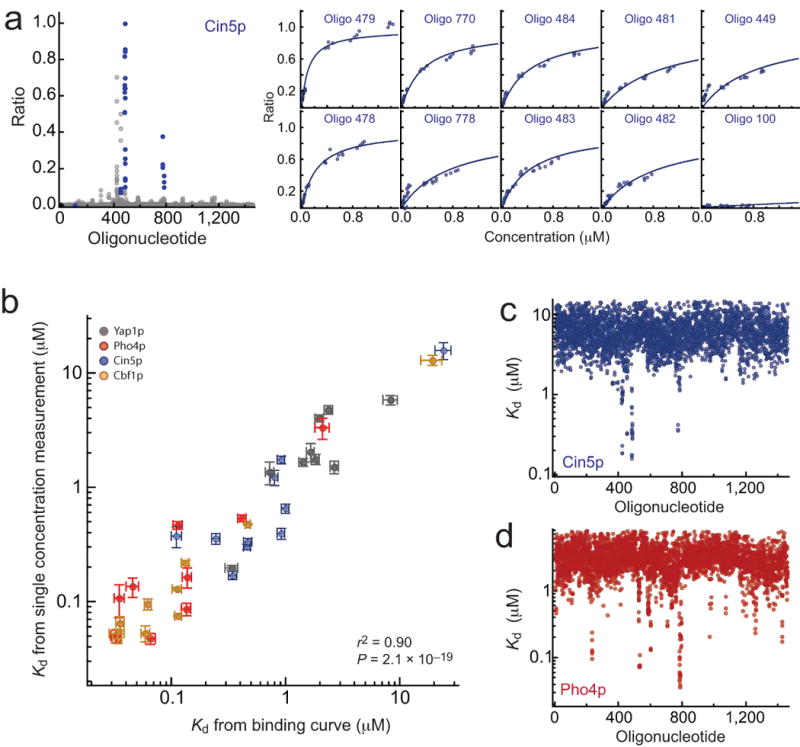
Comparison between Kd values derived from direct measurements of concentration-dependent binding and Kd values calculated from ratio measurements at a single concentration. (a) Cin5p measurements. Measured ratio signals for all oligonucleotides (grey) and selected oligonucleotides (blue) (left); concentration-dependent binding for selected oligonucleotides fit to a single-site binding model (right). (b) Kd calculated from single-concentration measurements vs. Kd derived from fits concentration-dependent binding for Cin5p (blue), Pho4p (red), Yap1p (grey) and Cbf1p (green). (c) Calculated Kd values for all oligonucleotides for Cin5p. (d) Calculated Kd values for all oligonucleotides for Pho4p.
Next, we calculated Kd values for the exact same oligonucleotides from single-concentration measurements. The low DNA concentration used for these measurements prevented direct determination of rmax, a parameter that depends on quantities that vary between experiments (e.g. amount and intensity of BODIPY and Cy5 dyes incorporated during protein and DNA library production, respectively), and must be empirically determined. Kd values from concentration-dependent binding can be used to “calibrate” the appropriate rmax value (Supplementary Information). Single-concentration Kd values calculated using calibrated rmax values were in excellent agreement with those derived from concentration-dependent binding (r2 = 0.90, p = 2.1 × 10-19) (Fig. 3b). Furthermore, once calibrated, rmax values can be used to calculate Kd values for all oligonucleotides with signals above background, providing absolute affinities for all 1457 oligonucleotides with only a few additional measurements (Fig. 3c,d; Supplementary Fig. 10). The range of Kd values calculated here for Pho4p and Cbf1p agree with those measured in previous studies (∼ 10 nM to 10 μM)20, validating our approach. Relative differences in binding affinities between oligonucleotides (the Gibbs free energy upon binding, ΔΔG) can also be calculated using these calibrated rmax values (Supplementary Fig. 11).
Even in the absence of additional information to calibrate rmax values, however, measured intensity ratios provide accurate information about binding affinity. To demonstrate this, we assumed an rmax value of 1 for all TFs and again compared measured and calculated Kd values. Kd measurements were well correlated (r2 = 0.67, p = 1.8 × 10-10), although precise values were somewhat offset (Supplementary Fig. 12a). ΔΔG describes relative affinity differences between oligonucleotides and is therefore less sensitive to these offsets, with stronger correlations (r2 = 0.76, p = 8.0 × 10-13) (Supplementary Fig. 12b).
Measured intensity ratios reflect interaction affinities between a given TF and a 70-bp oligonucleotide. Identifying TF target sites requires determination of the precise subsequences responsible for TF binding within each oligonucleotide. Traditionally, analysis of TF binding requires designation of sequences into bound and unbound populations, followed by a search for sequences overrepresented in the bound population, which ignores relative strengths of binding interactions, and can be sensitive to the precise threshold used to delineate populations. Here, we used a pipeline that incorporates all intensity information for all oligonucleotides to generate a position-specific affinity matrix (PSAM)23 describing the change in binding affinity upon mutation of a specific position within a consensus sequence (Supplementary Fig. 13). Notably, PSAMs describe actual binding affinities for any combination of nucleotides and can used to calculate predicted affinities to arbitrary sequences.
First, we analyzed all measured intensity ratios using fREDUCE, an enumerative algorithm that searches for sequences whose occurrence within oligonucleotides correlates strongly with their measured signal24. For all 28 proteins, fREDUCE returned sequences whose appearance within an oligonucleotide correlated strongly with measured intensity ratios (Fig. 5, Supplementary Table 6, Supplementary Fig. 14).
Next, the highest-correlated 7-and 8-bp fREDUCE sequences were converted to PSAMs using MatrixREDUCE23, an algorithm that fits all measured intensity ratios with a statistical mechanical model assessing the effects of individual base pair substitutions on binding affinity. Investigations of MatrixREDUCE performance have recommended the use of initial seed sequences derived from enumerative analysis to ensure optimization of global minima24 therefore, the fREDUCE sequences were used as seeds. MatrixREDUCE assumes that the free energy contributions of each position in the binding site are independent; although this is known to be false in some instances, we employ linear motifs here to compare our results with the largest possible set of previous literature.
To choose the single PSAM that best explains measured binding, we compared occupancies predicted by each PSAM for all oligonucleotides in the DNA library with measured intensity ratios (Supplementary Fig. 15). Predicted and measured values were well-correlated for almost all TFs (Supplementary Table 7). For all 26 TFs with described motifs, the final recovered motif was in agreement with those previously reported in the literature (Fig. 4) 14,15,22. We also derive PSAMs for two TFs, Msn1p and Nrg2p, that were previously resistant to characterization, establishing significantly enhanced sensitivity over both ChIP-based and PBM techniques.
Figure 4.

Comparison between motifs found for all 28 S. cerevisiae TFs and previous literature results (SWISS: SwissRegulon30, ChIP-chip: Harbison library22, PBM1′: protein binding microarray14, and PBM2: protein binding microarray15). For ChIP-chip data, boxes shaded in grey represent literature-derived motifs. For PBM2 results, white boxes represent proteins applied to arrays that did not yield motifs; boxes shaded in grey represent proteins that did not express sufficiently to be applied to arrays. fREDUCE Seeds: 7-and 8-bp fREDUCE motifs that correlate most strongly with measured intensities; Optimized PSAM: MatrixREDUCE PSAM represented as an AffinityLogo; r2: Pearson correlation coefficient between all measured ratio values and protein occupancies predicted by the optimized PSAM.
Two well-characterized basic helix-loop-helix (bHLH) proteins (Pho4p and Cbf1p) provide a test of the ability to detect both high-and low-affinity target sequences. Pho4p binds both high-affinity (5′-CACGTG-3′) and low-affinity (5′-CACGTT-3′) sites25 Cbf1p binds to a degenerate ‘5-RTCACRTG-3’ motif20,26. For both proteins, we recover the expected motif variants (Fig. 4, Supplementary Fig. 15).
Detailed analysis of differences between measured and calculated binding profiles can provide additional information about binding preferences. For example, oligonucleotides with high measured intensity ratios but low predicted occupancies, could indicate binding to additional motifs. In addition, this comparison allows investigation of whether free energy contributions at each position within the sequence are truly independent.
For most TFs, optimized PSAMs successfully described gross binding properties (e.g. Pho4p, Cin5p, Msn2p, and Sko1p; Supplementary Fig. 16), albeit with outliers at weak binding energies that may represent cooperative interactions between base pair substitutions. For a few transcription factors (Rpn4p, Cup9p, Cad1p, Matα2p, and Pdr3p), correlations between measured and predicted binding were much weaker (r2 < 0.25). To determine if low correlations resulted from binding to additional target sequences, we used BioPROSPECTOR27, MDScan27, MEME28, and WEEDER29 to scan for overrepresented sequences within oligonucleotides with high measured intensity ratios (Z-score > 25 or 75) but low predicted occupancies (Z-score < 3).
For Rpn4p, although both PBM studies and our first analysis identified binding to a 5′-GCCACC-3′ motif, ChIP and expression data suggest a T-rich 5′ extension of this motif upstream of Rpn4p target genes. Strikingly, analysis of the 13 oligonucleotides with discordant measured and predicted binding returned this precise extension, establishing that unexpected binding data can yield biologically relevant results (Supplementary Fig. 17).
The Cup9p optimized PSAM also agreed with previous PBM15 results (Fig. 4); however, 14 sequences showed stronger-than-predicted binding (Supplementary Fig. 18). Analysis of these sequences yielded motifs similar to the optimized PSAM, but with an ‘ACGT’ core (Supplementary Fig. 18, grey box). To assess the affinity of Cup9p for this candidate alternate motif, we measured concentration-dependent binding of Cup9p to the primary motif, candidate secondary motif, and several related motifs (Supplementary Fig. 19a). A random 2 bp substitution abolished binding, but mutating these bases or the entire second half of the motif to the candidate secondary motif reduced affinity only ∼ 20-fold (Supplementary Fig. 19b), confirming weak-affinity binding. Interestingly, this motif is found only 29 times in the genome outside of coding regions, primarily at the boundary of subtelomeric repeats and upstream of genes regulated by iron depletion, metal toxicity, or oxidative stress (Supplementary Table 8). While the physiological role of these putative binding sites is unknown, these results demonstrate the ability of MITOMI 2.0 to detect weak but potentially biologically relevant TF binding sites.
For the remaining 3 TFs (Cad1p, Matα2p, and Pdr3p), low correlations between predicted and measured binding likely result from experimental variability and not binding to additional motifs. Correlations between technical replicates across the device were relatively low (Supplementary Table 3) due to either binding to a limited number of oligonucleotides (Cad1p, Supplementary Fig. 3) or large variations in protein coverage (for Matα2p and Pdr3p). Consistent with this, these TFs do not bind any oligonucleotides with stronger-than-expected affinity.
The data presented here demonstrate increased sensitivity over current state-of-the-art techniques, detecting sequence-specific binding for several proteins that have failed to yield results in multiple experiments (Cad1p, Msn1p, Nrg2p, Sko1p, Yap7p, and Pdr3p). Moreover, these data represent the most comprehensive investigation of biophysical binding affinities to date, including ΔΔG values for 28 TFs and Kd values for 4 TFs from 2 different families (Cbf1p, Cin5p, Pho4p, and Yap1p) binding to 1457 individual sequences. These data can be used to test basic assumptions underlying current models of TF-DNA specificity and more accurately model cooperativity between nucleotide binding sites (‘non-additivity’).
The DNA library used here is not organism-specific, making this technique useful for a wide range of organisms, including higher eukaryotes and pathogens. In addition, the programmable nature of MITOMI 2.0 allows subsequent detailed examination of unexpected binding phenomena or systematic mutational analysis of candidate motifs through direct observations of concentration-dependent binding. Although these experiments probed TF binding to double-stranded DNA, MITOMI 2.0 can be used with only minimal changes to investigate single-stranded DNA binding and RNA binding. When paired with advances in rapid whole genome sequencing, we anticipate that MITOMI 2.0 characterization of all recognizable TFs in a given proteome will allow transcriptional networks and regulons to be quickly identified and ultimately modeled.
Materials and Methods
Oligonucleotide sequence files and data for all TFs are available for download at http://derisilab.ucsf.edu.
DNA library and transcription factor production
All possible 65, 536 8-bp DNA sequences were assembled into a maximally compact de Bruijn sequence that was subsequently divided over 1457 oligonucleotides. Sequences were hybridized to a Cy5-labeled oligonucleotide and extended using Klenow fragment (exo-) (NEB) to produce Cy5-labeled dsDNA. Cy5-labeled dsDNA was diluted to a final concentration of 1.25 μM in 3X SSC with polyethylene glycol (PEG) (Fluka) and D-(+)-trehalose dihydrate (Fluka) (for enhanced subsequent solubility) and printed onto custom 2″×3″ ThermoFisher Scientific SuperChip Epoxysilane slides (ThermoFisher Scientific) using a DeRisi lab custom microarrayer.
A two-step PCR reaction was used to amplify TF coding sequences and add appropriate upstream and downstream sequences for efficient transcription and translation in rabbit reticulocyte lysate (Promega) (Supplementary Fig. 2).
Microfluidic device fabrication and experimental procedure
Flow and control molds were fabricated on 4″ silicon wafers using positive (SPR 220-7.0) and negative (SU-8) photoresists, respectively. PDMS devices were produced and the MITOMI experimental procedure was performed as described previously20.
Initial data analysis and normalization
Median Cy5 and BODIPY fluorescence intensities varied somewhat between experiments. To facilitate comparisons between TFs, Cy5 intensity distributions were fit to a Gaussian and this Gaussian mean was subtracted from all measurements to center the background distribution around zero. Fluorescence intensity ratios were calculated by dividing Cy5 fluorescence intensities by BODIPY fluorescence intensities; ratios were similarly normalized such that the background was centered around zero, and further normalized such that the maximum measured intensity was 1.
Motif finding pipeline
We searched for 7-and 8-bp sequences that correlated most strongly with measured intensity ratios using fREDUCE. Both doubly- (R, Y, S, W, K, M) and triply- (B, D, H, V) degenerate IUPAC bases were included, and both the forward sequence and its reverse complement were analyzed. The highest-correlated 7-bp and 8-bp sequences were then used as seeds for MatrixREDUCE analysis, with additional unspecified base pairs added to either side of the 7-bp seed to standardize length.
Occupancy profile calculations
We calculated predicted occupancy profiles from PSAMs using a slight modification of the MatrixREDUCE formalism to reflect the fact that, in our assay, transcription factors are surface-immobilized and DNA sequences are in solution (Supplementary Information).
Supplementary Material
Acknowledgments
P.M.F. was supported by a Howard Hughes Medical Institute/Helen Hay Whitney Foundation Postdoctoral Fellowship. JDR, SRQ, and this work were supported by the Howard Hughes Medical Institute.
We thank Anastasia Potanina for assistance with fabrication of microfluidic devices, Oliver Homann for implementation of PSAM functionality with MochiView, and Dave Breslow, Florencia Caro, Stirling Churchman, Michelle Dimon, Toby Kiers, Amy Kistler, Chris Nelson, Katherine Sorber, Ellen Yeh, and Ignacio Zuleta for careful reading of the manuscript.
Footnotes
Author Contributions: P.M.F. designed experiments, designed, created, and printed the DNA library, made linear expression templates, fabricated microfluidic devices, performed microfluidic experiments assessing concentration-dependent binding and binding to the 8mer library, analyzed data, and wrote the manuscript. D.G. designed experiments, designed and fabricated microfluidic devices, and performed microfluidic experiments assessing binding to the 8mer library. D.T. fabricated microfluidic devices, and performed microfluidic experiments assessing binding to the 8mer library. J.Z. and H.L. analyzed data. S.R.Q. designed experiments, analyzed data and wrote the manuscript. JDR designed experiments, assisted with printing the DNA library, analyzed data, and wrote the manuscript.
Note: Supplementary information is available on the Nature Biotechnology website.
Bibliography
- 1.Tanay A. Extensive low-affinity transcriptional interactions in the yeast genome. Genome Research. 2006;16:962–972. doi: 10.1101/gr.5113606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, Gaul U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–540. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- 3.Badis G, et al. Diversity and Complexity in DNA Recognition by Transcription Factors. Science. 2009 doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim HD, O'Shea EK. A quantitative model of transcription factor-activated gene expression. Nat Struct Mol Biol. 2008;15:1192–1198. doi: 10.1038/nsmb.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Segal E, Widom J. From DNA Sequence to Transcriptional Behavior: A Quantitative Approach. Nature reviews Genetics. 2009;10:443. doi: 10.1038/nrg2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gertz J, Siggia ED, Cohen BA. Analysis of combinatorial cis-regulation in synthetic and genomic promoters. Nature. 2009;457:215–218. doi: 10.1038/nature07521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuh CH, Bolouri H, Davidson EH. Cis-regulatory logic in the endo16 gene: switching from a specification to a differentiation mode of control. Development. 2001;128:617. doi: 10.1242/dev.128.5.617. [DOI] [PubMed] [Google Scholar]
- 8.Iyer VR, et al. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- 9.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 10.Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic acids research. 1981;9:3047. doi: 10.1093/nar/9.13.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galas DJ, Schmitz A. DNAse footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 1978;5:3157–3170. doi: 10.1093/nar/5.9.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jost JP, Munch O, Andersson T. Study of protein-DNA interactions by surface plasmon resonance (real time kinetics) Nucleic Acids Res. 1991;19:2788. doi: 10.1093/nar/19.10.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng X, Brodsky MH, Wolfe SA. A bacterial one-hybrid system for determining the DNA-binding specificity of transcription factors. Nat Biotechnol. 2005;23:988–994. doi: 10.1038/nbt1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Badis G, et al. A Library of Yeast Transcription Factor Motifs Reveals a Widespread Function for Rsc3 in Targeting Nucleosome Exclusion at Promoters. Molecular Cell. 2008;32:878–887. doi: 10.1016/j.molcel.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu C, et al. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res. 2009;19:556–566. doi: 10.1101/gr.090233.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berger MF, et al. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nature biotechnology. 2006;24:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger M, et al. Variation in Homeodomain DNA Binding Revealed by High-Resolution Analysis of Sequence Preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Silva EK, et al. Specific DNA-binding by apicomplexan AP2 transcription factors. Proc Natl Acad Sci U S A. 2008;105:8393–8398. doi: 10.1073/pnas.0801993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonham AJ, Neumann T, Tirrell M, Reich NO. Tracking transcription factor complexes on DNA using total internal reflectance fluorescence protein binding microarrays. Nucleic Acids Research. 2009 doi: 10.1093/nar/gkp424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maerkl SJ, Quake SR. A Systems Approach to Measuring the Binding Energy Landscapes of Transcription Factors. Science. 2007;315:233–237. doi: 10.1126/science.1131007. [DOI] [PubMed] [Google Scholar]
- 21.Ralston A. De Bruijn Sequences-A Model Example of the Interaction of Discrete Mathematics and Computer Science. Mathematics Magazine. 1982;55:131–143. [Google Scholar]
- 22.Harbison CT, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foat BC, Morozov AV, Bussemaker HJ. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics. 2006;22 doi: 10.1093/bioinformatics/btl223. [DOI] [PubMed] [Google Scholar]
- 24.Wu R, Chaivorapol C, Zheng J, Li H, Liang S. fREDUCE: Detection of degenerate regulatory elements using correlation with expression. BMC Bioinformatics. 2007;8:399. doi: 10.1186/1471-2105-8-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vogel K, Horz W, Hinnen A. The two positively acting regulatory proteins PHO2 and PHO4 physically interact with PHO5 upstream activation regions. Molecular and Cellular Biology. 1989;9:2050. doi: 10.1128/mcb.9.5.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wieland G, et al. Determination of the binding constants of the centromere protein Cbf1 to all 16 centromere DNAs of Saccharomyces cerevisiae. Nucleic Acids Res. 2001;29:1054–1060. doi: 10.1093/nar/29.5.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, et al. A suite of web-based programs to search for transcriptional regulatory motifs. Nucleic Acids Res. 2004;32:W204–207. doi: 10.1093/nar/gkh461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pavesi G, et al. MoD Tools: regulatory motif discovery in nucleotide sequences from co-regulated or homologous genes. Nucleic Acids Res. 2006;34:W566–570. doi: 10.1093/nar/gkl285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pachkov M, Erb I, Molina N, Van Nimwegen E. SwissRegulon: a database of genome-wide annotations of regulatory sites. Nucleic Acids Research. 2006 doi: 10.1093/nar/gkl857. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.