

Abstract
Advances have been made in defining the mechanisms for the control of allergic airway inflammation in response to inhaled antigens. Several genes, including ADAM33, DPP10, PHF11, GPRA, TIM-1, PDE4D, OPN3, and ORMDL3 have been implicated in the pathogenesis and susceptibility to atopy and asthma. Growing evidence associates asthma with a systemic propensity for allergic type 2 T-cell cytokines. Disordered coagulation and fibrinolysis also exacerbate asthma symptoms. Balance among functionally distinct dendritic cell subsets contribute to the outcome of T cell-mediated immunity. Allergen-specific T-regulatory cells play a pivotal role of in the development of tolerance to allergens and immune suppression. Major emphasis on immunotherapy for asthma during the past decade has been to direct the immune response to a type 1 response or immune tolerance. In this review article, we discuss the current information on the pathogenesis of allergic airway inflammation and potential immunotherapy, which could be beneficial in the treatment of airway inflammation, allergy, and asthma.
Introduction
Asthma is a disease of chronic airway inflammation characterized by reversible airway obstruction, airway hyperresponsiveness (AHR), infiltration of eosinophils and type 2 T cells into the airway submucosa, mucus hypersecretion, and airway remodeling (1). Allergic asthma is classified as a type 1 hypersensitivity reaction. This involves allergen-specific immunoglobulins of the IgE class bound to high-affinity Fcε receptors on the surfaces of basophils and mast cells present in the subepithelial layer of the airways. Cross-linking of these bound IgE molecules results in an immediate release of mediators, including leukotrienes, prostaglandins, and histamine, that are capable of contracting airway smooth muscle cells and induce edema and mucus secretion, leading to narrowed, constricted airways. Locally-produced chemokines stimulate the recruitment of eosinophils, macrophages, neutrophils, and T lymphocytes (1). Once present, effector cells, such as eosinophils, release a collection of toxic granules, which in turn cause prolonged bronchoconstriction and damage epithelial layers. This damage, coupled with profibrotic cytokines also released by eosinophils and epithelial cells, may lay the groundwork for the process of airway remodeling to begin (2). Cytokines released at the time of mast cell degranulation can have more global effects. These include the recruitment of eosinophils from bone marrow and peripheral sources in addition to encouraging their survival (primarily via interleukin [IL]-5 and granulocyte-macrophage colony-stimulating factor [GM-CSF]) and the stimulation and continued production of IgE by B cells, as well as the induction of vascular cell adhesion molecule-1 (VCAM-1) by endothelial cells (IL-4) (1). Cytokines such as IL-4, IL-5, IL-6, and IL-13 ensure that this cycle of allergic inflammation persists (Table 1). The prevalence of asthma has been increasing steadily for several decades. Although there is an appreciable genetic component (1), external influences may regulate/influence the immune system by affecting the differentiation and activation of T lymphocytes. Therapeutic approaches targeting both intrinsic and extrinsic factors have been under extensive investigation.
Table 1.
Release of cytokines and other mediators and their effects from various cells in involved allergic airway inflammation
| Cells differentiated and infiltrated in the lung | Cytokines/mediators | Biological effects |
|---|---|---|
| Cells that exacerbate inflammation and asthma | ||
| Eosinophils | Toxic granules; MBP, EDN, EPO, cytokines | Prolonged bronchoconstriction, damaged epithelium, airway remodeling |
| TH2 cells | IL-4, IL-5, IL-13 | Humoral antibody production, chemotaxis and survival of eosinophils |
| Th17 cells | IL-17, IL-17F, IL-22, IL-26 | Neutrophilia, AHR, airway remadeling |
| Dendritic cells | IL-4, GM-CSF (cholera toxin, PGE2, histamine may potentiate the effect) | Induction of TH2 cells; suppression of TH1 cells |
| CD8+ T cells | IL-5, IL-13 | AHR, eosinophilia |
| Cells that suppress allergic and asthmatic response | ||
| Regulatory T cells | IL-10, TGF-β | Prevention of T-cell expansion |
| CD8+ T cells | IFN-γ | Suppression of allergic immune response |
| TH1 cells | IL-2, IFN-γ, lymphotoxin | Enhanced cellular response; suppression of TH2 cells |
| Plasmacytoid DCs and Regulatory Dendritic cells | IL-10, IL-12 (LPS, bacterial CpG, CpR oligonucleotides) | Induction of TH1 cells and T cell suppression |
AHR–airway hyperresponsiveness; CpG–cytosine-phosphorothiolated guanine; CpR–cytosine-phosphorothiolated 2′-dioxy-7-deazaguanosine; EDN–eosinophil-derived neurotoxin; EPO–erythropoietin; GM-CSF–granulocyte-macrophage colony-stimulating factor; IFN–interferon; IL–interleukin; MBP–myelin basic protein; TGF–transforming growth factor; TH–T helper.
Th1/Th2 Polarized Immunity
It is now generally accepted that allergic respiratory disease in adults is associated with active T-cell immune responses to inhaled allergens that are skewed toward the Th2 phenotype, in contrast with a Th1-skewed immunity in normal, healthy subjects. Helper T cells of the type 1 variety (Th1) secrete interferon (IFN)-γ, IL-12, and lymphotoxin (TNF-β), whereas T cells of the type 2 phenotype (Th2) secrete IL-4, IL-5, IL-9, and IL-13 (Fig. 1). TH1 cells enhance cellular immune responses; Th2 cells favor humoral antibody production (IgE), such as allergic asthmatic response. The improved hygiene results in a decreased stimulation of a type 1 response and leads, therefore, to a greater stimulation of type 2 responses and a consequent predisposition to allergic diseases. Unequal apoptosis of Th1 andTh2 effector cells in atopic patients lead to preferential deletion of circulating memory or effector Th1 cells (3), especially the high IFN-γ-producing Th1 cells (4), which contributes to skewing the immune response toward surviving Th2 cells. New effector T cell lineages have been identified recently. Th17 cells, which differentiate from naïve CD4+ T cell under the influence of IL-6/IL-21/IL23 and TGF-β via STAT3-RORγt pathway, are mainly responsible for neutrophilia in allergic asthma (5) (Fig. 1). In the presence of IL-4 and TGF-β, Th2 cells can be reprogrammed to a new T cell lineage expressing IL-9 and IL-10, namely Th9 cells (6) (Fig. 1).
Figure 1. Differentiation of CD4+ T helper cell and CD8+ cytotoxic T cells in allergic asthma.
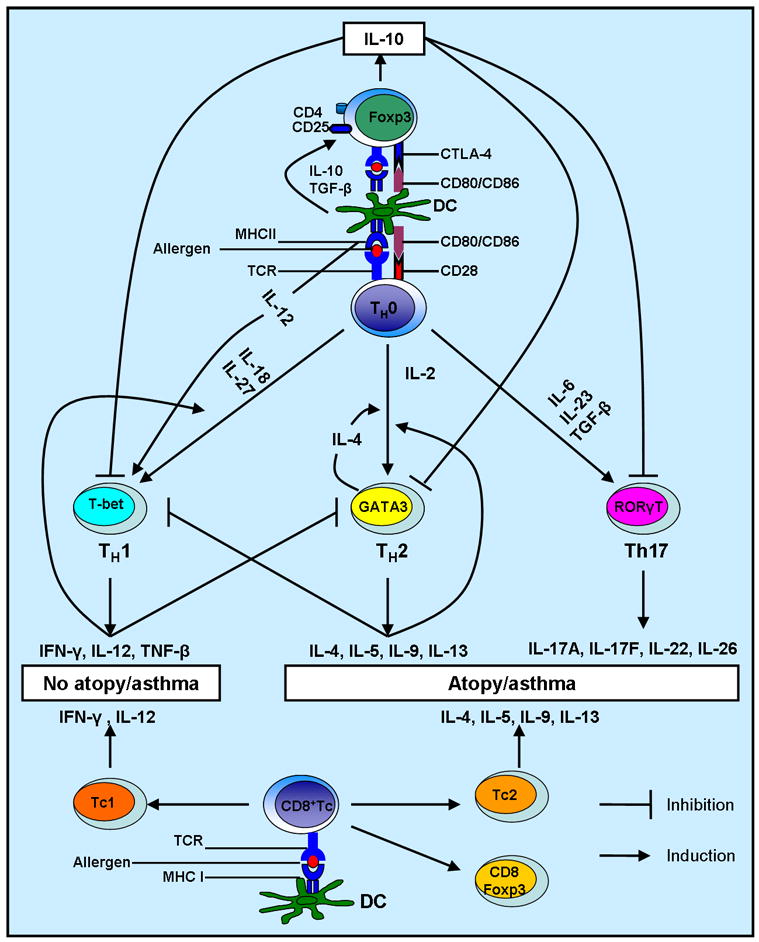
Differentiation of CD4+ T helper cells to effector Th1, Th2, Th17 cells or Treg cells depends upon environmental cytokine profile. Cytokines released from Th1 cells can antagonize differentiation and function of Th2 cells, and vice-versa. Undifferentiated Th0 cells can mature into Th1, Th2, Th17, or Treg cells in peripheral lymph organs, depending upon the costimulatory signals presented to them, along with antigen, by antigen-loaded dendritic cells. Functionally, CD8+ effector cytotoxic T cells (Tc) contain Tc1, Tc2, and CD8+Foxp3+ regulatory cells. Immunomodulators, such as Flt-3 ligand or CpR oligonucleotides, may preferentially increase the number of regulatory dendritic cells and/or T-regulatory cells inducing Th1 response, T cell anergy/tolerance to allergen. This could result in the prevention and/or reversal of atopy and/or asthma symptoms.
Transcription factors responsible for the Th1/Th2 dichotomy
The determination of T helper lineage fates of Th1 or Th2 is accompanied by a differential activation, expression and functionality of transcription factors in different T cell lineages, which accordingly induce and suppress, respectively, lineage-related and non-lineage-related cytokine secretion. It is becoming evident that cytokines differentially secreted by various DC subsets (7–9) play an essential role in driving T cell differentiation (Fig. 1). Th1 master regulator, transcription factor T-bet (T-box), is extensively expressed in polarized Th1 cells and its expression and activity are induced by IL-12 via STAT4 or by IFN-γ via STAT1 (10). STAT4 also influences Th1 commitment via a T-bet-independent manner (11). The transcription factor, Runx3, is also upregulated in Th1 cells in a T-bet-dependent manner. T-bet and Runx3 demonstrate cooperative effect on the production of IFN-γ and silencing of the gene encoding IL-4 in Th1 cells (12). IL-4drives differentiation of IL-4–producingTh2 cells through STAT6, which is necessary and sufficient for the induction of the Th2 master regulator, GATA3 (13). GATA3, through an autocrine pathway, upregulates its own expression in a STAT6-independent manner (14). Upon T cell receptor (TCR) stimulation, TCR-inducible transcription factor NFAT1 is also involved in regulating GATA3 expression (14). Transcription factor, c-Maf, is selectively expressed in Th2 cells as a downstream effector of IL4/IL4R/STAT6 signal transduction pathway and primarily regulates IL-4 expression in Th2 cells. The cross talk between Th1 and Th2 transcription factors are also involved in the regulation of T cell lineage commitment. STAT6 inhibits IL-12-STAT4 signaling pathway without involving downstream transcription factor T-bet (15). In fact, GATA3 and T-bet target a number of common genes and exert the opposing effect on their transcription, which consequently influences the choice between Th1 and Th2 lineage commitment (16). Therefore, both T-bet and GATA3 work concomitantly with other transcription factors, including nuclear factor of activated T cells (NF-AT), activator protein-1 (AP-1), nuclear factor (NF)-κB, and CCAAT/enhancer binding protein b (C/EBPb). The counterpart of GATA3 and T-bet in Th17 cells is RORγt, which is required to activate IL17 production in Th17 cells and directly/indirectly regulates production of IL-17F and IL-22 in Th17 cells (17).
Lung Dendritic Cells (DC)
An emerging concept is that different lung DC subsets induce different immune responses, such as immunity versus tolerance, or Th1 versus Th2. Lung CD11c+CD11bhiB220−Gr-1− myeloid DCs play a major role in inducing allergic airway inflammation in response to allergen challenge (9). CD11cintGr-1+B220+ DCs identified in mouse lymph nodes produce type 1 interferon and demonstrate tolerogenic potential, which leads to the notion that they are the murine counterpart of human plasmacytoid DCs (18). De Heer and colleagues (19) demonstrated that lung CD11cintGr-1+B220+ plasmacytoid DCs suppressed T cell division and effector T cell generation induced by myeloid DCs, confirming the involvement of plasmacytoid DC in regulating lung inflammation. Additionally, IL-10-producing CD80+CD86+CD40+MHCII+CD8− lung DCs induce pulmonary tolerance, which establishes the role of IL-10 in DC biology in conjunction with the fact that IL-10-treated DCs reverse allergic airway inflammation (20).
Lung CD11chighCD11blowCD103+ DCs have been recently discovered and their function appears to be related to increased IL-12 production (21), CD8+ T cell stimulation, and particulate antigen uptake (22). Thus, lung DCs demonstrate highly flexible phenotype and often accomplish diverse and opposing functions. We have identified two functionally and phenotypically distinct lung DC subsets, namely CD11chighCD11blow and CD11clowCD11bhigh in a mouse model of allergic airway inflammation (9). Under condition of allergen challenge, CD11clowCD11bhigh lung DC subset is rapidly expanded and more prone to induce robust Th2 response as compared to a Th1-prone response induced by CD11chighCD11blow lung DCs (9). They also differ in migratory and antigen uptake patterns (23).
In human, two subsets of blood DCs, myeloid and plasmacytoid DCs, were identified based on the expression of CD11c, a β2 integrin. Human blood myeloid DCs are further divided into mDC1 and mDC2, which uniquely express CD1c (BDCA-1) and CD141 (BDCA-3), respectively. Conversely, blood plasmacytoid DCs express CD123 and CD303 (BDCA-2) but not CD11c (24). The three DC subsets are present in human lung digest and bronchoalveolar lavage fluid (BALF) and are able to stimulate T cell proliferation (25). Functionally, human lung pDCs secret more IFN-α and have lower expression of MHC and costimulatory molecules than mDCs (26). As DCs mature in the lungs, the phenotypic characteristics of lung DCs might not follow the exactly same pattern as blood DCs. Possibly as a result, the blood pDC-specific marker CD123 is also expressed on a sub-population of lung mDCs, and this distinction is reflected by a subtle difference in the cytokine/chemokine secretion pattern between CD123+ and CD123− lung mDCs (26). Similar to murine allergic asthma model, human lung mDCs appear to play a more important role in the induction of asthma since an enhanced influx of functionally active antigen-presenting mDCs into the respiratory tract is observed following endotoxin administration (27). However, another study involving human subjects that analyzed BALF DCs suggests that both pDCs and mDCs are recruited into the asthmatic lungs in asthma patients upon allergen challenge with a greater amount of pDC recruited (28). The identification of human lung DC subset that can specifically direct inflammation to Th2 suppression will provide an effective approach in the treatment of asthma. Additional studies are warranted to examine functional role of each DC subtype in allergic airway inflammation and asthma.
T regulatory cells (Treg) in allergic airway inflammation
Tregs are a heterogeneous group of cells that play a central role in maintaining homeostasis of pulmonary immunity by establishing immune tolerance to non-harmful antigen or suppressing effector T cell immunity. Naturally-occurring CD4+CD25+ Tregs (NTreg) constitutively express CD25, a transmembrane protein and an α-chain of the receptor for IL-2, and suppress autoimmuneT-cell responses and maintain peripheral tolerance. They also constitutively express transcription factor Foxp3, which prevents deviation of Treg cells into effector T cells. NTregs are generated in the thymus by positive selection in a process mediated by class II major histocompatibility complex (MHC)-positive thymic cortical epithelium or dendritic cells (29). This development requires a higher affinity T-cell receptivity (TCR) and IL-2 signal (29). IL-2 presumably activates downstream effector STAT5 to regulate Foxp3 expression (30). Peripheral differentiation of Tregs, namely inducible-Tregs (iTreg) that secret IL-10, requires a combination of strong TCR signal, high levels of TGF-β (31) and/or IL-10 (Fig. 1). In TGF-β-mediated induction of iTregs, CTLA-4, a competitive receptor of CD28 for the costimulatory molecules, CD80 and CD86, is required (32). The activity of Tregs is associated with the development of asthma. Adoptive transfer of antigen-specific CD4+CD25+ T regulatory cells attenuates acute allergic airway inflammation, AHR, and airway remodeling in an IL-10-dependent manner (33). We recently reported that both NTregs and iTregs reverse the established cockroach antigen-induced allergic asthma, but the effect of iTregs depend on higher levels of transforming growth factor–β,IL-10, IFN-γ, and elevated levels of PD-1 than in NTregs to differentiate into IL-10-producing iTregs in the lung to exert their suppressive activity (34). Although TGF-β plays a detrimental role in promoting airway remodeling, the cooperation between IL-10 and TGF-β seems to be important in the iTreg-mediated immune suppression. These data suggest that one of the treatment options would be to enhance CD4+CD25+ Treg cells in addition to targeting decreased Th2 populations.
The role of CD8+ T cells in Allergic Airway Inflammation
The investigations on the contribution of cytotoxic CD8+ T cells in the development of allergic airway inflammation remain inconclusive. On one hand, a body of evidence has suggested that CD8+ T cells, which play a key role in cellular immunity by secreting IFN-γ and cytolytic factors, has suppressive effect on allergic airway inflammation. CD8+ T cells inhibit the allergen sensitization in a rodent animal model (35). The late allergic response and airway inflammation induced by adoptive transfers of CD4+ T cell is abolished by the resident CD8+ T cells. MHC class-restricted allergen-specific CD8+ T cells are generated in draining lymph nodes upon allergen challenge and rapidly infiltrate into the lung. These cells suppress the features of allergic airway inflammation by inducing IL-12 production (36). The Th2-suppressive effect of antigen-specific CD8+ T cells in an animal model immunized with OVA-CLDCvaccine is dependent on IFN-γ production (37). Two types of CD8+ T cells classified by TCR αβ and γδ have functional distinction. CD8+ T cells withγδ TCR are inhibitory in allergic asthma (38). The mediator of this suppressive effect on late allergic airway responses and eosinophilia has been proved to be IFN-γ as well (38) (Fig. 1). On the other hand, CD8+ T cell appear to be actively involved in the induction of allergic airway inflammation as demonstrated by the use of knockout mice deficient in CD8+ T cells. These mice demonstrated a decreased AHR and airway inflammation and lower IL-13 production in the BALF in response to allergen challenge compared with the wild type (37). This finding suggests that CD8+ T cells are required for full development of these responses and this process is IL-13-dependent. A later study has revealed that although CD8+ T cell is not absolutely required at the initiation stage of allergic airway inflammation unlike CD4+ Th2 cells, its absence appears to contribute to a less degree of AHR, airway eosinophilia, inflammatory cytokine production in BALF and goblet cell metaplasia in a long term sensitization protocol, suggesting a more active role of CD8+ T cell in chronic phase of allergic airway inflammation as compared to its importance in acute allergic reaction (37, 39). This notion is supported by the fact that depletion of CD8α-expressing cells worsens the airway remodeling, one of the main features of chronic asthma (40). A subset of CD8+ T cells, named as Tc2 cells, can produce Th2 cytokines such as IL-4, IL-5, and IL-13, are increased in BALF of allergic asthmatic patients (41) (Fig. 1). This leads to a concept that different types of CD8+ T cells function in such a way that favors a specific direction of immune response. In fact, two different antigen experienced T-cell subsets have been described, and they can be distinguished by their ability to home to lymphoid organs (central memory cells with a phenotype of high levels of CD62 ligand and CCR7) or non-lymphoid tissues and site of inflammation and acquire effector cell function more rapidly (effector cells with a phenotype of lower levels of CD62 ligand and CCR7) (42). It is effector memory CD8+ T cells but not central memory CD8+ T cells that are essential for the development of AHR and airway allergic inflammation in adoptive transfer models (37, 43). This may be due to the preferential localization of the effector cells to the lungs. Additionally, the CD8+-mediated AHR and airway inflammation is Th2 cells and IL-4 dependent (44). The concept of CD8+ T regulator cells have been raised and such T cell phenotypes as in vitro-generated CD8+CD28−Foxp3+ suppressor T cells (45), Tonsillar Foxp3+CD8+ T cells (46), LN/spleen CD45RClowCD8+Foxp3+CTLA-4+ T cells (47), and CD8+CD25+ T cells (48) have been shown to have regulatory properties in different experimental settings. Whether or not they are present and involved in the regulation of allergic inflammation is yet to be completely elucidated. Thus, CD8+ T cells exhibit functional and phenotypic plasticity.
Allergic Airway Inflammation and Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are enzymes that participate in extracellular matrix remodeling and degradation. Alteration in the regulation of MMPs is implicated in inflammatory processes. Their proteolytic cleavage of chemokines alters the chemokine gradient and inflammatory cell recruitment. Elevated levels of MMP-2 and MMP-9 have been observed upon allergen challenge and they are considered crucial for the infiltration of inflammatory cells such eosinophils (49), and MMP-9 is associated with airway remodeling including peribronchial fibrosis (50). However, other studies have shown that MMP-9-deficent mice develop enhanced pulmonary inflammation and airway hyperactivity (51). This is explained by its dominant role in controlling the resolution and egression of inflammatory cells in conjunction with MMP-2 (52). MMP-2 and MMP-9 differentially regulate chemotaxis of airway inflammatory cells by proteolytic processing of chemoattractant in the airways and facilitate the clearance of these cells, and one of the sources of MMP-9 is activated airway neutrophils (53). In addition, MMP-12 has been shown to be involved in the accumulation and survival of the airway eosinophils, infiltration of neutrophil and macrophages but not T cells in a cockroach-sensitized mouse model (54). MMP-7 is also expressed in asthmatic lungs and its deficiency results in attenuated allergic airway inflammation. MMP-7 is shown to plays a pro-inflammatory role by activating IL-25 and inhibiting retinoid-dependent development of regulatory T cells (55). Some attempts have been made to target MMPs in asthmatic condition. The tissue inhibitor of metalloproteinase-1 and a synthetic MMP inhibitor, Marimastat, could have therapeutic effect in the treatment of asthma (56).
Asthma and Genes
Asthma is a polygenetic disorder. Susceptibility to asthma depends on differences in a number of genetic loci. Positional cloning has made it possible to identify a few susceptibility genes. The T-cell immunoglobulin domain, mucin-like domain (TIM) gene family (57), and a disintegrin and metalloproteinase domain 33 (ADAM33) gene (58) found on chromosome 20p13 are associated with asthma and AHR. The subsequent studies using anti-TIM-1 in allergic airway inflammation animal model achieved expected effects (59). ADAM33 is important in the development, disease progression, and airway remodeling of asthma (60). Study has revealed susceptibility genes GPRA (G protein–coupled receptor for asthma susceptibility) for asthma found on chromosome 7p (61). However, an epidemiologic study reveals that PTGDR gene is not a significant risk factor for asthma among Puerto Ricans, Mexicans, or African Americans (62). DPP (dipeptidyl peptidase) 10 and PHD fingerprotein 11 (PHF 11) (63) have been linked with asthma and related phenotypes. A recent genome-wide association (GWA) study of asthma identified cAMP-specific (phosphodiesterase E3 dunce homolog, Drosophila) gene (PDE4D) as a asthma-susceptibility gene and PDE4 inhibitors have been developed as medications for asthma (64). In addition, genetic epidemiologic studies have shown that ORMDL3 locus is a risk factor for asthma (65) and the OPN3 on chromosome 1qter is an asthma susceptibilitygene (66). It should be noted that the importance of an asthma-susceptibility gene should be considered within a specific ethnic context because of the genetic difference among various ethnic groups.
The Role of the Coagulation System in Airway Inflammation
Interaction between inflammation-dependent tissue injury and thrombin formation, fibrin deposition, and impaired fibrinolysis has been shown in several pathologic conditions. Inflammatory cytokines, such as TNF-α, eotaxin, and RANTES (regulated on activation, normal T-cell expressed and secreted) decrease the expression and activity of activated protein C, which plays a protective role of in lung and airway remodeling (67). Allergic airway inflammation disturbs the balance in blood coagulation and fibrinolysis and result in the accumulation of extravascular fibrin, plasma exudates, and inflammatory cells, leading to airway closure. Increased concentrations of tissue plasminogen activator antigen, plasmin-antiplasmin complex, and fibrinogen/fibrin degradation products have been observed in patients with untreated asthma. Hataji et al. (67) observed a significantly decreased ratio of activated protein C and thrombin, and decreased ratio of activated protein C and protein C with increased concentrations of soluble thrombomodulin in the sputum of asthmatic patients as compared with healthy volunteers. In another study, Wagers et al. (68) reported the deposition of fibrin along the luminal surface of distal airways in a patient who died of status asthmaticus. This suggests an increased activity of the fibrinolysis system in asthmatics. Aerosolized tissue plasminogen activator, a fibrinolytic agent, has been observed to diminish AHR in a mouse model of allergic airway inflammation (68). Furthermore, treatment with activated protein C significantly inhibited the development of lung fibrosis in bleomycin-induced lung injury and the development of AHR and allergic airway inflammation in a mouse model of asthma (69). Overall, these studies suggest the role of disordered coagulation and fibrinolysis in the pathogenesis of asthma.
Novel Therapeutic Modalities
Currently asthma treatment still largely relies on anti-inflammatory corticosteroids and bronchodilators such as β2-adrenergic receptor agonist. In fact, numerous immune cells and mediators that contribute to the exacerbations and progress of allergy and asthma can be potential modalities to treat the disease. The counter-regulation relationship of Th1 and Th2 implies that manipulating the Th1/Th2 balance may be a potential approach to treat asthma, but the role of Th1 activation in allergic airway inflammation seems to be controversial. Adoptive transfer of Th1 cells causes an IFN-γ-mediated anti-eosinophilic effect (70). The coexistence of adoptively transferred antigen specific Th1 and Th2 cells reversed bronchial hyperresponsiveness and bronchoalveolar lavage eosinophilia in a IFN-γ-dependent fashion (71). Furthermore, Th1 cytokine treatment in both human (72) and animal asthmatic mouse models (73), has consistently shown a Th2-suppressive effect, including reduced AHR, airway eosinophilia and IgE production. More recently, some known Th1-prone antigen such as Bacille Calmette-Guérin (BCG) (74) and CpG oligodeoxynucleotides (ODN) (75) have been studied as novel therapeutic strategies for the prevention and treatment of atopic conditions with very promising therapeutic effects. The opposite findings (76, 77), however, weakens the validity of using Th1 cells/Th1 cytokines as an effective anti-asthma treatment (78). In fact, although there has not been a consensus as to neutrophilia in Th1 cell-induce airway inflammation (79), Th1 cells indeed induce hyperresponsiveness and lung fibrosis that are associated with IL-18 and antigen challenge (80, 81). In a chronic allergic asthma model, IFN-γ and IFN-γ-producing CD4+ cells in peripheral lymph nodes maintain at high levels. Therefore, the controversial role of Th1 cytokine IFN-γ in allergic airway inflammation leads to the caution in the application of Th1 to counteracting Th2-dominant allergic airway inflammation (82).
Administration of certain toll-like receptor (TLR) ligands activates innate immunity and induce high levels of indoleamine 2,3-dioxygenase, the rate-limiting enzyme of tryptophan catabolism in various organs. Pulmonary indoleamine 2,3-dioxygenase activity in the lung cells in response to TLR9 activation has been shown to inhibit Th2-driven allergic airway inflammation and AHR (83). These data suggest the pivotal role of the activation of innate immunity in inhibiting allergen-specific immune responses due to type 2 cytokines.
The role of calcium-activated potassium channel in the development and induction of asthma has currently drawn much attention due to their suitability for regulating membrane potential in cells with non-excitable membrane, such as immune cells, epithelial and endothelial cells, under inflammatory conditions. A growing body of evidence has linked the KCa3.1 activity to the migration, activation, and proliferation of the key cells in allergic inflammation, including T lymphocytes (84), mast cell (85), macrophages (86–88), and smooth muscle cells (89). Our data (unpublished) also suggest that KCa3.1 activity is associated with migration and antigen uptake of lung immunogenic DC subset. The application of a low-toxic KCa3.1 specific blocker TRAM-34 in a mouse model has been proved to be effective and safe in preventing formation of atherosclerosis in a mouse model (88).
The novel immunomodulators, Flt3-ligand (Flt3-L) suppresses Th2 responses in a mouse model of allergic asthma. The therapeutic effect of Flt3-L is achieved through facilitating the generation of regulatory CD11chighCD11blow lung DC subset, impairing migration of immunogenic CD11clowCD11bhigh DC subset to draining lymph node and antigen uptake in immunogenic CD11clowCD11bhigh DC subset (9, 90–92), and increasing the density of CD4+CD25+Foxp3+ICOS+ T-regulatory cells (93) in the lungs of asthmatic mice. Thus, Flt3-L may prove to be a novel adjuvant therapy in bronchial asthma. Likewise, compounds that can balance the function of dendritic cells and increase the activity of Foxp3-expressing Tregs have been under development. Local administration of the sphingosine 1-phosphate receptor agonist, FTY720, to lung attenuates experimental asthma by inhibiting lung DC function (94), but a recent study has indicated that FTY720 potentially inhibits T regulatory cells proliferation as well (95). Early study showed that prostaglandin D2 impaired the activation, differentiation, and migration of human dendritic cells (96). A recent study reveals that the activation of the D prostanoid 1 receptor by a selective agonist effectively suppresses asthma through regulating DC function and the generation of T regulatory cell (97). Obviously, more careful studies would confirm/refute their potential therapeutic effects in allergic airway inflammation and asthma.
Conclusions
Asthma is a multifactorial disease characterized by chronic airway inflammation and increased bronchoconstrictory response to nonspecific stimuli. Although there is a propensity for type 2 cytokines in asthmatic lungs, there is evidence suggesting the predominant role of local immune events in allergic airway inflammation. Allergen-specific DCs and T-regulatory cells appear to play a pivotal role in balancing tolerance versus immunologic response to allergens. Therapy with immunomodulators that do not elicit autoimmune response but enhance tolerance to allergen and increase T-regulatory cells would be most effective in the treatment of allergy and asthma.
Acknowledgments
This work was supported by the National Institute of Health Grants HL070885, AI075315, and HL086680.
Reference and Recommended Reading
Papers of particular interest, published recently, have been highlighted as:
• of importance
•• of major importance
- 1.Broide DH. Molecular and cellular mechanisms of allergic disease. J Allergy Clin Immunol. 2001;108:S65–71. doi: 10.1067/mai.2001.116436. [DOI] [PubMed] [Google Scholar]
- 2.Vignola AM, Kips J, Bousquet J. Tissue remodeling as a feature of persistent asthma. J Allergy Clin Immunol. 2000;105:1041–1053. doi: 10.1067/mai.2000.107195. [DOI] [PubMed] [Google Scholar]
- 3.Akdis M, Trautmann A, Klunker SI, et al. T helper (Th) 2 predominance in atopic diseases is due to preferential apoptosis of circulating memory/effector Th1 cells. Faseb J. 2003;17:1026–1035. doi: 10.1096/fj.02-1070com. [DOI] [PubMed] [Google Scholar]
- 4•.Akkoc T, de Koning PJ, Ruckert B, et al. Increased activation-induced cell death of high IFN-gamma-producing T(H)1 cells as a mechanism of T(H)2 predominance in atopic diseases. J Allergy Clin Immunol. 2008;121:652–658. e651. doi: 10.1016/j.jaci.2007.12.1171. This study reveals one of the mechanisms underlying the counteractive effect of Th1 and Th2 responses. [DOI] [PubMed] [Google Scholar]
- 5•.Louten J, Boniface K, de Waal Malefyt R. Development and function of TH17 cells in health and disease. J Allergy Clin Immunol. 2009;123:1004–1011. doi: 10.1016/j.jaci.2009.04.003. This article comprehensively reviews the available information for Th17 cells. [DOI] [PubMed] [Google Scholar]
- 6•.Veldhoen M, Uyttenhove C, van Snick J, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9:1341–1346. doi: 10.1038/ni.1659. This study demonstrates the pivotal role of TGF-β in the induction of Th9 cells. [DOI] [PubMed] [Google Scholar]
- 7.Feili-Hariri M, Falkner DH, Morel PA. Polarization of naive T cells into Th1 or Th2 by distinct cytokine-driven murine dendritic cell populations: implications for immunotherapy. J Leukoc Biol. 2005;78:656–664. doi: 10.1189/jlb.1104631. [DOI] [PubMed] [Google Scholar]
- 8.Gordon JR, Li F, Nayyar A, et al. CD8 alpha+, but not CD8 alpha−, dendritic cells tolerize Th2 responses via contact-dependent and -independent mechanisms, and reverse airway hyperresponsiveness, Th2, and eosinophil responses in a mouse model of asthma. J Immunol. 2005;175:1516–1522. doi: 10.4049/jimmunol.175.3.1516. [DOI] [PubMed] [Google Scholar]
- 9••.Shao Z, Bharadwaj AS, McGee HS, et al. Fms-like tyrosine kinase 3 ligand increases a lung DC subset with regulatory properties in allergic airway inflammation. J Allergy Clin Immunol. 2009;123:917–924. e912. doi: 10.1016/j.jaci.2009.01.052. This study reveals the mechanisms of Flt3-L-induced therapeutic effect in allergic airway inflammation and the presence of the functional distinct lung DC subsets in the mouse lung. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lighvani AA, Frucht DM, Jankovic D, et al. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci U S A. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furuta S, Kagami S, Tamachi T, et al. Overlapping and distinct roles of STAT4 and T-bet in the regulation of T cell differentiation and allergic airway inflammation. J Immunol. 2008;180:6656–6662. doi: 10.4049/jimmunol.180.10.6656. [DOI] [PubMed] [Google Scholar]
- 12.Djuretic IM, Levanon D, Negreanu V, et al. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. 2007;8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 13.Zhu J, Guo L, Watson CJ, et al. Stat6 is necessary and sufficient for IL-4’s role in Th2 differentiation and cell expansion. J Immunol. 2001;166:7276–7281. doi: 10.4049/jimmunol.166.12.7276. [DOI] [PubMed] [Google Scholar]
- 14.Scheinman EJ, Avni O. Transcriptional regulation of GATA3 in T helper cells by the integrated activities of transcription factors downstream of the interleukin-4 receptor and T cell receptor. J Biol Chem. 2009;284:3037–3048. doi: 10.1074/jbc.M807302200. [DOI] [PubMed] [Google Scholar]
- 15.Tamachi T, Takatori H, Fujiwara M, et al. STAT6 inhibits T-bet-independent Th1 cell differentiation. Biochem Biophys Res Commun. 2009;382:751–755. doi: 10.1016/j.bbrc.2009.03.101. [DOI] [PubMed] [Google Scholar]
- 16.Jenner RG, Townsend MJ, Jackson I, et al. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc Natl Acad Sci U S A. 2009;106:17876–17881. doi: 10.1073/pnas.0909357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang XO, Pappu BP, Nurieva R, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakano H, Yanagita M, Gunn MD. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J Exp Med. 2001;194:1171–1178. doi: 10.1084/jem.194.8.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Heer HJ, Hammad H, Soullie T, et al. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med. 2004;200:89–98. doi: 10.1084/jem.20040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koya T, Matsuda H, Takeda K, et al. IL-10-treated dendritic cells decrease airway hyperresponsiveness and airway inflammation in mice. J Allergy Clin Immunol. 2007;119:1241–1250. doi: 10.1016/j.jaci.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 21.Sung SS, Fu SM, Rose CE, Jr, et al. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol. 2006;176:2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 22.Dunne PJ, Moran B, Cummins RC, Mills KH. CD11c+CD8{alpha}+ Dendritic Cells Promote Protective Immunity to Respiratory Infection with Bordetella pertussis. J Immunol. 2009;183:400–410. doi: 10.4049/jimmunol.0900169. [DOI] [PubMed] [Google Scholar]
- 23•.Shao Z, Makinde TO, McGee HS, et al. Flt3 Ligand Regulates Migratory pattern and antigen uptake of lung dendritic cell subsets in a murine model of allergic airway inflammation. J Immunol. 2009;183(11):00–00. doi: 10.4049/jimmunol.0901341. (In press). This study further reveals the mechanisms underlying Flt3 ligand-mediated therapeutic effect in allergic asthma also involves regulating lung DC migration and antigen uptake. [DOI] [PubMed] [Google Scholar]
- 24.Wu L, Liu YJ. Development of dendritic-cell lineages. Immunity. 2007;26:741–750. doi: 10.1016/j.immuni.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 25.Demedts IK, Brusselle GG, Vermaelen KY, Pauwels RA. Identification and characterization of human pulmonary dendritic cells. Am J Respir Cell Mol Biol. 2005;32:177–184. doi: 10.1165/rcmb.2004-0279OC. [DOI] [PubMed] [Google Scholar]
- 26.Masten BJ, Olson GK, Tarleton CA, et al. Characterization of myeloid and plasmacytoid dendritic cells in human lung. J Immunol. 2006;177:7784–7793. doi: 10.4049/jimmunol.177.11.7784. [DOI] [PubMed] [Google Scholar]
- 27•.Schaumann F, Muller M, Braun A, et al. Endotoxin augments myeloid dendritic cell influx into the airways in patients with allergic asthma. Am J Respir Crit Care Med. 2008;177:1307–1313. doi: 10.1164/rccm.200706-870OC. Studies in reference 26 and 27 confirm the presence of dendritic cell subsets with similar phenotype to blood DCs in the lungs. Functionally, they are actively involved in the development of allergic airway inflammation in humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bratke K, Lommatzsch M, Julius P, et al. Dendritic cell subsets in human bronchoalveolar lavage fluid after segmental allergen challenge. Thorax. 2007;62:168–175. doi: 10.1136/thx.2006.067793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009;30:616–625. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burchill MA, Yang J, Vogtenhuber C, et al. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 31.Shevach EM, Tran DQ, Davidson TS, Andersson J. The critical contribution of TGF-beta to the induction of Foxp3 expression and regulatory T cell function. Eur J Immunol. 2008;38:915–917. doi: 10.1002/eji.200738111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng SG, Wang JH, Stohl W, et al. TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J Immunol. 2006;176:3321–3329. doi: 10.4049/jimmunol.176.6.3321. [DOI] [PubMed] [Google Scholar]
- 33.Kearley J, Robinson DS, Lloyd CM. CD4+CD25+ regulatory T cells reverse established allergic airway inflammation and prevent airway remodeling. J Allergy Clin Immunol. 2008;122:617–624. e616. doi: 10.1016/j.jaci.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34•.McGee HS, Agrawal DK. Naturally occurring and inducible T-regulatory cells modulating immune response in allergic asthma. Am J Respir Crit Care Med. 2009;180:211–225. doi: 10.1164/rccm.200809-1505OC. This study demonstrated the localization of inducible T regulatory cells to the asthmatic lungs by adoptive transfer of GFP-expressing inducible T regulatory cells intravenously. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stock P, Kallinich T, Akbari O, et al. CD8(+) T cells regulate immune responses in a murine model of allergen-induced sensitization and airway inflammation. Eur J Immunol. 2004;34:1817–1827. doi: 10.1002/eji.200324623. [DOI] [PubMed] [Google Scholar]
- 36.Wells JW, Cowled CJ, Giorgini A, et al. Regulation of allergic airway inflammation by class I-restricted allergen presentation and CD8 T-cell infiltration. J Allergy Clin Immunol. 2007;119:226–234. doi: 10.1016/j.jaci.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 37.Takeda K, Dow SW, Miyahara N, et al. Vaccine-induced CD8+ T cell-dependent suppression of airway hyperresponsiveness and inflammation. J Immunol. 2009;183:181–190. doi: 10.4049/jimmunol.0803967. [DOI] [PubMed] [Google Scholar]
- 38.Isogai S, Athiviraham A, Fraser RS, et al. Interferon-gamma-dependent inhibition of late allergic airway responses and eosinophilia by CD8+ gammadelta T cells. Immunology. 2007;122:230–238. doi: 10.1111/j.1365-2567.2007.02632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koya T, Matsuda H, Matsubara S, et al. Differential effects of dendritic cell transfer on airway hyperresponsiveness and inflammation. Am J Respir Cell Mol Biol. 2009;41:271–280. doi: 10.1165/rcmb.2008-0256OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsuchiya K, Isogai S, Tamaoka M, et al. Depletion of CD8+ T cells enhances airway remodelling in a rodent model of asthma. Immunology. 2009;126:45–54. doi: 10.1111/j.1365-2567.2008.02876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho SH, Stanciu LA, Holgate ST, Johnston SL. Increased interleukin-4, interleukin-5, and interferon-gamma in airway CD4+ and CD8+ T cells in atopic asthma. Am J Respir Crit Care Med. 2005;171:224–230. doi: 10.1164/rccm.200310-1416OC. [DOI] [PubMed] [Google Scholar]
- 42.Weninger W, Crowley MA, Manjunath N, von Andrian UH. Migratory properties of naive, effector, and memory CD8(+) T cells. J Exp Med. 2001;194:953–966. doi: 10.1084/jem.194.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taube C, Miyahara N, Ott V, et al. The leukotriene B4 receptor (BLT1) is required for effector CD8+ T cell-mediated, mast cell-dependent airway hyperresponsiveness. J Immunol. 2006;176:3157–3164. doi: 10.4049/jimmunol.176.5.3157. [DOI] [PubMed] [Google Scholar]
- 44.Koya T, Miyahara N, Takeda K, et al. CD8+ T cell-mediated airway hyperresponsiveness and inflammation is dependent on CD4+IL-4+ T cells. J Immunol. 2007;179:2787–2796. doi: 10.4049/jimmunol.179.5.2787. [DOI] [PubMed] [Google Scholar]
- 45.Manavalan JS, Kim-Schulze S, Scotto L, et al. Alloantigen specific CD8+CD28- FOXP3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. Int Immunol. 2004;16:1055–1068. doi: 10.1093/intimm/dxh107. [DOI] [PubMed] [Google Scholar]
- 46.Siegmund K, Ruckert B, Ouaked N, et al. Unique phenotype of human tonsillar and in vitro-induced FOXP3+CD8+ T cells. J Immunol. 2009;182:2124–2130. doi: 10.4049/jimmunol.0802271. [DOI] [PubMed] [Google Scholar]
- 47.Xystrakis E, Dejean AS, Bernard I, et al. Identification of a novel natural regulatory CD8 T-cell subset and analysis of its mechanism of regulation. Blood. 2004;104:3294–3301. doi: 10.1182/blood-2004-03-1214. [DOI] [PubMed] [Google Scholar]
- 48.Gilliet M, Liu YJ. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J Exp Med. 2002;195:695–704. doi: 10.1084/jem.20011603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maisi P, Sorsa T, Raulo SM, et al. Increased matrix metalloproteinase (MMP)-9 in the airway after allergen challenge. Am J Respir Crit Care Med. 2001;164:1740. doi: 10.1164/ajrccm.164.9.correspondence_b. discussion 1740–1741. [DOI] [PubMed] [Google Scholar]
- 50.Lim DH, Cho JY, Miller M, et al. Reduced peribronchial fibrosis in allergen-challenged MMP-9-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2006;291:L265–271. doi: 10.1152/ajplung.00305.2005. [DOI] [PubMed] [Google Scholar]
- 51.McMillan SJ, Kearley J, Campbell JD, et al. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J Immunol. 2004;172:2586–2594. doi: 10.4049/jimmunol.172.4.2586. [DOI] [PubMed] [Google Scholar]
- 52.Corry DB, Rishi K, Kanellis J, et al. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Page K, Ledford JR, Zhou P, Wills-Karp M. A TLR2 agonist in German cockroach frass activates MMP-9 release and is protective against allergic inflammation in mice. J Immunol. 2009;183:3400–3408. doi: 10.4049/jimmunol.0900838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warner RL, Lukacs NW, Shapiro SD, et al. Role of metalloelastase in a model of allergic lung responses induced by cockroach allergen. Am J Pathol. 2004;165:1921–1930. doi: 10.1016/S0002-9440(10)63244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goswami S, Angkasekwinai P, Shan M, et al. Divergent functions for airway epithelial matrix metalloproteinase 7 and retinoic acid in experimental asthma. Nat Immunol. 2009;10:496–503. doi: 10.1038/ni.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sands MF, Ohtake PJ, Mahajan SD, et al. Tissue inhibitor of metalloproteinase-1 modulates allergic lung inflammation in murine asthma. Clin Immunol. 2009;130:186–198. doi: 10.1016/j.clim.2008.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McIntire JJ, Umetsu DT, DeKruyff RH. TIM-1, a novel allergy and asthma susceptibility gene. Springer Semin Immunopathol. 2004;25:335–348. doi: 10.1007/s00281-003-0141-3. [DOI] [PubMed] [Google Scholar]
- 58.Van Eerdewegh P, Little RD, Dupuis J, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418:426–430. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- 59.Sizing ID, Bailly V, McCoon P, et al. Epitope-dependent effect of anti-murine TIM-1 monoclonal antibodies on T cell activity and lung immune responses. J Immunol. 2007;178:2249–2261. doi: 10.4049/jimmunol.178.4.2249. [DOI] [PubMed] [Google Scholar]
- 60.Jongepier H, Boezen HM, Dijkstra A, et al. Polymorphisms of the ADAM33 gene are associated with accelerated lung function decline in asthma. Clin Exp Allergy. 2004;34:757–760. doi: 10.1111/j.1365-2222.2004.1938.x. [DOI] [PubMed] [Google Scholar]
- 61.Laitinen T, Polvi A, Rydman P, et al. Characterization of a common susceptibility locus for asthma-related traits. Science. 2004;304:300–304. doi: 10.1126/science.1090010. [DOI] [PubMed] [Google Scholar]
- 62•.Tsai YJ, Choudhry S, Kho J, et al. The PTGDR gene is not associated with asthma in 3 ethnically diverse populations. J Allergy Clin Immunol. 2006;118:1242–1248. doi: 10.1016/j.jaci.2006.07.045. This study suggests that asthma-susceptibility genes have to be assessed in the context of a particular ethnic group. [DOI] [PubMed] [Google Scholar]
- 63.Allen M, Heinzmann A, Noguchi E, et al. Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat Genet. 2003;35:258–263. doi: 10.1038/ng1256. [DOI] [PubMed] [Google Scholar]
- 64.Himes BE, Hunninghake GM, Baurley JW, et al. Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am J Hum Genet. 2009;84:581–593. doi: 10.1016/j.ajhg.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Galanter J, Choudhry S, Eng C, et al. ORMDL3 gene is associated with asthma in three ethnically diverse populations. Am J Respir Crit Care Med. 2008;177:1194–1200. doi: 10.1164/rccm.200711-1644OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White JH, Chiano M, Wigglesworth M, et al. Identification of a novel asthma susceptibility gene on chromosome 1qter and its functional evaluation. Hum Mol Genet. 2008;17:1890–1903. doi: 10.1093/hmg/ddn087. [DOI] [PubMed] [Google Scholar]
- 67.Hataji O, Taguchi O, Gabazza EC, et al. Activation of protein C pathway in the airways. Lung. 2002;180:47–59. doi: 10.1007/s004080000080. [DOI] [PubMed] [Google Scholar]
- 68.Wagers SS, Norton RJ, Rinaldi LM, et al. Extravascular fibrin, plasminogen activator, plasminogen activator inhibitors, and airway hyperresponsiveness. J Clin Invest. 2004;114:104–111. doi: 10.1172/JCI19569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tanaka A, Minoguchi K, Chen X, et al. Activated protein C attenuates leukocyte elastase-induced lung injury in mice. Shock. 2008;30:153–158. doi: 10.1097/SHK.0b013e31815dd570. [DOI] [PubMed] [Google Scholar]
- 70.Cohn L, Herrick C, Niu N, et al. IL-4 promotes airway eosinophilia by suppressing IFN-gamma production: defining a novel role for IFN-gamma in the regulation of allergic airway inflammation. J Immunol. 2001;166:2760–2767. doi: 10.4049/jimmunol.166.4.2760. [DOI] [PubMed] [Google Scholar]
- 71.Huang TJ, MacAry PA, Eynott P, et al. Allergen-specific Th1 cells counteract efferent Th2 cell-dependent bronchial hyperresponsiveness and eosinophilic inflammation partly via IFN-gamma. J Immunol. 2001;166:207–217. doi: 10.4049/jimmunol.166.1.207. [DOI] [PubMed] [Google Scholar]
- 72.Bryan SA, O’Connor BJ, Matti S, et al. Effects of recombinant human interleukin-12 on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2149–2153. doi: 10.1016/S0140-6736(00)03497-8. [DOI] [PubMed] [Google Scholar]
- 73.Wild JS, Sigounas A, Sur N, et al. IFN-gamma-inducing factor (IL-18) increases allergic sensitization, serum IgE, Th2 cytokines, and airway eosinophilia in a mouse model of allergic asthma. J Immunol. 2000;164:2701–2710. doi: 10.4049/jimmunol.164.5.2701. [DOI] [PubMed] [Google Scholar]
- 74.Ahrens B, Gruber C, Rha RD, et al. BCG priming of dendritic cells enhances T regulatory and Th1 function and suppresses allergen-induced Th2 function in vitro and in vivo. Int Arch Allergy Immunol. 2009;150:210–220. doi: 10.1159/000222673. [DOI] [PubMed] [Google Scholar]
- 75.Kline JN. Immunotherapy of asthma using CpG oligodeoxynucleotides. Immunol Res. 2007;39:279–286. doi: 10.1007/s12026-007-0083-2. [DOI] [PubMed] [Google Scholar]
- 76.Hansen G, Berry G, DeKruyff RH, Umetsu DT. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J Clin Invest. 1999;103:175–183. doi: 10.1172/JCI5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Randolph DA, Stephens R, Carruthers CJ, Chaplin DD. Cooperation between Th1 and Th2 cells in a murine model of eosinophilic airway inflammation. J Clin Invest. 1999;104:1021–1029. doi: 10.1172/JCI7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stephens R, Eisenbarth SC, Chaplin DD. T helper type 1 cells in asthma: friend or foe? Curr Opin Allergy Clin Immunol. 2002;2:31–37. doi: 10.1097/00130832-200202000-00006. [DOI] [PubMed] [Google Scholar]
- 79.Cui J, Pazdziorko S, Miyashiro JS, et al. TH1-mediated airway hyperresponsiveness independent of neutrophilic inflammation. J Allergy Clin Immunol. 2005;115:309–315. doi: 10.1016/j.jaci.2004.10.046. [DOI] [PubMed] [Google Scholar]
- 80.Sugimoto T, Ishikawa Y, Yoshimoto T, et al. Interleukin 18 acts on memory T helper cells type 1 to induce airway inflammation and hyperresponsiveness in a naive host mouse. J Exp Med. 2004;199:535–545. doi: 10.1084/jem.20031368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81•.Hayashi N, Yoshimoto T, Izuhara K, et al. T helper 1 cells stimulated with ovalbumin and IL-18 induce airway hyperresponsiveness and lung fibrosis by IFN-gamma and IL-13 production. Proc Natl Acad Sci U S A. 2007;104:14765–14770. doi: 10.1073/pnas.0706378104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar RK, Webb DC, Herbert C, Foster PS. Interferon-gamma as a possible target in chronic asthma. Inflamm Allergy Drug Targets. 2006;5:253–256. doi: 10.2174/187152806779010909. [DOI] [PubMed] [Google Scholar]
- 83.Hayashi T, Beck L, Rossetto C, et al. Inhibition of experimental asthma by indoleamine 2,3-dioxygenase. J Clin Invest. 2004;114:270–279. doi: 10.1172/JCI21275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol Rev. 2009;231:59–87. doi: 10.1111/j.1600-065X.2009.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cruse G, Duffy SM, Brightling CE, Bradding P. Functional KCa3.1 K+ channels are required for human lung mast cell migration. Thorax. 2006;61:880–885. doi: 10.1136/thx.2006.060319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grgic I, Wulff H, Eichler I, et al. Blockade of T-lymphocyte KCa3.1 and Kv1.3 channels as novel immunosuppression strategy to prevent kidney allograft rejection. Transplant Proc. 2009;41:2601–2606. doi: 10.1016/j.transproceed.2009.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tharp DL, Bowles DK. The intermediate-conductance Ca2+-activated K+ channel (KCa3.1) in vascular disease. Cardiovasc Hematol Agents Med Chem. 2009;7:1–11. doi: 10.2174/187152509787047649. [DOI] [PubMed] [Google Scholar]
- 88•.Toyama K, Wulff H, Chandy KG, et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest. 2008;118:3025–3037. doi: 10.1172/JCI30836. This study carefullytested the effect and toxicity of KCa3.1 specific blocker TRAM-34 in a murine model and provides solid pre-clinical evidence for the application of TRAM-34 in treating diseases related to KCa3.1 activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shepherd MC, Duffy SM, Harris T, et al. KCa3.1 Ca2+ activated K+ channels regulate human airway smooth muscle proliferation. Am J Respir Cell Mol Biol. 2007;37:525–531. doi: 10.1165/rcmb.2006-0358OC. [DOI] [PubMed] [Google Scholar]
- 90.Edwan JH, Perry G, Talmadge JE, Agrawal DK. Flt-3 ligand reverses late allergic response and airway hyper-responsiveness in a mouse model of allergic inflammation. J Immunol. 2004;172:5016–5023. doi: 10.4049/jimmunol.172.8.5016. [DOI] [PubMed] [Google Scholar]
- 91.Edwan JH, Agrawal DK. Flt3-ligand plasmid prevents the development of pathophysiological features of chronic asthma in a mouse model. Immunol Res. 2007;37:147–159. doi: 10.1007/BF02685896. [DOI] [PubMed] [Google Scholar]
- 92.Bharadwaj AS, Agrawal DK. Flt3 ligand generates morphologically distinct semimature dendritic cells in ovalbumin-sensitized mice. Exp Mol Pathol. 2007;83:17–24. doi: 10.1016/j.yexmp.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McGee HS, Edwan JH, Agrawal DK. Flt3-L Increases CD4+CD25+Foxp3+ICOS+ Cells in the Lung of Cockroach-sensitized and Challenged Mice. Am J Respir Cell Mol Biol. 2009 doi: 10.1165/rcmb.2008-0397OC. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94•.Idzko M, Hammad H, van Nimwegen M, et al. Local application of FTY720 to the lung abrogates experimental asthma by altering dendritic cell function. J Clin Invest. 2006;116:2935–2944. doi: 10.1172/JCI28295. This study is the first attempt to target regulating DC function using a chemical compound in the treatment of asthma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wolf AM, Eller K, Zeiser R, et al. The sphingosine 1-phosphate receptor agonist FTY720 potently inhibits regulatory T cell proliferation in vitro and in vivo. J Immunol. 2009;183:3751–3760. doi: 10.4049/jimmunol.0901011. [DOI] [PubMed] [Google Scholar]
- 96.Gosset P, Pichavant M, Faveeuw C, et al. Prostaglandin D2 affects the differentiation and functions of human dendritic cells: impact on the T cell response. Eur J Immunol. 2005;35:1491–1500. doi: 10.1002/eji.200425319. [DOI] [PubMed] [Google Scholar]
- 97.Hammad H, Kool M, Soullie T, et al. Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. J Exp Med. 2007;204:357–367. doi: 10.1084/jem.20061196. [DOI] [PMC free article] [PubMed] [Google Scholar]