

Abstract
Squamous cell carcinomas (SCC) are sun-induced skin cancers that are particularly numerous and aggressive in patients taking T cell immunosuppressant medications. Imiquimod is a topical immune response modifier and TLR7 agonist that induces the immunologic destruction of SCC and other skin cancers. TLR7 activation by imiquimod has pleiotropic effects on innate immune cells but its effects on T cells remain largely uncharacterized. Because tumor destruction and formation of immunologic memory are ultimately T cell mediated effects, we studied the effects of imiquimod therapy on effector T cells infiltrating human SCC. SCC treated with imiquimod prior to excision contained dense T cell infiltrates associated with tumor cell apoptosis and histologic evidence of tumor regression. Effector T cells from treated SCC produced more IFN-γ, granzyme and perforin and less IL-10 and TGF-β than T cells from untreated tumors. Treatment of normal human skin with imiquimod induced activation of resident T cells and reduced IL-10 production but had no effect on IFN-γ, perforin or granzyme, suggesting that these latter effects arise from recruitment of distinct populations of T cells into tumors. Thus, imiquimod stimulates tumor destruction by recruiting cutaneous effector T cells from blood and by inhibiting tonic anti-inflammatory signals within the tumor.
INTRODUCTION
Over 100,000 squamous cell carcinomas (SCC) of the skin are diagnosed in the United States each year (Diepgen and Mahler, 2002). The treatment of non-melanoma skin cancer, of which SCC is the second most frequent type, accounts for 4.5% of all Medicare cancer costs (Housman et al., 2003). Actinic keratoses (AK), the premalignant precursors to SCC, are the third most common reason for consulting a dermatologist (Feldman et al., 1998). Over 5.2 million physician visits are made annually for the treatment of actinic keratoses, at a cost of over $900 million (Warino et al., 2006).
Recipients of solid organ transplants on immunosuppressive medications have a 65- to 250-fold increased risk of SCC (Berg and Otley, 2002). SCC in these patients are aggressive; nearly 10% of these tumors metastasize and the majority of patients die as a result (Berg and Otley, 2002; Euvrard et al., 2003). The development of SCC in transplant recipients is linked to the use of medications that suppress T cell function (Euvrard et al., 2003).
We have found that blood vessels in human SCC vessels lack E-selectin, allowing the tumor to exclude skin-homing effector T cells, while at the same time recruiting regulatory T cells (Treg), a cell type that can suppress immune responses (Clark et al., 2008; Sakaguchi et al., 1995). We found that the TLR7 agonist imiquimod acted indirectly to address both of these mechanisms of immune evasion, inducing E-selectin on blood vessels, allowing the influx of skin homing T cells and reducing the number and activity of tumor Treg (Clark et al., 2008).
Effector T cells are critical for both the destruction of SCC and the establishment of long-term, protective immunologic memory. Despite their importance, comprehensive studies of the phenotype and function of these cells in SCC are lacking. Similarly, imiquimod has been shown to induce IFN-γ production in vitro from mouse and human T cells but the effect of this medication on tumor resident T cells has not been studied thoroughly (Wagner et al., 1999). We present here our findings that imiquimod therapy significantly enhances the number and effector function of T cells infiltrating human SCC of the skin.
RESULTS
Imiquimod treated SCC are infiltrated by CD8+ T cells that are associated with tumor cell apoptosis and histologic evidence of tumor regression
We have previously reported that untreated SCC are infiltrated by equal numbers of CD4 and CD8 T cells whereas tumors treated with imiquimod prior to excision contain >90% CD8 cytotoxic T cells (Clark et al., 2008). To study the distribution and characteristics of these cells, we co-stained cryosections of imiquimod treated SCC for CD8 and for markers of tumor cell proliferation and apoptosis (Figure 1). We found that imiquimod treated SCC had three clear histologic zones. First, an area of residual tumor cells, many of which were proliferating, was evident superficially (Figure 1a,b). A dense, band-like infiltrate of CD8 T cells was present beneath the tumor and a zone of fibrotic regression was present beneath the T cell infiltrates. The zone of fibrotic regression was always present at the base of the specimen and its width varied with the amount of residual tumor; tumors that had almost fully regressed had the thickest zones of fibrosis and those in the earlier stages of destruction had fibrosis limited to the deeper portions of the dermis. In all specimens, fibrosis was uniform across the base, suggesting that fibrosis was not the result of scarring from a prior biopsy procedure. Dense infiltrates of CD8 T cells surrounded nests of residual tumor, and their presence was associated with tumor cell apoptosis as evidenced by TUNEL staining (Figure 1d). Untreated SCC were infiltrated by fewer CD8 T cells and lacked both fibrotic regression and tumor cell apoptosis (data not shown).
Figure 1. Imiquimod treated SCC are heavily infiltrated by CD8 T cells that are associated with tumor regression and apoptosis of tumor cells.
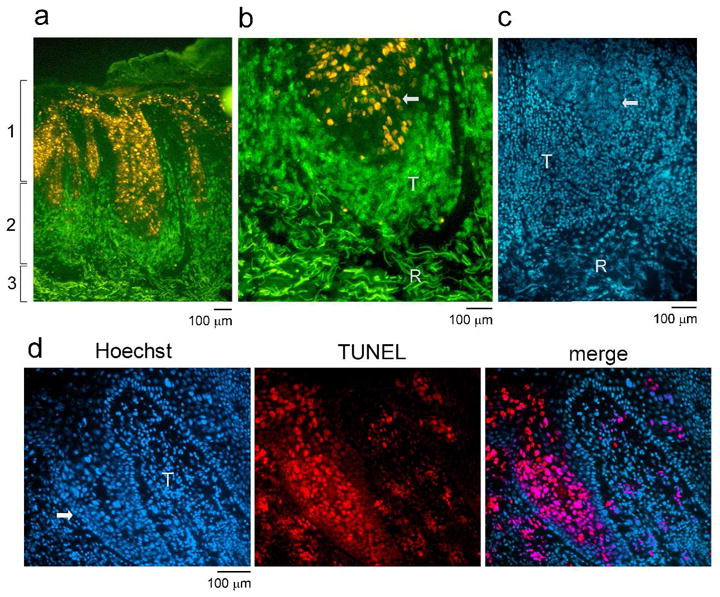
(a) Cryosections of SCC treated with imiquimod were stained for CD8 (green) and Ki-67 (to identify proliferating tumor cells). Treated SCC had three clear histologic zones: (1) residual tumor, (2) a dense CD8 T cell infiltrate and (3) a deeper fibrotic zone of tumor regression. (b) A higher power view is shown of the interface between the zones of T cell infiltration and tumor regression. Residual tumor (arrow) is surrounded by a dense T cell infiltrate (T) beneath which is a zone of fibrotic regression (R). (c) An additional imiquimod treated tumor stained with Hoechst nuclear dye is shown. Residual tumor (arrow) can be identified by the large atypical keratinocyte nuclei. A zone of dense T cell infiltrate (T) and an area of fibrotic regression (R) are also evident. (d) CD8 T cells infiltrates are associated with tumor cell apoptosis. Hoescht nuclear stain delineates a tumor nodule (arrow) surrounded by T cells (T). TUNEL staining demonstrates widespread tumor cell apoptosis in areas of dense T cell infiltrates. Three additional treated tumors showed similar results.
Production of IFN-γ, perforin and granzyme is markedly enhanced in T cells from imiquimod treated SCC
We isolated the T cells from untreated and imiquimod treated SCC and examined the production of cytokines and proteins associated with cytotoxic activity. Approximately 50% of T cells infiltrating untreated SCC are FOXP3+ Treg (Clark et al., 2008). For these studies, FOXP3 co-staining was used to exclude Treg from these analyses. Non-regulatory CD4 and CD8 T cells from imiquimod treated SCC produced markedly elevated levels of IFN-γ (Figure 2a,b). IFN-γ was produced by 69% of CD4 T cells (SD=3.1, N=3) and 79% of CD8 T cells (SD= 7.4, N=3) from treated tumors, compared with 8.2% of CD4 (SD=1.4, N=3) and 21% of CD8 T cells (SD=13, N=3) from untreated tumors. Few T cells from either treated or untreated SCC produced IL-17 (Figure 2a,b). The percentage of T cells producing granzyme and perforin, two molecules associated with T cell cytotoxicity, were also increased in imiquimod treated SCC (Figure 2c–f). 46% of CD4 T cells from imiquimod treated SCC produced granzyme (SD=11, N=3) and 43% produced perforin (SD=5.1, N=3). Of CD8 T cells from treated SCC, 81% produced granzyme (SD=6.1, N=3) and 73% produced perforin (SD=9.4, N=3).
Figure 2. Production of IFN-γ, perforin and granzyme is markedly enhanced in T cells from SCC treated with imiquimod.

IFN-γ is produced by the majority of both (a) CD4+ and (b) CD8+ T cells in SCC treated with imiquimod, compared with lower production in untreated tumors. IL-17 production was low in both treated and untreated tumors. (c) CD4 and CD8 T cells from SCC had increased co-expression of perforin after imiquimod therapy; CD4 T cells also showed enhanced granzyme production. Study of additional treated tumors confirmed enhanced production of perforin by both (d) CD4 and (e) CD8 T cells and increased production of granzyme by (d) CD4 T cells.
CD8 T cells do not proliferate locally within imiquimod treated tumors
We have observed a shift in the CD4:CD8 T cell ratio in untreated tumors from 1:1 in untreated tumors to 1:10 in imiquimod treated tumors and this shift is associated with a 3-fold increase in the total number of T cells infiltrating tumors (Clark et al., 2008). Increased numbers of CD8 T cells could result from either an influx of T cells from the blood or from local expansion of tumor-specific T cells already present within the tumor. To determine if CD8 T cells proliferate locally within imiquimod treated SCC, we co-stained cryosections of treated SCC with CD8 and the proliferation antigen Ki-67 (Figure 3). Although proliferating cells were readily detectable in stained sections, these were almost invariably tumor cells. Less than 5% of the CD8 T cells present in imiquimod treated SCC had recently proliferated, suggesting that the increased number of CD8 cells results from enhanced recruitment from the blood (Figure 3c).
Figure 3. CD8 T cells do not proliferate locally within imiquimod treated tumors.
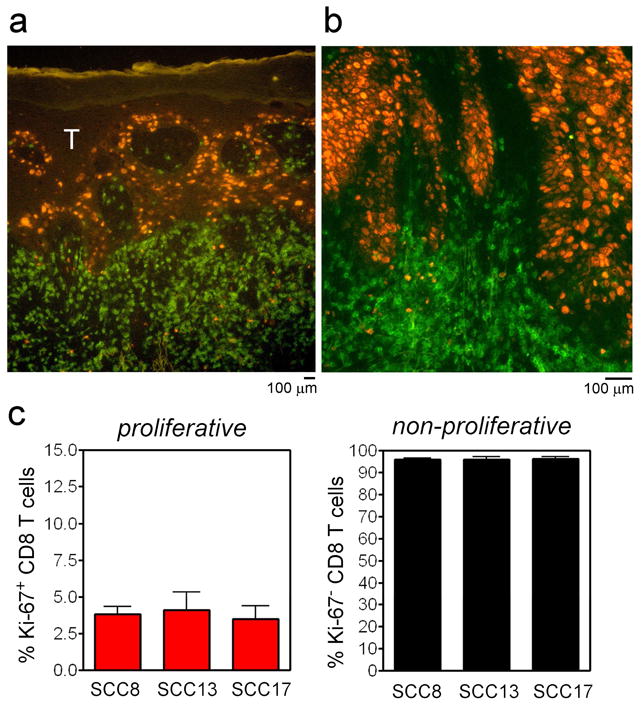
(a) Co-staining of cryosections of imiquimod treated SCC with CD8 and Ki-67 demonstrated proliferation of tumor cells but few CD8 T cells. (b) Higher power view of a second tumor with similar findings. (c) Quantification of CD8 T cell proliferation in three treated tumors.
Non-regulatory T cells from imiquimod treated SCC produce less IL-10 and TGF-β
We reported previously that imiquimod treatment is associated with decreased production of IL-10 and TGF-β by FOXP3+ Treg present in SCC (Clark et al., 2008). We studied the production of IL-10 and TGF-β by non-regulatory T cells and found that in untreated SCC, significant amounts of IL-10 were produced by both FOXP3+ regulatory and FOXP3− non-regulatory T cells (Figure 4a). Non-regulatory T cells from tumors treated with imiquimod produced less IL-10 (Figure 4b-d, mean 43%, SD=2.1, N=3 from untreated tumors vs. mean 20%, SD=4.1, N=3 from treated tumors). TGF-β was also produced by a mean 12% of non-regulatory T cells in untreated SCC and production was decreased to 2% in imiquimod treated tumors (Figure 4e, SD=1.5, N=3 from untreated tumors and SD=1.1, N=3 from treated tumors). These results demonstrate that non-regulatory T cells are also a significant source of immunosuppressive cytokines in SCC and that production of these cytokines is decreased by treatment with imiquimod.
Figure 4. Effector T cells from imiquimod treated SCC produce less IL-10 and TGF-β.

(a) Non-regulatory T cells in untreated SCC are a significant source of IL-10. IL-10 is also produced by tumor FOXP3+ Treg (Treg). (b,c) IL-10 production by non-regulatory T cells is reduced in tumors treated with imiquimod. (d) Summarized data from treated and untreated tumors. (e) TGF-β production by non-regulatory T cells is also reduced after imiquimod therapy.
IL-10 has been reported to be produced by SCC tumor cells (Kim et al., 1995). We immunostained cryosections of SCC and did observe detectable IL-10 production by tumor cells (Figure 5a). Also, intracellular cytokine analysis of the SCC cell line SCC13 demonstrated IL-10 production (Figure 5b).
Figure 5. Human SCC tumor cells also produce IL-10.

(a) Immunostaining of untreated SCC demonstrates that tumor cells, identified by their large, atypical nuclei on Hoechst stain (lower panels, blue), synthesize IL-10. The left panel (control) shows staining with secondary and tertiary antibodies alone and the right panel also includes the primary anti-IL-10 antibody. (b) The SCC cell line SCC13 produces IL-10. SCC13 tumor cell lines were analyzed for IL-10 production by intracellular cytokine analysis.
Treatment of normal human skin with imiquimod enhances T cell activation, and reduces IL-10 production but does not affect production of IFN-γ
Imiquimod could alter the biology of T cells in SCC by recruiting different T cell populations into the tumor, by altering the activity or function of T cells within the tumor, or by a combination of these two mechanisms. To separate the differential effects of recruitment vs. local T cell modulation, we studied the effects of imiquimod on the population of T cells resident in normal human skin. In order to focus on non-regulatory T cells, FOXP3+ Treg were excluded from all analyses.
For these studies, imiquimod was added to cultures of normal skin for one week prior to T cell isolation (Clark et al., 2006). T cells from imiquimod treated skin had increased expression of the early activation antigen CD69 but decreased levels of CD25 expression (Figure 6a). We found that a large population of T cells from normal human skin produced IL-10 (Figure 6b). Imiquimod treatment of normal skin T cells reduced the production of IL-10 but had no effect on the production of IFN-γ and IL-17 (Figure 6c,d). In contrast to a prior report that in vitro treatment with imiquimod induced perforin production in human CD8 T cells (Ambach et al., 2004), we observed no change in perforin and granzyme production in CD3+ T cells after imiquimod treatment (data not shown). TGF-β was produced at baseline by less than 2% of skin resident effector T cells; we were therefore unable to study the reduction of TGF-β production after treatment with imiquimod. Addition of imiquimod to purified skin T cells removed first from the skin microenvironment had no effect on the expression of activation antigens or cytokine production, confirming earlier work showing that the effect of imiquimod on T cells is indirect and requires the presence of other, imiquimod-responsive cell types (Wagner et al., 1999).
Figure 6. Treatment of skin effector T cells in vitro with imiquimod enhances activation and reduces IL-10 production but has no effect on IL-17 and IFN-γ.

(a) T cells isolated from human skin treated for 1 week with imiquimod expressed increased CD69 and decreased CD25. Imiquimod treatment of purified skin T cells removed first from the skin microenvironment had no effect (not shown). (b) IL-10 is produced by both CD4 and CD8 non-regulatory T cells from normal human skin. (c) Non-regulatory T cells isolated from imiquimod treated skin produced less IL-10 but (d) production of IL-17 and IFN-γ was unaffected. For all parts of this figure, only non-regulatory T cells are shown; Treg were removed by gating out FOXP3+ T cells prior to analysis.
DISCUSSION
The increased incidence of SCC in patients on T cell immunosuppressive medications suggests that T cells play a critical role in controlling these tumors. T cells are present within SCC but do not control tumor growth. We found previously that SCC exclude CLA+ skin homing T cells and instead recruit FOXP3+ Treg (Clark et al., 2008). Thus, tumors persist in part because of aberrant T cell homing.
Imiquimod is a topical immune response modifier that is effective in the treatment of basal cell carcinomas, SCC and AK (Hurwitz et al., 2003; Patel et al., 2006; Peris et al., 2006; Schon et al., 2003). Imiquimod induces tumor regression via both immunologic and non-immunologic mechanisms by serving as a TLR7 agonist and by binding to adenosine receptors (Gorden et al., 2005; Hemmi et al., 2002; Jurk et al., 2002; Lee et al., 2003; Schon et al., 2003).
Cells of the innate immune system are clearly critical to the initiation of immune reactions in response to TLR agonists such as imiquimod. Clinical response to topical imiquimod therapy has been associated with the migration of plasmacytoid dendritic cells (PDC) into the skin and subsequent cytokine production (Palamara et al., 2004; Urosevic et al., 2005). We have found that the effects of imiquimod on endothelial and T cells are all indirect and require the presence of other, imiquimod-responsive cell types (Clark et al., 2008).
Despite the importance of innate cells in the first stages of TLR7 agonist responses, tumor destruction and the subsequent formation of immune memory are ultimately T cell-mediated events. It is therefore critical to understand the downstream effects of TLR agonists such as imiquimod on the activity and effector functions of T cells.
Imiquimod therapy induced marked shifts in the T cell populations infiltrating SCC. Untreated SCC are populated by non-cutaneous central memory T cells, a cell type not usually found in the skin (Clark et al., 2008). These T cells are unlikely to be tumor specific and are aberrantly recruited into the tumor by unknown mechanisms. Topical therapy with imiquimod induced a massive recruitment of CLA+ skin homing CD8 effector T cells into SCC. Observation of multiple tumors at various stages of immune destruction allowed us to observe that these T cells formed a dense, band-like infiltrate that originated in the deep dermis and migrated up, inducing tumor cell death at the leading edge and leaving behind areas of fibrotic tumor regression (Fig. 1). We found little proliferation of CD8 T cells in SCC, suggesting that the increased number of these cells is a result of enhanced recruitment from the blood (Figure 3). This result also suggests that proliferation of tumor-specific T cells may occur instead in the draining lymph nodes. Immature DC are numerous in untreated SCC but imiquimod induces DC maturation and likely stimulates migration to the lymph nodes (Clark et al., 2008). Imiquimod therapy may therefore enhance antigen presentation and proliferation of tumor specific T cells in the draining lymph nodes.
T cells from imiquimod treated tumors produced large amounts of IFN-γ, a Th1 cytokine critical for anti-tumor immune responses (Nakajima et al., 2001). T cells from imiquimod treated tumors also showed enhanced production of perforin and granzyme, two proteins that mediate killing by cytotoxic T cells. Our results in SCC agree with prior gene profiling studies in BCC showing a marked upregulation of genes associated with cytotoxic T cell function following an initial phase of innate cell activation (Panelli et al., 2007).
In addition to these pro-inflammatory T cell changes, non-regulatory T cells from imiquimod treated SCC also had reduced production of the immunosuppressive cytokines IL-10 and TGF-β (Figure 4). Non-regulatory T cells were a significant source of IL-10 and TGF-β in untreated SCC and this production was significantly decreased in imiquimod treated tumors. Untreated SCC contain approximately 50% Treg and half of these Treg produce IL-10 (Clark et al., 2008). Approximately 45% of non-regulatory T cells in SCC also produce IL-10, suggesting that nearly half of the T cells in untreated SCC produce IL-10 and that contributions from Treg and non-regulatory T cells are roughly equivalent. The tumor cells of SCC have also been reported to produce IL-10 (Kim et al., 1995). We observed IL-10 production by tumor cells as shown by immunostaining of SCC tumors and intracellular cytokine analysis of the cell line SCC13 (Fig. 5). Thus, IL-10 is produced by at least 3 cell types in human SCC: tumor infiltrating lymphocytes (both regulatory and non-regulatory T cells) and the tumor cells themselves.
Imiquimod has marked effects on T cell recruitment into tumors but it may also locally affect the phenotype and function of T cells within the tumor. To separate out the differential effects of recruitment vs. local T cell modulation, we studied the effects of imiquimod on T cells isolated from normal skin samples treated in vitro with imiquimod. Imiquimod enhanced T cell activation as evidenced by increased expression of CD69 (Fig. 6a). We found that over 25% of FOXP3- CD4 T cells from normal human skin expressed IL-10 (Figure 6b,c). This population of IL-10-producing T cells has not been previously observed and may play a role in dampening immune responses in normal skin. Imiquimod treatment significantly decreased IL-10 production by both CD4 and CD8 T cells but had no effect on the production of IFN-γ or IL-17 (Figure 6c,d). Imiquimod may therefore alter the inflammatory tone in normal skin by inhibiting the tonic production of anti-inflammatory cytokines such as IL-10. The lack of effect on IFN-γ production suggests that the differences we observe in tumors treated in vivo reflect the recruitment of Th1-biased T cells into the tumor.
Based on our observations, we have separated the effects of imiquimod on tumor infiltrating T cells into those reflecting differential migration vs. local modulation of T cell activity (Figure 7). In this model, activation of the tumor endothelium plays a critical role in the recruitment of skin homing CLA+ T cells, which contain the subset of T cells specific for SCC (Clark et al., 2008). These T cells produce IFN-γ, granzyme and perforin, leading to tumor cell destruction. Prior to imiquimod therapy, approximately 50% of the T cells infiltrating SCC are FOXP3+ Treg (Clark et al., 2008). Treg can suppress the activation, cytokine production and cytotoxic activity of other T cells and their presence has the potential to inhibit local immune response to the tumor (Hori et al., 2003; Sakaguchi et al., 1995). Imiquimod mediated activation of the endothelium leads to an influx of CD8 T cells, diluting the % of tumor Treg to levels found in normal skin. In addition to this recruitment-mediated effect, imiquimod also inhibits the ability of Treg to suppress T cell responses. Imiquimod treated Treg produce less IL-10, TGF-β and have lower levels of expression of molecules critical for suppressive function including FOXP3, CD39 and CD73 (Clark et al., 2008). Imiquimod also acts locally to induce IL-6 production by non-regulatory T cells, likely rendering them resistant to suppression (Gibson et al., 1995; Gibson et al., 2002; Kono et al., 1994; Korn et al., 2007; Pasare and Medzhitov, 2003). We report here that imiquimod has additional effects on non-regulatory T cells, inducing their activation and inhibiting tonic expression of IL-10. Imiquimod therefore acts locally on both Treg and non-regulatory T cells to block tolerogenic signals within the tumor.
Figure 7. The effects of imiquimod on tumor associated T cells: consequences on T cell recruitment vs. local modulation of T cell function.

A summary of the current studies and our prior work (Clark et al., 2008) is shown. (a) Untreated SCC lack vascular expression of E-selectin and are populated by non-cutaneous central memory T cells, 50% of which are FOXP3+ Treg. (b) Imiquimod treatment of SCC induces vascular E-selectin and the recruitment of tumor-specific CLA+ skin homing T cells. These cells dilute out Treg resident in the tumor and their activation within the tumor leads to production of IFN-γ, perforin, granzyme, and to tumor cell destruction. (c) Imiquimod also acts locally on tumor T cells, indirectly inducing the production of IL-6 by non-regulatory effector T cells (Teff), likely rendering them resistant to suppression. Imiquimod also reduces Teff production of IL-10 and TGF-β, thereby reducing tonic inhibitory signals within the tumor. Imiquimod also acts on to regulatory T cells (Treg) to reduce their ability to suppress via cytokine production (IL-10, TGF-β) and contact suppression (CD39, CD73).
Imiquimod can induce the immunologic destruction of basal cell carcinomas, actinic keratoses and Bowen’s disease (Gupta et al., 2005; Patel et al., 2006; Schulze et al., 2005). In addition to treating existing actinic keratoses, imiquimod decreased the number of new and recurrent lesions in the year following therapy, suggesting a prolonged enhancement of immunologic memory (Krawtchenko et al., 2007). Imiquimod is increasingly being reported as an effective therapy for invasive SCC in normal or immunosuppressed patients who are poor surgical candidates (Hengge and Schaller, 2004; Konstantopoulou et al., 2006; Martin-Garcia, 2005; Nouri et al., 2003; Palungwachira et al., 2005; Peris et al., 2006). Our studies support the use of imiquimod as an adjunctive therapy to reduce tumor size prior to Moh’s surgery, to treat residual disease after surgery or as monotherapy for unresectable tumors.
SCC evade immune responses at least in part by excluding skin homing effector T cells and by recruiting Treg (Clark et al., 2008). Imiquimod neutralizes both of these defenses, restoring appropriate T cell homing and reducing the percentage and suppressive ability of Treg. Reduced vascular addressin expression and impaired T cell homing has been described in human melanoma, breast, gastric and lung cancers (Madhavan et al., 2002; Piali et al., 1995; Weishaupt et al., 2007). Treg inhibit immune responses to a large number of human cancers, including melanoma, lung, breast, ovarian and renal-cell cancers (Zou, 2006). Imiquimod’s efficacy in the treatment of SCC suggests that TLR7 agonists may have a role in the treatment of other malignancies that utilize similar mechanisms of immune evasion.
Although two short term trials have found that imiquimod is safe and effective in the prevention of SCC in transplant recipients, the long term effects of imiquimod on organ tolerance are unknown (Brown et al., 2005; Ulrich et al., 2007). Concerns about inducing widespread immune activation and graft rejection continue to limit the use of this medication in the treatment of transplant recipients, in whom SCC are a major cause of morbidity and mortality. The effects of imiquimod on vascular addressin expression and T cell function are indirect (Clark et al., 2008; Wagner et al., 1999). Once the cytokines or other molecules that mediate imiquimod’s effects have been identified, these agents could be incorporated into delayed release delivery systems and administered directly to tumors. Local release of these agents within the tumor microenvironment could induce immunologic destruction of tumors without the risk of widespread immune activation engendered by TLR agonists such as imiquimod. These agents would also obviate the requirement for TLR7 responder cells such as PDC to be present within tumors. A lack of PDC in tumors correlated with the lack of response to imiquimod in human BCC (Urosevic et al., 2005).
The skin is an accessible site in which to study human tumor immunity because immune reactions in the skin are visible, easily sampled and can be manipulated with topical medications. By studying SCC, we can observe, sample and modify the human immune response to cancer. Insights gained from these studies will likely be applicable to the treatment of both cutaneous and non-cutaneous malignancies.
MATERIALS AND METHODS
SCC samples
Tumor samples consisted of curetted tumor removed prior to excision of the first Moh’s section during Moh’s micrographic excision of biopsy-proven squamous cell carcinomas. Acquisition of tumor samples and all studies were approved by the Institutional Review Board, Dana Farber Cancer Institute and were performed in accordance with the Declaration of Helsinki. Tumors were divided into bread loaf sections. Adjacent sections were used for (a) immunofluorescence studies to confirm the presence of invasive tumor cells and (b) T cell isolation, as described below. Imiquimod treated tumors were obtained from patients treated qd with topical imiquimod for 10–14 days prior to excision.
Immunofluorescence studies
Freshly excised SCC tumors were embedded in OCT, frozen and stored at −80° C until use. 5 μm cryosections were cut, air dried, fixed for five minutes in acetone, rehydrated in PBS and blocked with 20 μg/ml of human IgG (Jackson) for 15 minutes at room temperature. Sections were incubated with primary antibody for 30 minutes, then rinsed three times in PBS/1% BSA for five minutes. Sections were stained with 0.5 μg/ml Hoechst stain for two minutes, rinsed briefly in PBS/1% BSA, mounted using Prolong anti-fade mounting medium (Invitrogen), and examined immediately by immunofluorescence microscopy. Antibodies were obtained from BD (CD8 FITC, 1:20 dilution; Ki-67 PE, 1:5 dilution). IL-10 staining: rat anti-human IL-10 (BD, 1:5), secondary FITC goat anti-rat IgG (Invitrogen, 1:50), tertiary rabbit polyclonal anti-FITC conjugated to Alexafluor 488 (Molecular probes, 1:50). In all studies, Hoechst nuclear stain was used to confirm the presence of invasive tumor. TUNEL staining was performed on sections from untreated and treated SCC using the ApopTag Red In Situ Apoptosis Detection Kit (Chemicon) according to the manufacturer’s instructions. Following the final wash step, sections were stained with Hoechst dye and mounted as described above. Sections were photographed using a Nikon Eclipse 6600 microscope (Nikon) equipped with a Nikon 40X/0.75 Plan Fluor objective lens. Images were captured with a SPOT RT model 2.3.1 camera (Diagnostic Instruments) and were acquired with SPOT 4.0.9 software (Diagnostic Instruments).
Isolation of T cells from SCC tumors and normal skin
The clinically evident portions of biopsy-proven invasive SCC tumors were obtained. The tumors were divided into bread loaf sections. One section was histologically examined to confirm the presence or absence of invasive SCC tumor. Sections without evidence of tumor were designated peritumoral. Tumor-infiltrating T cells were isolated from sections adjacent to those studied by histology. T cells were isolated from SCC tumors in the absence of exogenous cytokines as previously described (Clark et al., 2006). For normal skin studies, T cells from skin discarded after plastic surgery procedures were isolated from 3 week explant cultures maintained in the presence of 100 U/ml IL-2 (R&D systems) and 20 ng/ml of IL-15 (R&D systems) as previously described (Clark et al., 2006).
Flow cytometry studies
Flow cytometry analysis of T cells was performed using directly conjugated monoclonal antibodies obtained from: BD Biosciences (CD3, CD4, CD8, CD25 and CD69) and eBioscience (FOXP3, clone PCH101). Staining for FOXP3 and activated NF-KB p65 was performed as per manufacturer’s instructions. Analysis of flow cytometry samples was performed on Becton Dickinson FACScan or FACSCanto instruments and data was analyzed using FACSDiva software (V5.1).
Analyses of cytokine, granzyme and perforin production
T cells were stimulated with 50 ng/mL PMA (Sigma) and 750 ng/ml ionomycin (Invitrogen) for 6 hours, with inclusion of 10 μg/ml brefeldin-A (Calbiochem) for the last 5 hours of incubation. SCC13 tumor cells (kindly provided by Dr. James Rheinwald) were cultured and brefeldin-A was added to the cultures 12 hours prior to analysis. Cells were stained for surface marker expression, fixed, permeabilized and then stained with anti-cytokine, granzyme or perforin antibodies (IL-17, IL-10, IFN-γ, TGF-β, granzyme and perforin, all from BD Pharmingen) and examined by flow cytometry. We observed loss of cytokine staining in unstimulated samples lacking PMA and ionomycin (but treated with brefeldin-A) and in samples treated with PMA and ionomycin but lacking brefeldin-A, suggesting that T cell stimulation was required for cytokine production and that cytokines synthesized by T cells were secreted.
Imiquimod treatment of normal skin T cells
10,000x (30 mM) imiquimod stocks were made by solubilizing imiquimod cream in DMSO. Stocks were then diluted 1:10 in culture medium and 1 μl of this 1000X stock was added to each ml of culture medium (end concentration was 3 μM). For control medium samples, an equivalent amount of DMSO was added to the control culture medium (a 1:10,000 dilution). Normal skin explant cultures were set up and maintained in 100 U/ml IL-2 (R&D systems) and 20 ng/ml IL-15 (R&D systems) to produce an expanded population of skin resident T cells as previously described (Clark et al., 2006). Imiquimod or control DMSO was added to 2 week skin explant cultures and replenished with feeds for one week; cells were harvested on week 3. In separate experiments, T cells were harvested from 2 week skin explant cultures (grown in IL-2 and IL-15 for two weeks) and then cultured separately with IL-2 and IL-15 and with or without imiquimod for one week.
Acknowledgments
Dr. Thomas Cochran of the Boston Center for Plastic Surgery and Dr. Elof Eriksson of Brigham and Women’s Hospital generously provided normal human skin samples. Dr. Richard Miller and 3M Pharmaceuticals kindly provided imiquimod cream and powdered imiquimod. Dr. James Rheinwald generously provided the SCC13 cell line. This research was supported by NIH grants 1K08AI060890-01A1, a Translational Research Award from the Leukemia and Lymphoma Society (to R.A.C.), a Pilot & Feasibility grant from the Harvard Skin Disease Research Center (to R.A.C. from NIH grant P30 AR-42689-11, to T.S.K.), a Developmental project from the SPORE in Skin Cancer (to R.A.C., from NIH grant P50 CA-93683-04, to T.S.K.), a Clinical Investigator Award from the Damon Runyon Cancer Research Foundation (to R.A.C.) and a Howard Hughes Medical Institute Medical Research Training Fellowship (to S.J.H.).
Abbreviations used
- CLA
cutaneous lymphocyte antigen
- PDC
plasmacytoid dendritic cell
- SCC
squamous cell carcinoma
- Treg
regulatory T cells
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Ambach A, Bonnekoh B, Nguyen M, Schon MP, Gollnick H. Imiquimod, a Toll-like receptor-7 agonist, induces perforin in cytotoxic T lymphocytes in vitro. Mol Immunol. 2004;40:1307–1314. doi: 10.1016/j.molimm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Berg D, Otley CC. Skin cancer in organ transplant recipients: Epidemiology, pathogenesis, and management. Journal of the American Academy of Dermatology. 2002;47:1–17. doi: 10.1067/mjd.2002.125579. [DOI] [PubMed] [Google Scholar]
- Brown VL, Atkins CL, Ghali L, Cerio R, Harwood CA, Proby CM. Safety and efficacy of 5% imiquimod cream for the treatment of skin dysplasia in high-risk renal transplant recipients: randomized, double-blind, placebo-controlled trial. Archives of dermatology. 2005;141:985–993. doi: 10.1001/archderm.141.8.985. [DOI] [PubMed] [Google Scholar]
- Clark RA, Chong BF, Mirchandani N, Yamanaka K, Murphy GF, Dowgiert RK, et al. A novel method for the isolation of skin resident T cells from normal and diseased human skin. The Journal of investigative dermatology. 2006;126:1059–1070. doi: 10.1038/sj.jid.5700199. [DOI] [PubMed] [Google Scholar]
- Clark RA, Huang SJ, Murphy GF, Mollet IG, Hijnen D, Muthukuru M, et al. Human squamous cell carcinomas evade the immune response by down-regulation of vascular E-selectin and recruitment of regulatory T cells. The Journal of experimental medicine. 2008;205:2221–2234. doi: 10.1084/jem.20071190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diepgen TL, Mahler V. The epidemiology of skin cancer. The British journal of dermatology. 2002;146(Suppl 61):1–6. doi: 10.1046/j.1365-2133.146.s61.2.x. [DOI] [PubMed] [Google Scholar]
- Euvrard S, Kanitakis J, Claudy A. Skin Cancers after Organ Transplantation. The New England journal of medicine. 2003;348:1681–1691. doi: 10.1056/NEJMra022137. [DOI] [PubMed] [Google Scholar]
- Feldman SR, Fleischer AB, Jr, McConnell RC. Most Common Dermatologic Problems Identified by Internists, 1990–1994. 1998. pp. 726–730. [DOI] [PubMed] [Google Scholar]
- Gibson SJ, Imbertson LM, Wagner TL, Testerman TL, Reiter MJ, Miller RL, et al. Cellular requirements for cytokine production in response to the immunomodulators imiquimod and S-27609. J Interferon Cytokine Res. 1995;15:537–545. doi: 10.1089/jir.1995.15.537. [DOI] [PubMed] [Google Scholar]
- Gibson SJ, Lindh JM, Riter TR, Gleason RM, Rogers LM, Fuller AE, et al. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cellular immunology. 2002;218:74–86. doi: 10.1016/s0008-8749(02)00517-8. [DOI] [PubMed] [Google Scholar]
- Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, et al. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol. 2005;174:1259–1268. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Davey V, McPhail H. Evaluation of the effectiveness of imiquimod and 5-fluorouracil for the treatment of actinic keratosis: Critical review and meta-analysis of efficacy studies. Journal of cutaneous medicine and surgery. 2005;9:209–214. doi: 10.1007/s10227-005-0148-6. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, et al. Small antiviral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nature immunology. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- Hengge UR, Schaller J. Successful treatment of invasive squamous cell carcinoma using topical imiquimod. Archives of dermatology. 2004;140:404–406. doi: 10.1001/archderm.140.4.404. [DOI] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (New York, NY) 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Housman TS, Feldman SR, Williford PM, Fleischer AB, Jr, Goldman ND, Acostamadiedo JM, et al. Skin cancer is among the most costly of all cancers to treat for the Medicare population. Journal of the American Academy of Dermatology. 2003;48:425–429. doi: 10.1067/mjd.2003.186. [DOI] [PubMed] [Google Scholar]
- Hurwitz DJ, Pincus L, Kupper TS. Imiquimod: a topically applied link between innate and acquired immunity. Archives of dermatology. 2003;139:1347–1350. doi: 10.1001/archderm.139.10.1347. [DOI] [PubMed] [Google Scholar]
- Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nature immunology. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- Kim J, Modlin RL, Moy RL, Dubinett SM, McHugh T, Nickoloff BJ, et al. IL-10 production in cutaneous basal and squamous cell carcinomas. A mechanism for evading the local T cell immune response. J Immunol. 1995;155:2240–2247. [PubMed] [Google Scholar]
- Kono T, Kondo S, Pastore S, Shivji GM, Tomai MA, McKenzie RC, et al. Effects of a novel topical immunomodulator, imiquimod, on keratinocyte cytokine gene expression. Lymphokine Cytokine Res. 1994;13:71–76. [PubMed] [Google Scholar]
- Konstantopoulou M, Lord MG, Macfarlane AW. Treatment of invasive squamous cell carcinoma with 5-percent imiquimod cream. Dermatology online journal. 2006;12:10. [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nature medicine. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawtchenko N, Roewert-Huber J, Ulrich M, Mann I, Sterry W, Stockfleth E. A randomised study of topical 5% imiquimod vs. topical 5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. The British journal of dermatology. 2007;157(Suppl 2):34–40. doi: 10.1111/j.1365-2133.2007.08271.x. [DOI] [PubMed] [Google Scholar]
- Lee J, Chuang TH, Redecke V, She L, Pitha PM, Carson DA, et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6646–6651. doi: 10.1073/pnas.0631696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan M, Srinivas P, Abraham E, Ahmed I, Vijayalekshmi NR, Balaram P. Down regulation of endothelial adhesion molecules in node positive breast cancer: possible failure of host defence mechanism. Pathol Oncol Res. 2002;8:125–128. doi: 10.1007/BF03033721. [DOI] [PubMed] [Google Scholar]
- Martin-Garcia RF. Imiquimod: an effective alternative for the treatment of invasive cutaneous squamous cell carcinoma. Dermatol Surg. 2005;31:371–374. doi: 10.1111/j.1524-4725.2005.31093. [DOI] [PubMed] [Google Scholar]
- Nakajima C, Uekusa Y, Iwasaki M, Yamaguchi N, Mukai T, Gao P, et al. A role of interferon-gamma (IFN-gamma) in tumor immunity: T cells with the capacity to reject tumor cells are generated but fail to migrate to tumor sites in IFN-gamma-deficient mice. Cancer research. 2001;61:3399–3405. [PubMed] [Google Scholar]
- Nouri K, O’Connell C, Rivas MP. Imiquimod for the treatment of Bowen’s disease and invasive squamous cell carcinoma. J Drugs Dermatol. 2003;2:669–673. [PubMed] [Google Scholar]
- Palamara F, Meindl S, Holcmann M, Luhrs P, Stingl G, Sibilia M. Identification and characterization of pDC-like cells in normal mouse skin and melanomas treated with imiquimod. J Immunol. 2004;173:3051–3061. doi: 10.4049/jimmunol.173.5.3051. [DOI] [PubMed] [Google Scholar]
- Palungwachira P, Palungwachira P, Ogawa H. Treatment of multiple lesions of Bowen’s disease and squamous cell carcinoma with topical imiquimod. The Journal of dermatology. 2005;32:1005–1009. doi: 10.1111/j.1346-8138.2005.tb00891.x. [DOI] [PubMed] [Google Scholar]
- Panelli M, Stashower M, Slade H, Smith K, Norwood C, Abati A, et al. Sequential gene profiling of basal cell carcinomas treated with imiquimod in a placebo-controlled study defines the requirements for tissue rejection. 2007;8:R8. doi: 10.1186/gb-2007-8-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll Pathway-Dependent Blockade of CD4+CD25+ T Cell-Mediated Suppression by Dendritic Cells. 2003:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- Patel GK, Goodwin R, Chawla M, Laidler P, Price PE, Finlay AY, et al. Imiquimod 5% cream monotherapy for cutaneous squamous cell carcinoma in situ (Bowen’s disease): a randomized, double-blind, placebo-controlled trial. Journal of the American Academy of Dermatology. 2006;54:1025–1032. doi: 10.1016/j.jaad.2006.01.055. [DOI] [PubMed] [Google Scholar]
- Peris K, Micantonio T, Fargnoli MC, Lozzi GP, Chimenti S. Imiquimod 5% cream in the treatment of Bowen’s disease and invasive squamous cell carcinoma. Journal of the American Academy of Dermatology. 2006;55:324–327. doi: 10.1016/j.jaad.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Piali L, Fichtel A, Terpe HJ, Imhof BA, Gisler RH. Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. The Journal of experimental medicine. 1995;181:811–816. doi: 10.1084/jem.181.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- Schon M, Bong AB, Drewniok C, Herz J, Geilen CC, Reifenberger J, et al. Tumor-selective induction of apoptosis and the small-molecule immune response modifier imiquimod. Journal of the National Cancer Institute. 2003;95:1138–1149. doi: 10.1093/jnci/djg016. [DOI] [PubMed] [Google Scholar]
- Schulze HJ, Cribier B, Requena L, Reifenberger J, Ferrandiz C, Garcia Diez A, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from a randomized vehicle-controlled phase III study in Europe. 2005. pp. 939–947. [DOI] [PubMed] [Google Scholar]
- Ulrich C, Bichel J, Euvrard S, Guidi B, Proby CM, van de Kerkhof PC, et al. Topical immunomodulation under systemic immunosuppression: results of a multicentre, randomized, placebo-controlled safety and efficacy study of imiquimod 5% cream for the treatment of actinic keratoses in kidney, heart, and liver transplant patients. The British journal of dermatology. 2007;157(Suppl 2):25–31. doi: 10.1111/j.1365-2133.2007.08269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urosevic M, Dummer R, Conrad C, Beyeler M, Laine E, Burg G, et al. Disease-independent skin recruitment and activation of plasmacytoid predendritic cells following imiquimod treatment. Journal of the National Cancer Institute. 2005;97:1143–1153. doi: 10.1093/jnci/dji207. [DOI] [PubMed] [Google Scholar]
- Wagner TL, Ahonen CL, Couture AM, Gibson SJ, Miller RL, Smith RM, et al. Modulation of TH1 and TH2 cytokine production with the immune response modifiers, R-848 and imiquimod. Cellular immunology. 1999;191:10–19. doi: 10.1006/cimm.1998.1406. [DOI] [PubMed] [Google Scholar]
- Warino L, Tusa M, Camacho F, Teuschler H, Fleischer AB, Jr, Feldman SR. Frequency and cost of actinic keratosis treatment. Dermatol Surg. 2006;32:1045–1049. doi: 10.1111/j.1524-4725.2006.32228.x. [DOI] [PubMed] [Google Scholar]
- Weishaupt C, Munoz KN, Buzney E, Kupper TS, Fuhlbrigge RC. T-cell distribution and adhesion receptor expression in metastatic melanoma. Clin Cancer Res. 2007;13:2549–2556. doi: 10.1158/1078-0432.CCR-06-2450. [DOI] [PubMed] [Google Scholar]
- Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]