

Abstract
We investigated oxidative stress in human postmortem frontal cortex from individuals characterized as mild-cognitive impairment (n = 8), mild/moderate Alzheimer disease (AD) (n = 4), and late stage AD (n = 9). Samples from subjects with no cognitive impairment (n = 10) that were age- and postmortem interval-matched with these cases were used as controls. The short postmortem interval brain samples were processed for postmitochondrial supernatant (PMS), non-synaptic mitochondria, and synaptosome fractions. Samples were analyzed for several antioxidants (glutathione [GSH], glutathione peroxidase [GPx], glutathione reductase, glutathione-S-transferase [GST] glucose-6-phosphate dehydrogenase, superoxide dismutase [SOD], catalase) and the oxidative marker, thiobarbituric acid reactive substances. The tissue was also analyzed for possible changes in protein damage using neurochemical markers for protein carbonyls, 3-nitrotyrosine, 4-hydroxynonenal, and acrolein. All 3 neuropil fractions (PMS, mitochondrial, and synaptosomal) demonstrated significant disease-dependent increases in oxidative markers. The highest changes were observed in the synaptosomal fraction. Both mitochondrial and synaptosomal fractions had significant declines in antioxidants (GSH, GPx, GST, and SOD). Levels of oxidative markers significantly correlated with Mini-Mental Status Examination scores. Oxidative stress was more localized to the synapses, with levels increasing in a disease-dependent fashion. These correlations implicate an involvement of oxidative stress in AD-related synaptic loss.
Keywords: Alzheimer disease, Free radicals, Mild cognitive impairment, Mitochondria, Neurodegeneration, Oxidative stress, Synapses
INTRODUCTION
Oxidative stress is defined as a marked imbalance between the reactive oxygen species (ROS) and its removal by antioxidant system. This may originate from an overproduction of ROS or from a reduction in antioxidant defenses (1, 2). Brain tissue has multiple potential sources of ROS (3) and a large oxidative capacity, but its ability to combat oxidative stress is limited (4). Increasing evidence suggests that oxidative stress that is normally associated with aging is a prominent and early feature of Alzheimer disease (AD) and plays a role in its pathogenesis (5–7).
Glutathione (GSH) is a well-known antioxidant that is synthesized in the cytoplasm and is present in higher concentrations in the mitochondrial matrix. Since the majority of oxidized GSH (GSSG) forms are under oxidative stress (8), several different enzymes are necessary to reduce GSSG to maintain the GSH/GSSG ratio. Under severe conditions of oxidative stress, cells are not able to maintain the appropriate GSH/GSSG ratio, causing an accumulation of GSSG and resulting in protein modifications. The level of GSSG is a key factor in determining neuronal susceptibility to ROS/reactive nitrogen species (RNS)-mediated neuronal injury (9–11). The cytosolic GSH pool is critical for maintaining plasma membrane integrity and adenosine triphosphate (ATP) levels in synaptosomes (12).
The widespread occurrence of protein nitration in neurons is due to increased oxidative stress (13–15). Lipid peroxidation (LPO) generates various reactive aldehydes, such as acrolein, which rapidly incorporates into proteins to generate carbonyl derivatives. Protein-bound acrolein is a powerful marker of oxidative protein damage that plays an important role in the formation of neurofibrillary tangles (NFTs) in AD (16). Both acrolein and 4-hydroxynonenal (4-HNE), another product of LPO, can alter phospholipid asymmetry of membrane lipid bilayer, initiating apoptotic neuronal loss in mild cognitive impairment (MCI) and AD (17). The reactivity of LPO with key mitochondrial enzymes (18) increases free radical release into the cytoplasm (19), resulting in elevated oxidative stress that can affect synaptic function and neuronal death in AD (20, 21).
It is now clear that there are regional variations in the levels of oxidative stress in AD brain resulting from differences in the levels of antioxidant defenses and rates of oxygen consumption (22–25). Higher levels of oxidative DNA damage in mitochondria of the frontal, parietal, and temporal lobes suggests that mitochondrial oxidative stress may be an important contributor to the pathogenesis of AD (25). Transient or sustained mitochondrial dysfunction can deplete ATP levels, increase ROS generation, and initiate apoptosis leading to neurodegeneration (22, 26).
When neuronal DNA damage resulting from oxidative stress is not completely repaired, it can cause accumulated synaptic protein alterations (27, 28). Postsynaptic regions are subjected to particularly high levels of calcium influx and oxidative stress as a result of local activation of glutamate receptors; therefore, they are likely sites at which neurodegenerative processes are initiated in AD (29). Increased oxidative stress precedes the loss of several synaptic proteins (10, 30) and this may contribute to synaptic degeneration (31). It is well known that synaptic mitochondria differ enzymatically (32, 33) and may be more sensitive to ROS than nonsynaptic mitochondria (34, 35). Defects in synaptic mitochondria can alter synaptic functions, leading to alterations in cognitive function. It is unclear whether levels of oxidative stress are high in mitochondria as a function of disease progression and whether or not the levels are similar to those observed in synaptic terminals.
AD is a progressive dementing disorder characterized by both a loss of working and episodic memory. Although senile plaques and NFTs are considered hallmarks of the disease, many non-demented elderly individuals have these lesions, often at levels suggestive of early AD (36–38). There are also elderly individuals who manifest some cognitive deficits that are greater than expected of those observed in normal aging but are not clinically demented. These individuals are thought to be in a transitional stage between normal aging and dementia and have been termed MCI (39–41). The conversion rate from MCI to mild AD (mAD) is 10% to 15% per year (42).
Here, we analyzed multiple biochemical markers of oxidative stress and antioxidant defenses in postmitochondrial supernatant (PMS), mitochondrial, and synaptic fractions in age-matched noncognitively impaired (NCI), MCI, mAD, and AD subjects in the frontal cortex (FC), a cortical region known to be involved in AD.
MATERIALS AND METHODS
Reagents
All chemicals and reagents used were purchased from Sigma (St. Louis, MO) unless stated otherwise.
Subjects
Frozen FC samples from NCI (n = 10), MCI (n = 8), mAD (n = 4), and AD (n = 9) subjects were obtained from the University of Kentucky Alzheimer's Disease Center (UKADC) Rapid Autopsy Program. The diagnosis of probable AD was made according to criteria developed by the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (43). The diagnosis of MCI was identical to that used previously (44). The Human Investigations Committee of the University of Kentucky College of Medicine approved the studies. Individuals included in these studies agreed to an annual clinical evaluation and brain donation at the time of death. NCI subjects were without history of dementia or other neurological disorders and underwent annual mental status testing and semiannual physical and neurological exams as part of the UKADC normal volunteer longitudinal aging study (38). Additional demographic parameters of NCI, MCI, mAD, and AD patients available from medical records are provided in the Table. When selecting subjects, agonal state prior to death was taken into consideration. Standard criteria for exclusion were the presence of 1) significant cerebral stroke regardless of antemortem date, 2) large cortical infarcts identified in the postmortem neuropathological evaluation, 3) significant head trauma within 12 months prior autopsy, and 4) individuals on a respirator longer than 12 hours prior to death, 5) individuals in coma longer than 12 hours immediately prior to death, 6) individuals that had been under excessive medication for periods longer than 6 months prior to death, or 7) individuals currently undergoing radiation therapy for CNS tumor. As part of the rapid autopsy protocol we ask the individuals who inform us of a subject’s death what the conditions were for the 24 hours preceding death and if any significant events may have dramatically influenced the process. Questionable tissue was not included in the study.
TABLE 1.
Clinical and Pathological Features of Study Subjects
| Subjects/ Classificat ion |
Age, years | Sex | Brain Weight, g |
MMSE | CERAD Classificat ion |
Braak Score |
PMI, hours |
|---|---|---|---|---|---|---|---|
| NCI | 88 | F | 1,240 | 29 | Probable | V | 3 |
| NCI | 95 | F | 1,130 | 27 | Possible | 0 | 2 |
| NCI | 80 | M | 1,430 | 27 | None | IV | 3 |
| NCI | 70 | M | 1,530 | 30 | None | I | 3 |
| NCI | 90 | F | 1,110 | 29 | None | I | 4 |
| NCI | 89 | F | 1,210 | 30 | Probable | IV | 2 |
| NCI | 84 | F | 940 | 30 | None | III | 5 |
| NCI | 88 | F | 1,070 | 29 | None | III | 3 |
| NCI | 84 | F | 1,230 | 30 | None | 0 | 3 |
| NCI | 92 | M | 1,220 | 28 | Probable | 0 | 3.5 |
| Mean ± SD |
86.0 ± 7.1 | 1,211 ± 170 |
28.9 ± 1.2 | 3.1 ± 0.9 | |||
| MCI | 91 | F | 1,130 | 26 | Probable | V | 2.0 |
| MCI | 93 | F | 1,050 | 27 | Probable | III | 2.5 |
| MCI | 87 | M | 1,200 | 24 | Definite | IV | 3.5 |
| MCI | 88 | F | 1,080 | 17 | Definite | V | 5.0 |
| MCI | 99 | F | 930 | 20 | Possible | V | 2.0 |
| MCI | 84 | M | 1,350 | 24 | Probable | IV | 4.0 |
| MCI | 89 | M | 1,330 | 26 | Definite | I | 3.0 |
| MCI | 88 | F | 1,130 | 28 | Definite | III | 3.0 |
| Mean ± SD |
90.3 ± 4.8 | 1,150 ± 141 |
24.0 ± 3.7 | 3.1 ± 1.0 | |||
| mAD | 93 | M | 1,230 | 21 | Definite | IV | 4.0 |
| mAD | 89 | F | 1,190 | 20 | None | IV | 3.5 |
| mAD | 77 | F | 1,190 | 23 | Definite | V | 3.0 |
| mAD | 82 | M | 1,090 | 25 | Definite | II | 2.0 |
| Mean ± SD |
85.3 ± 7.1 | 1,175 ± 60 |
22.3 ± 2.2 | 3.1 ± 0.9 | |||
| AD | 84 | M | 1,150 | 14 | Definite | VI | 4.5 |
| AD | 86 | F | 1,030 | 7 | Definite | VI | 3.0 |
| AD | 68 | M | 1,380 | 11 | Probable | II | 4.0 |
| AD | 90 | M | 1,200 | 12 | Definite | VI | 3.0 |
| AD | 86 | F | 1,070 | 14 | Definite | VI | 5.0 |
| AD | 78 | F | 1,080 | 11 | Definite | VI | 3.5 |
| AD | 82 | F | 1,260 | 13 | Definite | VI | 4.5 |
| AD | 85 | M | 1,020 | 15 | Probable | VI | 3.0 |
| AD | 73 | M | 1,130 | 16 | Definite | VI | 2.0 |
| Mean ± SD |
81.3 ± 7.1 | 1,147 ± 117 |
12.6 ± 2.7 | 3.6 ± 1.0 |
AD, Alzheimer disease; CERAD, Consortium to Establish a Registry for Alzheimer Disease; F, female; M, male; mAD, mild Alzheimer disease; MCI, mild cognitive impairment; MMSE, Mini Mental State Examination score; NCI, no cognitive impairment; PMI, postmortem interval.
Isolation of Tissue Fractions
Subcellular fractions (synaptosome, mitochondria, and PMS) were isolated from FC using standard procedures with some modifications (45). Briefly, the brain tissue was gently homogenized with a Wheaton tissue homogenizer in ice-cold sucrose isolation buffer (250 mM sucrose, 10 mM Hepes, 1.0 mM EDTA, pH 7.2, 4.0 µg/ml leupeptin, 4.0 µg/ml pepstatin, 5.0 µg/ml aprotinin, 20.0 µg/ml trypsin inhibitor). The homogenates were subjected to centrifugation for 5 minutes at 1,330 g at 4°C. The collected supernatants were spun at 21,200 g at 4°C for 10 minutes. The PMS fraction was collected. Pellets were resuspended and layered over a discontinuous sucrose gradient (1.0 M, pH 8.0; and 1.18 M, pH 8.5) and spun at 85,500 g for 1 hour at 4°C. The purified synaptosomes were collected from the sucrose gradient interface and mitochondrial fraction collected from the bottom layer. Synaptosomes and mitochondrial fractions were diluted with isolation buffer and centrifuged at 32,000 g for 20 minutes and the collected pellet used for the analysis.
Antioxidants
Determination of antioxidants and associated enzymes were performed as described previously (9, 10, 30). For GSH and GSSG, changes in fluorescence at emission 420 nm were recorded by excitation at 350 nm. For the estimation of GPx, glutathione reductase (GR) enzyme activity, the disappearance of nicotinamide adenosine dinucleotide phosphate (NADPH) and glucose-6-phosphate dehydrogenase (G-6PD) activity with formation of NADPH at 340 nm was recorded at room temperature (RT). The enzyme activity was calculated as nmol NADPH oxidized/min/mg protein, using a molar extinction coefficient of 6.22 × 1.03 M−1 cm−1. Glutathione-S-transferase (GST) (EC 2.5.1.18) activity was measured by the method as previously described (10, 30). The reaction mixture consisted of 0.1 M phosphate buffer (pH 6.5), 1.0 mM reduced GSH, 1.0 mM CDNB, and 0.03 ml above mentioned sample in a final volume of 0.3 ml. The changes in absorbance were recorded at 340 nm and the enzyme activity calculated as nmol 1-chloro-2,4-dinitrobenzene conjugate formed/min/mg protein, using a molar extinction coefficient of 9.6×103M−1cm−1. For GST activity, the changes in absorbance were recorded at 340 nm and the enzyme activity calculated as nmol 1-chloro-2,4-dinitrobenzene conjugate formed/minutes/mg protein, using a molar extinction coefficient of 9.6 × 103 M−1 cm−1.
Superoxide Dismutase and Catalase Activity
SOD (EC 1.15.1.1) and catalase (CAT) activities were measured as previously described (10, 30, 46). Briefly, for SOD, triethylenetetramine was used as a copper/zinc (Cu/Zn)-SOD inhibitor to measure manganese-SOD activity. The absorbance was monitored for 5 minutes at 480 nm and activity calculated using a molar extinction coefficient of 4.02 × 103 M−1 cm−1 expressed as nmol of epinephrine protected from oxidation/min/mg protein. For CAT, changes in absorbance were recorded at 240 nm and activity was calculated in terms of nmol H2O2 consumed/min/mg protein, using a molar extinction coefficient of 43.6 M−1 cm−1.
Thiobarbituric Acid Reactive Substances
The estimation of thiobarbituric acid reactive substances (TBARS) was performed as described previously (30, 46). The rate of LPO was expressed as nmol of TBARS formed/30 min/mg protein, using a molar extinction coefficient of 1.56 × 105 M−1 cm−1.
Protein Carbonyls, 3-Nitrotyrosine, 4-Hydroxynonenal, and Acrolein
Protein carbonyls (PCs), 3-nitrotyrosine (3-NT), 4-HNE, and acrolein were assessed by following a standard previously described protocol (9, 10). For PC, 5 µl samples (normalized to 4 mg/ml) were incubated for 20 minutes at RT with 5 µl of 12% sodium dodecyl sulfate (SDS) and 10 µl of 20 mM 2,4-dinitrophenyl hydrazine, and neutralized with 7.5 µl neutralization solution (2 M Tris in 30% glycerol). For 3-NT, 4-HNE and acrolein 5 µl samples were incubated for 20 minutes at RT with 5 µl of 12% SDS and 5 µl of modified Laemmli buffer containing 0.125 M Tris base, pH 6.8, 4% (v/v) SDS, and 20% (v/v) glycerol. Each sample (250 ng) was loaded into a well on a nitrocellulose membrane in a slot-blot apparatus under vacuum. The membrane was blocked in buffer (3% bovine serum albumin) in PBS/Tween for 1 hour and incubated with a 1:100 anti-DNP, 1:2000 anti-3-NT, 1:5000 anti-4-HNE dilution of polyclonal antibody, and 1:5000 monoclonal ant-acrolein in PBS/Tween for 90 minutes. The membrane was washed in PBS/Tween for 5 minutes 3 times after incubation. The membrane was incubated for 1 hour after washing with an anti-rabbit, anti-Goat, and anti-mouse IgG alkaline phosphatase secondary antibody diluted 1:8,000 in PBS/Tween. The membrane was washed X3 in PBS/Tween for 5 minutes and developed in Sigma Fast tablets. Blots were dried, scanned with Adobe PhotoShop, and quantified with Scion Image as above. No nonspecific binding of antibody to the membrane was observed.
Protein Estimation
Total protein concentrations were measured using the Pierce BCA method (Sigma).
Ultrastructural Analysis of Mitochondrial Preparations
Random samples of both synaptosomal and non-synaptic mitochondrial preparations were analyzed with ultrastructural techniques to verify the relative purity of the preparations. Mitochondrial samples were fixed with 1% glutaraldehyde/4% paraformaldehyde in 0.1M sodium phosphate buffer (pH 7.4). Samples were postfixed in 1% osmium tetroxide (OsO4), stained en block with 0.5% uranyl acetate, dehydrated in a graded series of ethanol, treated with propylene oxide and embed in epoxy resin as previously described (44). Cured blocks were sectioned with an ultramicrotome and imaged with a Zeiss 902 electron microscope. Pictures were taken at an initial magnification of 4,400× and photographically enlarged to approximately 18,000×. Random pictures (n = 5) were taken from each preparation.
Statistical Analysis
Enzymatic and nonenzymatic oxidative stress markers are reported as group mean ± SD. Group means were evaluated with a one-way ANOVA coupled with a Student Newman-Keuls post-hoc test. Correlations were calculated using the Pearson product-moment correlation coefficient test (StatView 5.0, SAS Institute). For significance, alpha was set at 0.05.
RESULTS
Subject Demographics
The Table shows the characteristics of the sample population by diagnostic group. The NCI, MCI, mAD, and AD groups were similar in age, gender, brain weight, and PMI; they did not show any significant differences (p > 0.05) for any of these variables. An ANOVA revealed a significant difference in the MMSE between groups (F [3,27] = 61.922, p < 0.0001). Post hoc analysis revealed a statistically significant change between NCI and MCI and between MCI and AD but not with mAD. AD and mAD were also significantly different. The AD group had the highest incidence of Braak stage VI subjects. Subjects in all groups were highly educated with a mean education level of 16 ± 3 years and failed to reveal a significant difference between groups (F [3,27] = 2.277, p > 0.1).
Mitochondrial Preparations
As shown in Figure 1, the synaptosomal preparation contained primarily synaptosomes and the non-synaptic fraction contained primarily mitochondria and no synaptosomes. No precise quantitative measurements were made on either preparation or grading of relative purity was attempted. All assessments were qualitative.
Figure 1.

Electron photomicrographs of representative isolated mitochondrial (A) and (B) synaptosomal fractions from the frontal cortex of a human postmortem brain sample. Calibration bars: A, 2.0 µm, B, 0.5 µm.
Glutathione Levels
The level of internal antioxidant tri-peptide molecule GSH was measured in the PMS, mitochondrial, and synaptosomal fractions of the FC in NCI, MCI, mAD, and AD subjects. GSH levels demonstrated a significant change in the PMS (F [3, 27] = 6.335, p < 0.005), mitochondria (F [3, 27] = 7.761, p < 0.001), and synaptosome (F [3, 27] = 12.935, p < 0.0001) fractions. In all fractions, levels decreased with decline in cognitive status. The greatest change was observed in the synaptosomal fraction (MCI, 77.7%; mAD, 51.3%; AD, 51.2%) compared to NCI (Fig. 2A). Both AD groups were significantly lower (p < 0.05) than the MCI group but not significantly different from each other. While a similar trend was observed in the PMS and mitochondrial fractions, the cognitively impaired groups were not significantly (p > 0.05) different from each other. These results indicate that GSH decline is an early event in the progression of the disease. There was a significant correlation (p < 0.0005) between the subjects’ MMSE and the GSH levels in all 3 fractions (data not shown). Thus, with lower MMSE scores had lower levels of GSH.
Figure 2.

Changes in the glutathione (GSH) system (GSH, oxidized GSH [GSSG] and GSH/GSSG) analyzed in post mitochondrial supernatant (PMS), mitochondrial, and synaptosomal fractions of the frontal cortex from non-cognitively impaired (NCI), mild cognitively impaired (MCI) mild Alzheimer disease (mAD), and AD subjects. All 3 neuropil fractions showed significant declines in GSH (A) and significant increases in GSSG levels (B) with increased cognitive impairment. The changes in GSH and GSSG also resulted in significant changes in the GSH/GSSG ratios with disease progression (C). The synaptosomal fraction showed the greatest changes. Each bar represents the group mean ± SD. **p < 0.01, *p < 0.05 vs. NCI; αp < 0.05 vs. MCI.
Oxidized Glutathione Levels
Analysis of GSSG levels in the PMS, mitochondrial, and synaptosomal fractions demonstrated significant differences between the NCI, MCI, mAD, and AD groups (Fig. 2B). GSSG increased significantly in the PMS (F [3, 27] = 33.736, p < 0.0001), mitochondrial (F [3, 27] = 34.544, p < 0.0001) and synaptosomal (F [3, 27] = 10.622, p < 0.0001) fractions. In all fractions, the levels of GSSG increased with increased cognitive impairment; AD subjects had significantly higher levels than the MCI and mAD groups (p < 0.05) in the PMS and mitochondrial fractions. The greatest change was observed in the synaptosomes with increases correlating with decreased cognitive status: 257% (MCI), 343% (mAD), and 366% (AD).
Glutathione/Oxidized Glutathione Ratios
GSH/GSSG ratios significantly declined in the PMS (F [3, 27] = 69.409, p < 0.0001), mitochondrial (F [3, 27] = 48.169, p < 0.0001), and synaptosomal (F [3, 27] = 32.125, p < 0.0001) fractions compared to the NCI group (Fig. 2C). In the PMS and mitochondrial fractions, the levels of the AD group were significantly lower than MCI (p < 0.01) and mAD (p < 0.05) groups. The synaptosomal fraction was significantly decreased to about the same level in all cognitively impaired groups. These data suggest that a decline in antioxidants occurs early in the progression of the disease and that the free radical burden continues to increase with a decline in cognitive status. There was a significant correlation (p < 0.0001) between the subjects; MMSE and GSSG ratios in all 3 fractions (not shown), i.e. individuals with lower MMSE scores had lower GSH/GSSR ratios.
Glutathione Peroxidase
The activity of GPx in the mitochondrial fraction was significantly (F [3, 27] = 5.164, p < 0.01) altered only in the mAD (61%) and AD (57%) groups. While in synaptosomes, it was significantly (F [3, 27] = 6.342, p < 0.005) depleted in MCI (70%), mAD (58.39%), and AD (58%) compared to the NCI group. The PMS fraction (F [3, 27] = 0.769. p > 0.1) failed to show any significant change in GPx activity (Fig. 3A). The mAD group had levels equivalent to that seen in the more advanced AD group in the mitochondrial, synaptosomal, and PMS fractions.
Figure 3.

Different antioxidant enzymes were assessed in post mitochondrial supernatant (PMS), mitochondrial, and synaptosomal fractions of the frontal cortex from non-cognitively impaired (NCI), mild cognitive impairment (MCI,) mild Alzheimer disease (mAD), and AD subjects. (A) Glutathione peroxidase (GPx) activity was significantly changed in mitochondrial and synaptosomal fractions only, not in the PMS. (B) Glutathione reductase (GR) activity showed significant decline only in synaptosomes,. (C) Glutathione-S-transferase (GST) activity showed a significant decline in the mitochondrial and synaptosomal fractions from mAD, and AD, and a significant decline in MCI only in the synaptosomal fraction. (D) Significant alterations in glucose-6-phosphate dehydrogenase (G-6PD) activity were only detected in synaptosomes. Each bar represents the group mean ± SD. **p < 0.01, *p < 0.05 vs. NCI.
Glutathione Reductase
GR reduces GSSG into GSH to continue the antioxidant cycle during detoxification of oxidants. The activity of GR in synaptosomes was significantly (F [3, 27] = 7.126, p < 0.001) depleted in the MCI (66%), mAD (63.71%), and AD (60%) groups compared to the NCI cohort. These cognitively impaired groups were not significantly different from each other. The PMS (F [3,7] = 0.781, p > 0.1) and mitochondrial (F [3,27] = 2.030, p > 0.1) fractions failed to show any significant change in GR activity (Fig. 3B).
Glutathione-S-Transferase
GST is used for detoxification of external or internal toxicants. The activity of GST was significantly lower in the mitochondrial (F [3,27] = 4.868, p < 0.01), and synaptosomal (F [3, 27] = 12.760, p < 0.0001) fractions, but failed to reach significance in the PMS fraction (F [3,27] = 2.810, p > 0.1). The MCI group was only significantly different from the NCI group in the synaptosomal fraction (p < 0.01), whereas the AD groups were significantly lower in the mitochondrial and synaptosomal fractions (Fig. 3C). The mAD and AD groups demonstrated similar degrees of decline.
Glucose-6-Phosphate Dehydrogenase
G-6PD provides electrons for the reduction of oxidized nicotinamide adenine dinucleotides (NAD+/NADP+). The activity of G-6PD in synaptosomes was significantly (F [3, 27] = 5.297, p < 0.01) depleted in the MCI (67%), mAD (59%), and AD (65%) groups compared to the NCI subjects and these groups were not significantly different from each other. PMS fractions of the MCI, mAD and AD groups failed to show any significant (F [3,27] = 0.899, p > 0.1) change in G-6PD activity (Fig. 3D).
Superoxide Dismutase
SOD is an important enzyme that reduces the superoxide burden in tissue. Analysis of the SOD activity in the PMS (F [3, 27] = 4.927, p < 0.01), mitochondria (F [3, 27] = 5.322, p < 0.01), and synaptosomal (F [3, 27] = 5.612, p < 0.005) fractions demonstrated a significant decline in FC (Fig. 4A). The activity of SOD in PMS was significantly depleted in the mAD (62%), and AD (59%) groups compared to NCI. Analysis of the synaptosomal fraction revealed that SOD activity was significantly depleted in the MCI (70%), mAD (66%), and AD (61%) groups compared to the NCI subjects. The activity of manganese-SOD in the mitochondrial fraction was also significantly (F [3, 27] = 5.243, p < 0.01) reduced in the MCI (74%), mAD (65%), and AD (63%) groups compared to the NCI cohorts. In all fractions, the MCI, mAD, and AD cases were not significantly different from each other (p > 0.1). There was a significant correlation (p < 0.005) between the subjects’ MMSE and SOD levels in all 3 fractions (not shown), i.e. individuals with lower MMSE scores had lower levels of SOD.
Figure 4.

Superoxide dismutase (SOD) and catalase (CAT) activities in the post mitochondrial supernatant (PMS), mitochondrial, and synaptosomal fractions of frontal cortex in mild cognitive impairment (MCI) mild Alzheimer disease (mAD), and AD subjects. (A) There was significant depletion in SOD activity in mitochondrial and synaptosomal fractions of MCI, mAD, and AD; the PMS fraction of MCI was not significantly depleted. (B) CAT activity in the PMS and synaptosomal fractions was significantly decreased in MCI, mAD, and AD. Each bar represents the group mean ± SD. **p < 0.01, *p < 0.05 vs. NCI; αp < 0.05 vs. MCI; #p < 0.05 vs. mAD.
Catalase
CAT is an important enzyme that breaks down H2O2 into H2O and O2. Analysis of synaptosomes revealed a significant decline in CAT activity (F [3, 27] = 13.652, p < 0.0001) (Fig. 4B). The decline mirrored the change in cognitive status with the MCI group (68%) demonstrating a loss in activity that was not as severe as that observed in the mAD (44%), and AD (29%) groups. The mAD and AD group were not significantly different. The PMS fraction also showed significant depletion in CAT activity (F [3,27] = 30.792, p < 0.0001) with the loss in the AD group (31%) significantly greater (p < 0.01) compared to MCI (69%) and mAD (66%) subjects. CAT levels in the PMS and synaptosomal fractions revealed a significant (p < 0.0001) correlation with the subjects’ MMSE scores (not shown), i.e. lower levels of CAT were associated with lower MMSE scores.
Thiobarbituric Acid Reactive Substances
TBARS levels were evaluated as a measure of total LPO. The analysis demonstrated a significant increase in TBARS in the PMS (F [3, 27] = 21.067, p < 0.0001), mitochondria (F [3, 27] = 8.657, p < 0.0005), and synaptosomes (F [3, 27] = 19.038, p < 0.0001) fractions. In all fractions, the MCI, mAD, and AD groups were significantly elevated (p < 0.01) compared to NCI subjects. In the PMS and synaptosome fractions, the AD group showed the highest TBARS value and was significantly elevated compared to both the MCI and mAD groups. In the mitochondrial fraction, all 3 cognitively impaired groups showed similar increases compared to the NCI cohorts (Fig. 5). The subjects’ MMSE and TBARS levels were significantly correlated (p < 0.0001) in all 3 fractions (data not shown), i.e. individuals with lower MMSE scores had higher TBARS levels. Furthermore, when the subjects were grouped according to Braak scores (I-II, III-IV, V-VI), individuals with the highest scores showed the highest levels of TBARS in both the synaptosomal and PMS fractions (p < 0.05). The mitochondrial fraction did not show this relationship.
Figure 5.

Thiobarbituric acid reactive substances (TBARS) demonstrated significant increases in all fractions of the frontal cortex from mild cognitive impairment (MCI) mild Alzheimer disease (mAD), and AD subjects. TBARS formation was higher in synaptosomal fractions. Each bar represents the group mean ± SD. *p < 0.01 vs. NCI; αp < 0.05 vs. MCI; #p < 0.05 vs. mAD.
Protein Oxidation, Nitration, and Lipid Peroxidation Levels
Levels of protein oxidation and nitration products (PC and 3-NT) were evaluated using slot blots (Fig. 6). PCs demonstrated a significant increase in the PMS (F [3, 27] = 10.356, p < 0.0005), mitochondria (F [3, 27] = 8.261, p < 0.0005), and synaptosomal (F [3, 27] = 9.738, p < 0.0005) fractions (Fig. 7A). Although the PC values were significantly elevated in all fractions as a function of the degree of cognitive impairment, there were no significant differences between the cognitively impaired groups, suggesting that PC formation is an early event in the disease process.
Figure 6.
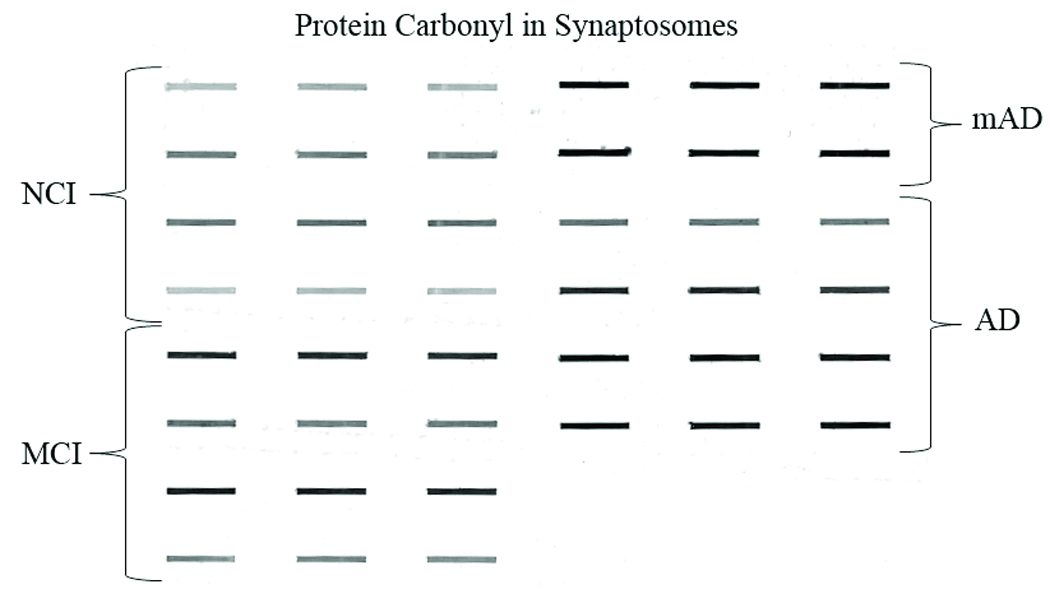
Representative Slot-blot for protein carbonyl (PCs) shows half of the synaptosomal fractions of the frontal cortex from non-cognitively impaired (NCI), mild cognitive impairment (MCI) mild Alzheimer disease (mAD), and AD subjects. The slot-blot shows prominent alterations in staining for the different cognitively impaired groups.
Figure 7.

Levels of protein modification were analyzed in post mitochondrial supernatant (PMS), mitochondrial, and synaptosomal fractions of the frontal cortex from non-cognitively impaired (NCI), mild cognitive impairment (MCI) mild Alzheimer disease (mAD), and AD subjects. (A) Protein carbonyl (PC) levels showed significant increases in all fractions of the frontal cortex with disease progression. (B) Significant changes in 3-nitrotyrosine levels mirrored the PC changes in all the fractions from MCI, mAD, and AD subjects. (C) 4-hydroxynonenal (4-HNE) and (D) acrolein levels showed significant increases in protein modification via lipid peroxidation. The significant increases in 4-HNE and acrolein were already present in MCI subjects, with the greatest elevations in the synaptosomal fractions. Each bar represents the group mean ± SD. **p < 0.01, *p < 0.05 vs. NCI; αp < 0.05 vs. MCI.
3-NT levels were also significantly elevated in the PMS (F [3, 27] = 8.953, p < 0.0005), mitochondrial (F [3, 27] = 8.543, p < 0.0005), and synaptosomal fractions (F [3, 27] = 8.123, p < 0.0005). Changes in individual cognitive groups closely mirrored those observed in the PC analysis and were not significantly different from each other (Fig. 7B).
Analysis of 4-HNE demonstrated a significant increase in the PMS (F [3, 27] = 11.390, p < 0.0001), mitochondria (F [3, 27] = 5.206, p < 0. 01), and synaptosomes (F [3, 27] = 20.672, p < 0.0001) fractions (Fig. 7C). Post hoc analysis revealed that for all fractions, the levels of 4-HNE were significantly elevated in cognitively impaired groups compared to NCI cohorts but were not significantly different from each other with the exception of the mitochondrial fraction in the AD group that was significantly higher than the MCI group. Together these data indicate that the increase in 4-HNE is an early and persistent event.
Significantly increased acrolein levels were also found in the PMS (F [3, 27] = 9.978, p < 0.0001), mitochondria (F [3, 27] = 6.594, p < 0.005), and synaptosomal fractions (F [3, 27] = 28.410, p < 0.0001). Changes as a function of cognitive status mirrored that observed in the 4-HNE analysis with the greatest increases observed in the AD groups (Fig. 7D). Both the mAD and AD groups were significantly elevated compared to the MCI group in the synaptosomal fraction. The levels of protein oxidation, nitration, and LPO in the synaptosomal fraction were analyzed for possible association with each individual’s MMSE score. There was a significant positive correlation with PCs (r = .536, p < 0.005), 3-NT (r = 0.560, p < 0.005), 4-HNE (r = 0.675, p < 0.0001) and acrolein (r = 0.786, p < 0.0001) (Fig. 8).
Figure 8.

Correlations of protein oxidation, nitration and lipid peroxidation in the synaptosomal fraction with the subjects’ Mini Mental State Examination (MMSE) scores. (A–D) There was a positive correlation for protein carbonyls (A), 3-nitrotyrosiine (B), 5-hydroxynonenal (C) and acrolein (D) of increasing levels of oxidative marker with MMSE decline. All cognitive test scores were obtained within 12 months prior to death. **p < 0.0001, *p < 0.005.
With one exception, no significant relationship between protein oxidation, nitration and LPO of the PMS, mitochondrial, and synaptosomal fractions with Braak scores was identified (p > 0.05). In the mitochondrial fraction, levels of PCs were significantly elevated in the subjects with the highest Braak scores (p < 0.05) (not shown).
DISCUSSION
Our results support and extend previous findings of enhanced oxidative stress in MCI and mAD. Numerous studies have established that MCI is a transition state between normal aging and dementia (39, 40, 47). There is also compelling evidence that oxidative stress is elevated in the brains of individuals with MCI (15, 48–51). We analyzed FC samples not only from subjects with MCI but also mild and late stage AD. For all cellular oxidative markers studied there was a stepwise increase in each of the subcellular fractions as a function of cognitive change, reinforcing the idea that oxidative stress is not only an early event but also an active, persistent process.
Oxidative stress has been implicated in many neurodegenerative disorders, including AD (24, 52–54), and is associated with ROS formation in neurons and synaptosomes (52, 55). While neuronal membranes are rich in polyunsaturated fatty acids (PUFA), a source of LPO reactions, their ability to combat oxidative stress is limited (4). The cortex from individuals with AD has increased PUFA oxidation, altered antioxidant defenses, high rates of oxygen consumption, and oxidative stress (22) with regional variations (23–25). Levels of oxidative DNA damage in the brains of individuals with AD are significantly elevated in the mitochondria from frontal, parietal, and temporal lobes (25). The attention on mitochondria is not only due to its ATP production, but also because of the many pro- and anti-apoptotic molecules associated with these organelles (56). Mitochondria regulate intracellular Ca2+ homeostasis and ROS formation (57, 58), and contribute to neurodegeneration (22, 25, 26).
All samples used were obtained from the superior FC in cases with relatively short postmortem intervals. Although the AD-related involvement of this region is well documented, the literature reporting possible oxidative stress is mixed. Early studies reported both significant elevations in TBARS (59, 60), or no change in late stage AD compared to age-matched controls (61–63). Lipid peroxidation changes as determined by F2-isoprostanes were only recently reported in the FC (50) in AD and in MCI. The levels were moderately elevated in AD compared to MCI. A more recent study using F2-isoprostanes reported significant FC increases in mAD but no significant differences in MCI (64). The levels also failed to correlate with the clinical diagnosis but were strongly correlated with AD pathology. In the present study, significant increases in all markers of oxidative stress were observed in the MCI and in both mAD and AD groups. The greatest changes were observed in the synaptosomal fraction with AD subjects having significantly greater LPO than both MCI and mAD cases. In all fractions, there were no significant differences between the MCI and mAD groups, supporting the idea that MCI is an early and perhaps progressive transition from a cognitively normal state.
A decrease in GSH levels and subsequent GSSG production has been linked to neuronal loss in AD (11, 65). The GSH/GSSG ratio is an indicator of oxidative and/or nitrosative burden in the system, which was decreased significantly in FC (all 3 fractions) of the MCI, mAD, and AD groups. The low levels of GSH may be directly related to increased ROS/RNS, lipid peroxides, and highly reactive hydroxyl radicals (9, 46, 66, 67). A low GSH/GSSG ratio could contribute to promoting free radical load and oxidative stress (10, 65).
To eliminate the peroxides, GSH works in conjunction with GPx and produces GSSG, which is reconverted to GSH by GR at the consumption of NADPH. A reduction in GSH may impair H2O2 clearance and promote OH formation, thus increasing the free radical load, which triggers oxidative stress (9, 10, 30, 46) and pathological pathways in AD (68). GSH also acts as a peroxynitrite reductant, thereby providing enzymatic defense against peroxynitrites. Reduced GPx activity in mitochondria and synaptosomes could directly affect the clearance of ROS and lipid peroxides, as indicated by elevated levels of oxidative markers. GST, another detoxification enzyme (9, 46, 69), can alleviate damage from 4-HNE and acrolein, by catalyzing its conjugation with GSH (70). The depletion of GST observed in all 3 FC fractions could result in increased protein modification/dysfunction leading to further oxidative stress and decline of GSH. Balancing the GSH/GSSG level requires an important cofactor, NADPH, which is produced from reactions catalyzed by G-6PD and 6-phosphogluconate dehydrogenase. Thus, GST, GR, and G-6PD are secondary antioxidant enzymes that play an important role in detoxifying ROS/RNS by maintaining a ready supply of intermediates such as GSH and NADPH (9, 10, 30, 71). In the present study, these 3 enzymes were significantly depleted in the synaptosomal fraction. Decreased activity of GR would directly affect GSH as would a reduction in the levels of GST, resulting in overall low levels of antioxidant defense, thus predicting the overpowering influences of ROS/RNS in AD (9, 72). Prior studies have reported increased levels of G-6PD in end stage AD subjects (73, 74), suggesting a complex role for some antioxidant enzymes.
The most abundant ROS superoxide radical (O2−•) has been implicated in memory function and synaptic plasticity (75, 76) and also in neuronal death (77, 78). The first enzymatic reaction in the reduction pathway is dismutation of 2 molecules of O2−• when they are converted into H2O2 and O2. The enzyme Cu/Zn-SOD and/or manganese-SOD protect neurons from high O2−• environment (78, 79). Moreover, GPx and CAT participate in the elimination of H2O2, which is one of the most toxic molecules in the brain (9, 46, 80). Depletion in CAT in synaptosomes and PMS fractions represents the loss of one of the major defense against ROS. Decreased activity of manganese-SOD in mitochondrial and synaptosomal fractions could lead to further oxidative stress and progressively enhance peroxynitrite production as part of the secondary damage cascade (78, 79, 81). Decreased activity of Cu/Zn-SOD also compromises defense capacities against oxidative stress (10, 30) in PMS fractions. In the 3 FC fractions analyzed, CAT and SOD levels were significantly decreased in MCI, mAD, and AD. It is unclear if this decline precedes the onset of oxidative stress or is in response to increased free radical production.
During oxidative stress, formation of carbonyls derived from proteins and lipids such as PCs, 4-HNE and acrolein cause damage to biomembranes and participate in the formation of NFTs in AD (16, 82, 83). These carbonyls (alkenals) form the immediate substrate for GSH and are involved in neuronal apoptosis in MCI and AD (17), which is seen as a consequence of GSH depletion. Elevated oxidative stress resulting from PC, 3-NT, 4-HNE, or acrolein can lead to delayed neuronal death in FC of MCI, mAD, and AD. Cortical synaptosomes show decreased ability to detoxify lipid peroxides, including 4-HNE (84), suggesting that decreases in antioxidants play a role in AD-related pathology (4). The present findings support the idea that the deleterious effects of increased oxidative stress may be the consequence of diminished antioxidant defenses. Levels of 4-HNE and acrolein were especially elevated in synaptosomal fractions of the FC for MCI, mAD, and AD, suggesting that the antioxidant defenses were not able to offset oxidative stress. The reactivity of 4-HNE with mitochondrial key enzymes (18) increases free radical release into the cytoplasm (19) and downstream effects of mitochondrial dysfunction yield to loss of synaptic function and neuronal death in AD (20, 21). Protein carbonyl, 3-NT, 4-HNE and acrolein levels were elevated throughout the MCI, mAD, and AD samples in this study signaling protein modification via oxidation, nitration and binding with the lipid byproducts acrolein and 4-HNE. Synaptic proteins also might be affected through these modifications. These results provide further insight into the relationship of synaptic and mitochondrial oxidative stress and the progression of cognitive change associated with AD. Oxidative stress related damage to nucleic acids have been suggested as a prime contributing factor in the disease progression as a result of mistakes in base pairings resulting in compromised transcription and translation (85).
Relatively recent studies have shown that β-amyloid (Aβ) plays a central role in AD and may be key in disease-related synaptic change (86). Aβ oligomers can induce AD-like pathology including oxidative stress (87) and play an important role in synaptic loss, which is linked to increased synaptic activity (88–90).
The nature of the terminal illness and the rapidity of death, as well as the methods used to harvest, prepare, and store tissue samples can play a critical role in determining postmortem changes, especially neurochemical parameters (91–93). Hypoxia is one of the key agonal events that can have a major impact on neurochemical analyses and tissue morphology. Prolonged agonal state with marked hypoxia increases tissue lactate levels, which lowers the brain pH, thus affecting protein integrity. Although we cannot control the agonal state or the various conditions of each patient in the period prior to death, we screened each case before including it in this study; cases in which there is an obvious event that would affect the tissue analyses were excluded. Therefore, we feel confident that our results were not influenced by differences in agonal states between groups.
This is the first study to report a strong correlation between levels of synaptic LPO, protein oxidation and nitration, and the subjects’ global cognitive status (Fig. 8). Changes in antioxidant levels (GSH, SOD, CAT) also strongly correlated with the MMSE score, supporting the idea that significant changes in oxidative stress is an early event that plays an important role in the progression of the disease. While it is unclear whether or not the decline in antioxidant level precedes the increase in oxidants, the levels certainly are not capable of neutralizing them. These finding suggests that efforts aimed at increasing brain antioxidants should have beneficial values if enacted early in the disease process or as a prophylactic measure.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health: AG27219, PO1 AG14449, AG028383.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no competing financial interests.
REFERENCES
- 1.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 2.Halliwell B. Role of free radicals in the neurodegenerative diseases: Therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18:685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- 3.Faraci FM. Reactive oxygen species: Influence on cerebral vascular tone. J Appl Physiol. 2006;100:739–743. doi: 10.1152/japplphysiol.01044.2005. [DOI] [PubMed] [Google Scholar]
- 4.Mantha AK, Moorthy K, Cowsik SM, et al. Neuroprotective role of neurokinin B (NKB) on beta-amyloid (25–35) induced toxicity in aging rat brain synaptosomes: Involvement in oxidative stress and excitotoxicity. Biogerontology. 2006;7:1–17. doi: 10.1007/s10522-005-6043-0. [DOI] [PubMed] [Google Scholar]
- 5.Markesbery WR, Lovell MA. DNA oxidation in Alzheimer's disease. Antioxid Redox Signal. 2006;8:2039–2045. doi: 10.1089/ars.2006.8.2039. [DOI] [PubMed] [Google Scholar]
- 6.Markesbery WR, Lovell MA. Damage to lipids, proteins, DNA, and RNA in mild cognitive impairment. Arch Neurol. 2007;64:954–956. doi: 10.1001/archneur.64.7.954. [DOI] [PubMed] [Google Scholar]
- 7.Moreira PI, Nunomura A, Nakamura M, et al. Nucleic acid oxidation in Alzheimer disease. Free Radic Biol Med. 2008;44:1493–1505. doi: 10.1016/j.freeradbiomed.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Starke DW, Chen Y, Bapna CP, et al. Sensitivity of protein sulfhydryl repair enzymes to oxidative stress. Free Radic Biol Med. 1997;23:373–384. doi: 10.1016/s0891-5849(97)00009-9. [DOI] [PubMed] [Google Scholar]
- 9.Ansari MA, Joshi G, Huang Q, et al. In vivo administration of D609 leads to protection of subsequently isolated gerbil brain mitochondria subjected to in vitro oxidative stress induced by amyloid beta-peptide and other oxidative stressors: Relevance to Alzheimer's disease and other oxidative stress-related neurodegenerative disorders. Free Radic Biol Med. 2006;41:1694–1703. doi: 10.1016/j.freeradbiomed.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med. 2008;45:443–452. doi: 10.1016/j.freeradbiomed.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benzi G, Moretti A. Age- and peroxidative stress-related modifications of the cerebral enzymatic activities linked to mitochondria and the glutathione system. Free Radic Biol Med. 1995;19:77–101. doi: 10.1016/0891-5849(94)00244-e. [DOI] [PubMed] [Google Scholar]
- 12.Martinez M, Ferrandiz ML, Diez A, et al. Depletion of cytosolic GSH decreases the ATP levels and viability of synaptosomes from aged mice but not from young mice. Mech Ageing Dev. 1995;84:77–81. doi: 10.1016/0047-6374(95)01644-f. [DOI] [PubMed] [Google Scholar]
- 13.Good PF, Werner P, Hsu A, et al. Evidence of neuronal oxidative damage in Alzheimer's disease. Am J Pathol. 1996;149:21–28. [PMC free article] [PubMed] [Google Scholar]
- 14.Smith MA, Richey Harris PL, Sayre LM, et al. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams TI, Lynn BC, Markesbery WR, et al. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer's disease. Neurobiol Aging. 2006;27:1094–1099. doi: 10.1016/j.neurobiolaging.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Calingasan NY, Uchida K, Gibson GE. Protein-bound acrolein: A novel marker of oxidative stress in Alzheimer's disease. J Neurochem. 1999;72:751–756. doi: 10.1046/j.1471-4159.1999.0720751.x. [DOI] [PubMed] [Google Scholar]
- 17.Bader Lange ML, Cenini G, Piroddi M, et al. Loss of phospholipid asymmetry and elevated brain apoptotic protein levels in subjects with amnestic mild cognitive impairment and Alzheimer disease. Neurobiol Dis. 2008;29:456–464. doi: 10.1016/j.nbd.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Floyd RA, Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002;23:795–807. doi: 10.1016/s0197-4580(02)00019-2. [DOI] [PubMed] [Google Scholar]
- 19.Cash AD, Perry G, Ogawa O, et al. Is Alzheimer's disease a mitochondrial disorder? Neuroscientist. 2002;8:489–496. doi: 10.1177/107385802236968. [DOI] [PubMed] [Google Scholar]
- 20.Onyango I, Khan S, Miller B, et al. Mitochondrial genomic contribution to mitochondrial dysfunction in Alzheimer's disease. J Alzheimer’s Dis. 2006;9:183–193. doi: 10.3233/jad-2006-9210. [DOI] [PubMed] [Google Scholar]
- 21.Reddy PH, Beal MF. Are mitochondria critical in the pathogenesis of Alzheimer's disease? Brain Res Brain Res Rev. 2005;49:618–632. doi: 10.1016/j.brainresrev.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Cardoso SM, Santana I, Swerdlow RH, et al. Mitochondria dysfunction of Alzheimer's disease cybrids enhances Aβ toxicity. J Neurochem. 2004;89:1417–1426. doi: 10.1111/j.1471-4159.2004.02438.x. [DOI] [PubMed] [Google Scholar]
- 23.Culmsee C, Landshamer S. Molecular insights into mechanisms of the cell death program: role in the progression of neurodegenerative disorders. Curr Alzheimer Res. 2006;3:269–283. doi: 10.2174/156720506778249461. [DOI] [PubMed] [Google Scholar]
- 24.Hensley K, Hall N, Subramaniam R, et al. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Xiong S, Xie C, et al. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 26.Zhu X, Smith MA, Perry G, et al. Mitochondrial failures in Alzheimer's disease. Am J Alzheimer’s Dis Other Demen. 2004;19:345–352. doi: 10.1177/153331750401900611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forero DA, Casadesus G, Perry G, et al. Synaptic dysfunction and oxidative stress in Alzheimer's disease: Emerging mechanisms. J Cell Mol Med. 2006;10:796–805. doi: 10.1111/j.1582-4934.2006.tb00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Urano S, Asai Y, Makabe S, et al. Oxidative injury of synapse and alteration of antioxidative defense systems in rats, and its prevention by vitamin E. Eur J Biochem. 1997;245:64–70. doi: 10.1111/j.1432-1033.1997.00064.x. [DOI] [PubMed] [Google Scholar]
- 29.Mattson MP, Partin J, Begley JG. Amyloid beta-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 1998;807:167–176. doi: 10.1016/s0006-8993(98)00763-x. [DOI] [PubMed] [Google Scholar]
- 30.Ansari MA, Roberts KN, Scheff SW. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma. 2008;25:513–526. doi: 10.1089/neu.2007.0451. [DOI] [PubMed] [Google Scholar]
- 31.Gilman CP, Chan SL, Guo Z, et al. p53 is present in synapses where it mediates mitochondrial dysfunction and synaptic degeneration in response to DNA damage, and oxidative and excitotoxic insults. Neuromolecular Med. 2003;3:159–172. doi: 10.1385/NMM:3:3:159. [DOI] [PubMed] [Google Scholar]
- 32.Faff-Michalak L, Albrecht J. The two catalytic components of the 2-oxoglutarate dehydrogenase complex in rat cerebral synaptic and nonsynaptic mitochondria: Comparison of the response to in vitro treatment with ammonia, hyperammonemia, and hepatic encephalopathy. Neurochem Res. 1993;18:119–123. doi: 10.1007/BF01474673. [DOI] [PubMed] [Google Scholar]
- 33.Gillardon F, Rist W, Kussmaul L, et al. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics. 2007;7:605–616. doi: 10.1002/pmic.200600728. [DOI] [PubMed] [Google Scholar]
- 34.Brown MR, Sullivan PG, Geddes JW. Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J Biol Chem. 2006;281:11658–11668. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- 35.Naga KK, Sullivan PG, Geddes JW. High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J Neurosci. 2007;27:7469–7475. doi: 10.1523/JNEUROSCI.0646-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldman WP, Price JL, Storandt M, et al. Absence of cognitive impairment or decline in preclinical Alzheimer's disease. Neurology. 2001;56:361–367. doi: 10.1212/wnl.56.3.361. [DOI] [PubMed] [Google Scholar]
- 37.Haroutunian V, Perl DP, Purohit DP, et al. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998;55:1185–1191. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- 38.Schmitt FA, Davis DG, Wekstein DR, et al. "Preclinical" AD revisited: Neuropathology of cognitively normal older adults. Neurology. 2000;55:370–376. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- 39.Flicker C, Ferris SH, Reisberg B. Mild cognitive impairment in the elderly: Predictors of dementia. Neurology. 1991;41:1006–1009. doi: 10.1212/wnl.41.7.1006. [DOI] [PubMed] [Google Scholar]
- 40.Morris JC, Storandt M, Miller JP, et al. Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol. 2001;58:397–405. doi: 10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- 41.Petersen RC. Mild cognitive impairment: Transition between aging and Alzheimer's disease. Neurologia. 2000;15:93–101. [PubMed] [Google Scholar]
- 42.Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol. 2001;58:1985–1992. doi: 10.1001/archneur.58.12.1985. [DOI] [PubMed] [Google Scholar]
- 43.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 44.Scheff SW, Price DA, Schmitt FA, et al. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 45.Garey RE, Harper JW, Heath RG. Postmortem isolation of synaptosomes from human brain. Brain Res. 1974;82:151–162. doi: 10.1016/0006-8993(74)90900-7. [DOI] [PubMed] [Google Scholar]
- 46.Ahmad AS, Ansari MA, Ahmad M, et al. Neuroprotection by crocetin in a hemi-parkinsonian rat model. Pharmacol Biochem Behav. 2005;81:805–813. doi: 10.1016/j.pbb.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 47.Luis CA, Loewenstein DA, Acevedo A, et al. Mild cognitive impairment: directions for future research. Neurology. 2003;61:438–444. doi: 10.1212/01.wnl.0000080366.90234.7f. [DOI] [PubMed] [Google Scholar]
- 48.Butterfield DA, Reed T, Perluigi M, et al. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2006;397:170–173. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 49.Keller JN, Schmitt FA, Scheff SW, et al. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 50.Markesbery WR, Kryscio RJ, Lovell MA, et al. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann Neurol. 2005;58:730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- 51.Pratico D, Sung S. Lipid peroxidation and oxidative imbalance: early functional events in Alzheimer's disease. J Alzheimers Dis. 2004;6:171–175. doi: 10.3233/jad-2004-6209. [DOI] [PubMed] [Google Scholar]
- 52.Butterfield DA, Boyd-Kimball D. The critical role of methionine 35 in Alzheimer's amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity. Biochim Biophys Acta. 2005;1703:149–156. doi: 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 53.Perry G, Castellani RJ, Smith MA, et al. Oxidative damage in the olfactory system in Alzheimer's disease. Acta Neuropathol. 2003;106:552–556. doi: 10.1007/s00401-003-0761-7. [DOI] [PubMed] [Google Scholar]
- 54.Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Chem Res Toxicol. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- 55.Cardoso SM, Pereira C, Oliveira CR. The protective effect of vitamin E, idebenone and reduced glutathione on free radical mediated injury in rat brain synaptosomes. Biochem Biophys Res Commun. 1998;246:703–710. doi: 10.1006/bbrc.1998.8563. [DOI] [PubMed] [Google Scholar]
- 56.Antonsson B. Mitochondria and the Bcl-2 family proteins in apoptosis signaling pathways. Mol Cell Biochem. 2004;256–257:141–155. doi: 10.1023/b:mcbi.0000009865.70898.36. [DOI] [PubMed] [Google Scholar]
- 57.Marlatt M, Lee HG, Perry G, et al. Sources and mechanisms of cytoplasmic oxidative damage in Alzheimer's disease. Acta Neurobiol Exp (Wars) 2004;64:81–87. doi: 10.55782/ane-2004-1493. [DOI] [PubMed] [Google Scholar]
- 58.Sheehan JP, Swerdlow RH, Miller SW, et al. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer's disease. J Neurosci. 1997;17:4612–4622. doi: 10.1523/JNEUROSCI.17-12-04612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Balazs L, Leon M. Evidence of an oxidative challenge in the Alzheimer's brain. Neurochem Res. 1994;19:1131–1137. doi: 10.1007/BF00965146. [DOI] [PubMed] [Google Scholar]
- 60.Subbarao KV, Richardson JS, Ang LC. Autopsy samples of Alzheimer's cortex show increased peroxidation in vitro. Journal of Neurochemistry. 1990;55:342–345. doi: 10.1111/j.1471-4159.1990.tb08858.x. [DOI] [PubMed] [Google Scholar]
- 61.Lovell MA, Ehmann WD, Butler SM, et al. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer's disease. Neurology. 1995;45:1594–1601. doi: 10.1212/wnl.45.8.1594. [DOI] [PubMed] [Google Scholar]
- 62.Lyras L, Cairns NJ, Jenner A, et al. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer's disease. J Neurochem. 1997;68:2061–2069. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- 63.Palmer AM, Burns MA. Selective increase in lipid peroxidation in the inferior temporal cortex in Alzheimer's disease. Brain Res. 1994;645:338–342. doi: 10.1016/0006-8993(94)91670-5. [DOI] [PubMed] [Google Scholar]
- 64.Forman MS, Mufson EJ, Leurgans S, et al. Cortical biochemistry in MCI and Alzheimer disease: lack of correlation with clinical diagnosis. Neurology. 2007;68:757–763. doi: 10.1212/01.wnl.0000256373.39415.b1. [DOI] [PubMed] [Google Scholar]
- 65.Bains JS, Shaw CA. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res Brain Res Rev. 1997;25:335–358. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- 66.Ansari MA, Ahmad AS, Ahmad M, et al. Selenium protects cerebral ischemia in rat brain mitochondria. Biol Trace Elem Res. 2004;101:73–86. doi: 10.1385/BTER:101:1:73. [DOI] [PubMed] [Google Scholar]
- 67.Beckman JS, Beckman TW, Chen J, et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perez-De La Cruz V, Gonzalez-Cortes C, Galvan-Arzate S, et al. Excitotoxic brain damage involves early peroxynitrite formation in a model of Huntington's disease in rats: Protective role of iron porphyrinate 5,10,15,20-etrakis (4-sulfonatophenyl)porphyrinate iron (III) Neuroscience. 2005;135:463–474. doi: 10.1016/j.neuroscience.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 69.Xie C, Lovell MA, Markesbery WR. Glutathione transferase protects neuronal cultures against four hydroxynonenal toxicity. Free Radic Biol Med. 1998;25:979–988. doi: 10.1016/s0891-5849(98)00186-5. [DOI] [PubMed] [Google Scholar]
- 70.Baez S, Segura-Aguilar J, Widersten M, et al. Glutathione transferases catalyse the detoxication of oxidized metabolites (o-quinones) of catecholamines and may serve as an antioxidant system preventing degenerative cellular processes. Biochem J. 1997;324:25–28. doi: 10.1042/bj3240025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shah ZA, Vohora SB. Antioxidant/restorative effects of calcined gold preparations used in Indian systems of medicine against global and focal models of ischaemia. Pharmacol Toxicol. 2002;90:254–259. doi: 10.1034/j.1600-0773.2002.900505.x. [DOI] [PubMed] [Google Scholar]
- 72.Ishrat T, Khan MB, Hoda MN, et al. Coenzyme Q10 modulates cognitive impairment against intracerebroventricular injection of streptozotocin in rats. Behav Brain Res. 2006;171:9–16. doi: 10.1016/j.bbr.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 73.Russell RL, Siedlak SL, Raina AK, et al. Increased neuronal glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch Biochem Biophys. 1999;370:236–239. doi: 10.1006/abbi.1999.1404. [DOI] [PubMed] [Google Scholar]
- 74.Martins RN, Harper CG, Stokes GB, et al. Increased cerebral glucose-6-phosphate dehydrogenase activity in Alzheimer's disease may reflect oxidative stress. J Neurochem. 1986;46:1042–1045. doi: 10.1111/j.1471-4159.1986.tb00615.x. [DOI] [PubMed] [Google Scholar]
- 75.Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal. 2007;9:233–244. doi: 10.1089/ars.2007.9.ft-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tejada-Simon MV, Serrano F, Villasana LE, et al. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol Cell Neurosci. 2005;29:97–106. doi: 10.1016/j.mcn.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harman D. Alzheimer's disease: role of aging in pathogenesis. Ann N Y Acad Sci. 2002;959:384–395. doi: 10.1111/j.1749-6632.2002.tb02109.x. discussion 463-65. [DOI] [PubMed] [Google Scholar]
- 78.Resende R, Moreira PI, Proenca T, et al. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic Biol Med. 2008;44:2051–2057. doi: 10.1016/j.freeradbiomed.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 79.Callio J, Oury TD, Chu CT. Manganese superoxide dismutase protects against 6-hydroxydopamine injury in mouse brains. J Biol Chem. 2005;280:18536–18542. doi: 10.1074/jbc.M413224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dringen R, Pawlowski PG, Hirrlinger J. Peroxide detoxification by brain cells. J Neurosci Res. 2005;79:157–165. doi: 10.1002/jnr.20280. [DOI] [PubMed] [Google Scholar]
- 81.Bayir H, Kagan VE, Clark RS, et al. Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J Neurochem. 2007;101:168–181. doi: 10.1111/j.1471-4159.2006.04353.x. [DOI] [PubMed] [Google Scholar]
- 82.Sayre LM, Zelasko DA, Harris PL, et al. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 83.Smith MA, Sayre LM, Anderson VE, et al. Cytochemical demonstration of oxidative damage in Alzheimer disease by immunochemical enhancement of the carbonyl reaction with 2,4-dinitrophenylhydrazine. J Histochem Cytochem. 1998;46:731–735. doi: 10.1177/002215549804600605. [DOI] [PubMed] [Google Scholar]
- 84.Sidell KR, Montine KS, Picklo MJ, Sr, et al. Mercapturate metabolism of 4-hydroxy-2-nonenal in rat and human cerebrum. J Neuropathol Exp Neurol. 2003;62:146–153. doi: 10.1093/jnen/62.2.146. [DOI] [PubMed] [Google Scholar]
- 85.Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 86.Marcello E, Epis R, Di Luca M. Amyloid flirting with synaptic failure: Towards a comprehensive view of Alzheimer's disease pathogenesis. Eur J Pharmacol. 2008;585:109–118. doi: 10.1016/j.ejphar.2007.11.083. [DOI] [PubMed] [Google Scholar]
- 87.De Felice FG, Velasco PT, Lambert MP, et al. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 88.Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 89.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Knobloch M, Mansuy IM. Dendritic spine loss and synaptic alterations in Alzheimer's disease. Mol Neurobiol. 2008;37:73–82. doi: 10.1007/s12035-008-8018-z. [DOI] [PubMed] [Google Scholar]
- 91.Arranz B, Blennow K, Ekman R, et al. Brain monoaminergic and neuropeptidergic variations in human aging. J Neural Transm. 1996;103:101–115. doi: 10.1007/BF01292620. [DOI] [PubMed] [Google Scholar]
- 92.Dodd PR, Hambley JW, Cowburn RF, et al. A comparison of methodologies for the study of functional transmitter neurochemistry in human brain. J Neurochem. 1988;50:1333–1345. doi: 10.1111/j.1471-4159.1988.tb03013.x. [DOI] [PubMed] [Google Scholar]
- 93.Palmer AM, Lowe SL, Francis PT, et al. Are post-mortem biochemical studies of human brain worthwhile? Biochem Soc Trans. 1988;16:472–475. doi: 10.1042/bst0160472. [DOI] [PubMed] [Google Scholar]