

Abstract
The dynamic alterations in the cardiac extracellular matrix following myocardial infarction not only determine the mechanical properties of the infarcted heart, but also directly modulate the inflammatory and reparative response. During the inflammatory phase of healing, rapid activation of matrix metalloproteinases (MMP) causes degradation of the cardiac extracellular matrix. Matrix fragments exert potent pro-inflammatory actions, while MMPs process cytokines and chemokines altering their biological activity. In addition, vascular hyperpermeability results in extravasation of fibronectin and fibrinogen leading to formation of a plasma-derived provisional matrix that serves as a scaffold for leukocyte infiltration. Clearance of the infarct from dead cells and matrix debris is essential for resolution of inflammation and marks the transition to the proliferative phase. The fibrin-based provisional matrix is lysed and cellular fibronectin is secreted. ED-A fibronectin, mechanical tension and Transforming Growth Factor (TGF)-β are essential for modulation of fibroblasts into myofibroblasts, the main collagen-secreting cells in the wound. The matricellular proteins thrombospondin-1 and -2, osteopontin, tenascin-C, periostin, and secreted protein acidic and rich in cysteine (SPARC) are induced in the infarct regulating cellular interactions and promoting matrix organization. As the infarct matures, matrix cross-linking results in formation of a dense collagen-based scar. At this stage, shielding of fibroblasts from external mechanical tension by the mature matrix network may promote deactivation and cellular quiescence. The components of the extracellular matrix do not passively follow the pathologic alterations of the infarcted heart but critically modulate inflammatory and reparative pathways by transducing signals that affect cell survival, phenotype and gene expression.
Keywords: myocardial infarction, remodeling, Matrix Metalloproteinases, hyaluronan, collagen, matricellular proteins, myofibroblast, cytokine, chemokines, TGF-β, wound healing
1. Introduction: the cardiac extracellular matrix
The cardiac extracellular matrix is critical to maintaining the structural integrity of the heart. Disruption of the matrix network results in alterations of the ventricular geometry and is associated with both systolic and diastolic dysfunction [1], [2], [3]. Perhaps the most dramatic changes in the composition of the cardiac extracellular matrix occur in the setting of acute myocardial infarction. Cardiomyocyte death triggers an intense inflammatory reaction leading to infiltration of the infarct with activated leukocytes. The pro-inflammatory environment is associated with protease activation and matrix degradation. As professional phagocytes clear the wound from dead cells and matrix debris, the reparative phase begins. Myofibroblasts accumulate in the infarcted area and produce large amounts of extracellular matrix proteins. This collagen-based matrix will ultimately become a mature scar that provides mechanical support to the infarcted heart. These dynamic alterations in the composition of the matrix in the infarcted heart are critical determinants of outcome following myocardial infarction. Excessive early degradation of the cardiac matrix network and defective or delayed formation of newly-synthesized matrix proteins may play an important role in the pathogenesis of cardiac rupture, a dramatic and fatal complication of acute myocardial infarction. In the later stages of healing, defects in extracellular matrix composition alter the mechanical properties of the heart resulting in enhanced ventricular dilation and increased sphericity of the ventricle. These geometric changes, termed post-infarction remodeling, are associated with increased mortality and a higher incidence of arrhythmias, and are linked with the development of chronic heart failure [4]. Beyond their obvious contribution in providing mechanical support to the infarcted heart, extracellular matrix proteins also act as key regulators of the reparative response by modulating cellular interactions. Matrix components are critically involved in inflammatory pathways and modulate fibroblast phenotype and gene expression, thus playing an important role in the cell biology of cardiac repair. The current review focuses on the dynamic role of extracellular matrix proteins in regulation of inflammatory and fibrotic signals in the infarcted heart.
2. How does the extracellular matrix modulate the inflammatory and reparative response?
Because the heart has negligible regenerative capacity, myocardial infarction ultimately results in replacement of dead cardiomyocytes with a collagen-based scar. From a descriptive viewpoint, the cellular and molecular events involved in the reparative response can be divided in three overlapping phases (Figure 1). Cardiomyocyte death rapidly triggers cytokine, chemokine and adhesion molecule expression initiating the inflammatory phase and leading to recruitment of leukocytes in the infarct. As the wound is cleared from dead cells, activation of “stop signals” (such as Interleukin (IL)-10 and Transforming Growth Factor (TGF)-β) suppresses the inflammatory reaction leading to transition to the proliferative phase. Fibroblasts proliferate and acquire specialized contractile features differentiating into contractile cells, termed myofibroblasts. These phenotypically modulated fibroblasts secrete large amounts of extracellular matrix proteins in the infarct. The maturation phase follows, during which extracellular matrix proteins are cross-linked and a mature scar is formed, while most fibroblasts and vascular cells undergo apoptosis. Understanding the role of matrix components in regulation of the cellular events associated with cardiac repair requires knowledge of the dynamic changes in the extracellular matrix network over the course of infarct healing.
Figure 1.
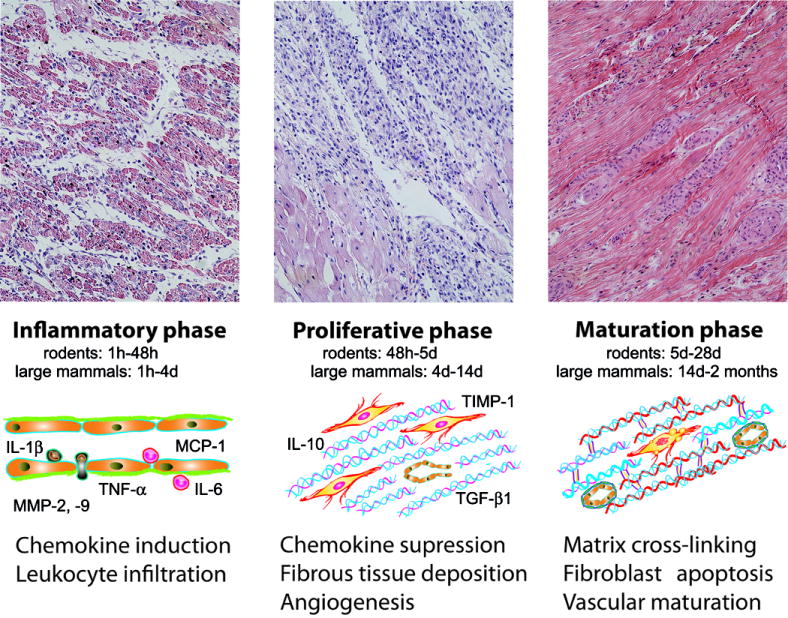
The phases of infarct healing. Myocardial infarction triggers a reparative response that can be divided in three overlapping phases. During the inflammatory phase, chemokine, cytokine and adhesion molecule expression is upregulated leading to leukocyte recruitment in the infarct. As the acute inflammatory response is suppressed, mesenchymal cells infiltrate the infarct marking the transition to the proliferative phase. Myofibroblasts deposit extracellular matrix while a rich capillary network is formed. During the maturation phase the cellularity of the infarct decreases and the matrix is cross-linked forming a dense collagen-based scar. The images show hematoxylin/eosin-stained sections of reperfused canine infarcts after 1 h coronary occlusion followed by 3 days (inflammatory phase), 7 days (proliferative phase) and 2 months (maturation phase) of reperfusion. The time intervals for each phase are shown for both rodent and large animal models of reperfused infarction. In comparison to large mammals, rodents exhibit an accelerated inflammatory and reparative response following myocardial infarction [50], [19]. Symbols: MCP, Monocyte Chemoattractant Protein-1; IL, Interleukin; MMP, Matrix Metalloproteinase; TNF, Tumor Necrosis Factor; TGF, Transforming Growth Factor; TIMP, Tissue Inhibitor of Metalloproteinases.
Extensive evidence suggests that inflammatory pathways play an important role in extracellular matrix metabolism. During the inflammatory phase of infarct healing induction of pro-inflammatory cytokines, such as Tumor Necrosis Factor (TNF)-α and IL-1β, enhances Matrix Metalloproteinase (MMP) expression and activity promoting matrix degradation [5], [6]. During the proliferative phase, repression of pro-inflammatory signals and induction of inhibitory cytokines, such as IL-10 [7] and TGF-β [8], induce Tissue Inhibitor of Metalloproteinase (TIMP) expression enhancing matrix deposition and preservation. However, the interaction between the matrix and the inflammatory and reparative response is reciprocal. The dynamic alterations in the extracellular matrix composition critically regulate inflammatory, fibrogenic and angiogenic pathways in the healing infarct. During the inflammatory phase of healing extracellular matrix fragments enhance cytokine and chemokine synthesis, but may also induce fibroblast activation. In addition, deposition of a plasma-derived provisional matrix provides a scaffold for leukocyte infiltration and may transduce pro-inflammatory signals. During the proliferative phase the temporally and spatially restricted expression of fibronectin and matricellular proteins critically regulates cellular phenotype, survival and gene expression. Finally during the maturation phase collagen cross-linking may promote fibroblast quiescence leading to formation of a mature scar.
3. The cardiac matrix during the inflammatory phase of infarct healing (Figure 2)
Figure 2.

Alterations of the extracellular matrix during the inflammatory phase of infarct healing. Protease activation results in degradation of matrix proteins. MMPs also process inflammatory cytokines and chemokines modulating their activity. Generation of collagen and low molecular weight hyaluronan (H) fragments exerts potent pro-inflammatory effects. In addition, fibrinogen (F) and plasma fibronectin (pFN) are extravasated through the hyperpermeable vasculature resulting in formation of a fibrin-based provisional matrix that serves as a scaffold for migration of inflammatory cells (M, mononuclear cell; N, neutrophil).
Cardiomyocyte death triggers rapid activation of the complement system, generates free radicals and activates Toll-Like Receptor (TLR)-mediated pathways. These overlapping pathways activate Nuclear Factor (NF)-κB in resident myocardial cells inducing expression of cytokines, chemokines and adhesion molecules [9]. Adhesive interactions between leukocytes and activated endothelial cells ultimately result in infiltration of the infarcted myocardium with neutrophils and mononuclear cells. During the inflammatory phase of healing activation of cytokine and chemokine signaling induces a protease-rich environment leading to extensive degradation of the cardiac matrix. Increased permeability of the cardiac microvasculature results in extravasation of fibrinogen and plasma fibronectin forming a provisional matrix network. The cardiac extracellular matrix does not passively follow the changes in the cellular environment. Rather, the early alterations in matrix composition facilitate leukocyte infiltration and critically regulate inflammatory pathways.
3.1 MMP activation and matrix degradation
Myocardial infarction results in early disruption of the cardiac extracellular matrix network [10], [11]. Early activation of latent collagenases may be responsible for the rapid degradation of matrix in the infarcted heart. MMP activation is noted in the cardiac interstitium within 10 minutes after coronary occlusion [12] resulting in marked reduction in collagen content in the infarcted heart [13]. After the latent pool of interstitial collagenases has been depleted, new synthesis of MMP-1 promotes collagenolytic activity in the infarcted area [14]. Collagenases cleave collagens at unique sites generating 75 and 25 kD fragments called gelatins. The gelatins are further degraded into amino acids and oligopeptides by the gelatinases MMP-2 and MMP-9 [15] and by serine proteases [16], [17]. The appearance of type I collagen fragments in the serum within 15-30 minutes after coronary occlusion [18] reflects the extensive matrix degradation in the infarcted heart. Fragmentation of extracellular matrix constituents during the early stages following infarction is not limited to fibrillar collagen; glycosaminoglycans, such as hyaluronan also undergo degradation leading to generation of low molecular weight fragments with pro-inflammatory properties [19], [20].
3.2 MMPs as regulators of inflammatory pathways
Beyond their effects on matrix metabolism, MMPs are capable of modulating inflammatory pathways by processing cytokines, chemokines and growth factors. Several MMPs are capable of releasing active TNF-α from the cell surface enhancing its biological activity [21]. The effects of MMPs on cytokine signaling are not limited to TNF-α, as MMP-mediated actions on IL-1 and TGF-β activity have been reported [22]. MMPs also process chemokines exerting diverse and often contradictory actions. MMP-mediated proteolysis may inactivate a chemokine, may generate antagonistic derivatives capable of binding to a chemokine receptor without inducing a chemotactic response, or may result in formation of a truncated chemokine with more potent activity [22]. In addition, MMPs may interfere with chemokine binding to glycosaminoglycans, a molecular step critical for their chemotactic effects. Detailed reviews of the role of MMPs and TIMPs are provided in the manuscripts authored by Drs Merry Lindsey and Stephane Heymans in this issue of the journal.
3.3 Generation of matrix fragments and their role in the inflammatory reaction
Emerging evidence suggests that generation of matrix fragments may play an important role in leukocyte recruitment linking tissue injury with the inflammatory response [23]. Non-specific collagen fragments and elastin-derived peptides are capable of inducing neutrophil, monocyte and fibroblast chemotaxis [24]. Weathington and co-workers demonstrated that a specific collagen-derived peptide, acetylated proline-glycine-proline (AcPGP) acts as a neutrophil chemoattractant in endotoxin-induced pulmonary inflammation by signaling through the chemokine receptors CXCR1 and CXCR2 [25]. Generation of PGP appears to be a multistep process involving MMP-8, MMP-9 and members of the serine protease family [26]. Although activation of proteolytic enzymes in the healing infarct results in matrix degradation, the significance of specific fragments in triggering the inflammatory and reparative response has not been investigated.
Glycosaminoglycan fragments are also generated upon tissue injury and may be involved in regulation of the inflammatory response. Hyaluronan, exists in a high molecular weight form under physiologic conditions, but undergoes processing to low molecular weight species after tissue injury. Hyaluronan fragments induce the expression of a variety of inflammatory genes by endothelial cells and macrophages [27], including chemokines and cytokines, and may play an important role in regulating the inflammatory processes. Furthermore, clearance of hyaluronan fragments from the injured tissue is essential for resolution of chronic inflammation [28]. Hyaluronan participates in induction and resolution of inflammation through interactions with the transmembrane adhesion molecule CD44 [29], a ubiquitously distributed glycoprotein that mediates a wide variety of cell-cell and cell-matrix interactions. Teder et al. demonstrated that CD44 is a key factor in resolution of pulmonary inflammation through removal of matrix breakdown products and clearance of apoptotic neutrophils [28]. We have recently investigated the significance of CD44-mediated interactions in myocardial infarction [20]. We found disruption of the cardiac hyaluronan network in the infarcted area and demonstrated that CD44 expression was markedly induced in the infarct and was predominantly localized on infiltrating leukocytes, wound myofibroblasts and vascular cells. In comparison with wildtype mice, CD44 -/- animals showed enhanced and prolonged neutrophil and macrophage infiltration and increased expression of pro-inflammatory cytokines following myocardial infarction. CD44 absence was also associated with reduced recruitment of myofibroblasts and markedly decreased collagen deposition in the center of the infarct. Impaired healing in CD44 null mice resulted in accentuated adverse remodeling. Whether the healing defects observed in CD44 deficient animals are due to impaired clearance of hyaluronan fragments from the infarcted myocardium remains unknown.
3.4 The role of the plasma-derived provisional matrix in leukocyte infiltration
As the original cardiac matrix network is degraded, a fibrin-based provisional matrix is formed [19]. Extravasation of plasma proteins results in generation of a complex and dynamic matrix network based on fibrin and plasma fibronectin. In addition to its hemostatic role, the fibrin-based provisional matrix serves as a scaffold for migration and proliferation of infiltrating inflammatory cells, endothelial cells and fibroblasts [30]. The migrating cells use integrin receptors to interact with fibrin and plasma fibronectin within the matrix network; these interactions also provide signals that modulate cellular phenotype and gene expression [31]. Although investigations using dermal endothelial cells and fibroblasts suggest that fibrin and fibronectin critically alter cell behavior, studies examining the role of the provisional matrix in modulating the cardiac reparative response are lacking.
A recent investigation suggested that fibrinogen -/- mice have decreased infarct size in a Langendorf model of myocardial ischemia [32] and demonstrated that fibrinogen-derived fragments contribute to the pathology of ischemic injury. Administration of a naturally occurring peptide that competes with the fibrin fragment N-terminal disulfide knot-II (an analog of the fibrin E1 fragment) for binding to vascular endothelial cadherin prevented leukocyte infiltration in the ischemic myocardium reducing infarct size in experimental models [32]. In a small clinical trial peptide treatment in patients with acute ST-elevation myocardial infarction did not affect the size of the infarct but significantly reduced the necrotic core zone on magnetic resonance imaging [33]. Although these findings suggest that fibrin-mediated interactions may be important in early injury following myocardial infarction, the significance of the plasma protein-derived provisional matrix in infarct healing and cardiac remodeling has not been investigated. In skin wounds, the absence of fibrinogen has only minor consequences on the reparative process: although fibrinogen -/- animals have reduced cutaneous wound tensile strength and delayed wound closure compared with wildtype mice, cutaneous repair is ultimately completed in time with a comparable outcome [34]. However, in contrast to skin wounds, decreased tensile strength in healing myocardial infarcts may result in enhanced left ventricular remodeling and significant ventricular dysfunction.
4. The cardiac extracellular matrix during the proliferative phase of infarct healing (Figure 3)
Figure 3.

Alterations of the extracellular matrix during the proliferative phase of infarct healing. As the provisional matrix is lysed and matrix fragments are cleared, fibroblasts (F) and macrophages (Ma) secrete cellular fibronectin (cFN) and hyaluronan (H) forming a “second order” provisional matrix. ED-A fibronectin, TGF-β and mechanical tension due to loss of the shielding effects of the normal matrix, contribute to myofibroblast (MF) differentiation and activation. The matricellular proteins TSP-1, OPN, SPARC and tenascin-C are transiently induced in the infarcted heart and modulate cellular interactions and matrix assembly.
The transition from the inflammatory to the proliferative phase is associated with activation of pathways that inhibit pro-inflammatory signals while promoting fibrous tissue deposition and angiogenesis. The infarct environment becomes highly cellular; activated myofibroblasts proliferate and secrete extracellular matrix proteins while endothelial cells form microvessels that provide oxygen and nutrients to the healing wound. The matrix alterations during the proliferative phase of healing provide essential signals for myofibroblast activation, matrix organization, and repression of the inflammatory reaction.
4.1 Clearance of the plasma-derived provisional matrix
The plasma-derived provisional matrix is lysed by proteolytic enzymes produced by granulation tissue cells, and is replaced by an organized cell-derived provisional matrix containing cellular fibronectin and hyaluronan [19]. Clearance of the plasma protein-based provisional matrix is mediated by the plasminogen system and appears to be an essential step in the reparative process. Creemers and coworkers demonstrated that infarct healing was abolished in plasminogen -/- mice. In the absence of plasminogen, migration of inflammatory cells into the infarcted myocardium was markedly reduced, necrotic cardiomyocytes were not removed and granulation tissue formation was delayed. However, defective infarct healing was not associated with accentuation of the remodeling process [35]. As fibrin and plasma fibronectin are cleared from the infarcted area, cellular fibronectin is secreted by fibroblasts and macrophages and serves as a “second order” provisional matrix [36]. Deposition of fibronectin is essential for myofibroblast transdifferentiation and activation.
4.2 The role of cellular fibronectin in the reparative process
Expression of cellular fibronectin in myocardial infarcts is maximally induced during the proliferative phase of healing [37] and is produced primarily by macrophages and fibroblasts. Fibronectin is essential for modulation of fibroblasts toward myofibroblasts, an important process in the reparative response. Myofibroblast transdifferentiation results from the combined action of three distinct factors: a) mechanical tension, b) TGF-β and c) the ED-A splice variant of cellular fibronectin [38], [39], [40]. The ED-A splice segment is included in the fibronectin mRNA in tissue repair leading to de novo expression of ED-A fibronectin. The interaction between the ED-A segment and the cell surface is essential for TGF-β1-mediated myofibroblast transdifferentiation [40]; however, the molecular pathways responsible for this effect remain unknown [41]. Activated myofibroblasts play an important role in wound contraction and are the main source of collagen in the healing infarct [14]. In addition, evidence suggests that fibronectin plays an important role in organization and stabilization of the matrix. In vitro studies demonstrated that fibronectin polymerization into the extracellular matrix is required for the deposition of type I collagen and TSP-1 [42]. The significance of these interactions in cardiac repair has not been investigated.
4.3 Matricellular proteins as regulators of the reparative response
Matricellular proteins are a family of structurally diverse extracellular matrix proteins that do not play a direct structural role but act contextually modulating cell function and activity. They include thrombospondin (TSP)-1 and -2, tenascin-C and –X, Osteonectin/SPARC (Secreted protein, acidic and rich in cysteine), osteopontin (OPN) and periostin. Matricellular proteins are not expressed in normal hearts; however, their induction is a prominent feature of the proliferative phase of healing and appears to play an important role in regulating the dynamic cellular events in cardiac repair.
Tenascin-C is markedly but transiently upregulated during the proliferative phase of healing [43], is predominantly produced by fibroblasts [44] and is localized in the border zone between infarcted and viable remodeling myocardium. The mechanisms responsible for tenascin-C induction in the infarct remain unknown. Cytokines and growth factors, released in healing infarcts (such as TNF-α, TGF-β, and basic Fibroblast Growth Factor), are capable of upregulating fibroblast tenascin-C synthesis. In addition, angiotensin II, an important regulator of cardiac remodeling and fibrous tissue deposition, is also known to stimulate tenascin-C expression [45]. Tenascin-C expression virtually disappears in the mature infarct [43]. The role of tenascin-C in infarct healing and post-infarction remodeling remains unknown. Its selective expression in the infarct border zone suggests that it may be important in the dynamic events associated with cardiac remodeling following injury. Tenascin-C promotes a deadhesive state and may facilitate migration of fibroblasts and other granulation tissue cells in the infarct. Imanaka-Yoshida and co-workers suggested that tenascin-C may modulate adhesion of surviving cardiomyocytes in the infarct border zone facilitating tissue remodeling [44]. In addition, a recent study examined the effects of tenascin-C deficiency in a model of electrical cardiac injury [46] demonstrating that tenascin-C null mice exhibit delayed recruitment of myofibroblasts in the injured site.
TSP-1, a crucial TGF-β activator with potent angiostatic properties, is also strikingly upregulated in experimental models of infarction [47]. Our experiments demonstrated that TSP-1 mRNA and protein was markedly and selectively induced in the border zone of healing canine and murine myocardial infarcts. TSP-1 -/- mice exhibited enhanced and prolonged expression of chemokines in the infarcted heart and showed expansion of the inflammatory infiltrate into the non-infarcted area. Prolonged and expanded inflammation resulted in increased adverse remodeling of the ventricle [47]. These findings suggest an important role for TSP-1 in suppression and containment of the inflammatory reaction following myocardial infarction. Localized induction of TSP-1 in the infarct border zone may form a “barrier” preventing expansion of the inflammatory infiltrate in the non-infarcted area. The mechanisms involved in TSP-1-mediated inhibition of inflammation remain unknown. We noted decreased Smad2 phosphorylation in TSP-1 null infarcts, suggesting that TSP-1 may exert anti-inflammatory actions in the infarct border zone through TGF-β activation. However, TSP-1 may also regulate infarct healing through its angiostatic effects, by exerting direct anti-inflammatory actions, or by mediating MMP activation. TSP-2 also appears to be an important modulator of infarct healing. Myocardial infarction in TSP-2 -/-mice resulted in a high incidence of cardiac rupture [48], suggesting a crucial role for TSP-2 in formation and structural integrity of the remodeling matrix. The mechanisms responsible for these effects remain to be determined.
OPN upregulation is also consistently found in experimental models of myocardial infarction [49] [50]. In both murine and canine infarcts OPN is induced early following myocardial infarction and is predominantly localized in macrophages. OPN is markedly upregulated during monocyte to macrophage differentiation [51], thus its expression in the infarct may reflect differentiation and activation of infiltrating monocytes. OPN plays a pivotal role in infarct healing and cardiac remodeling [52]. Absence of OPN resulted in increased left ventricular dilatation associated with reduced collagen deposition in the infarcted myocardium [53]. These experiments suggested that OPN may play a protective role, attenuating adverse remodeling, by promoting collagen deposition in the infarct. The mechanisms responsible for the OPN-mediated effects on matrix remodeling remain poorly understood.
SPARC is also induced in healing infarcts. A recent investigation demonstrated that SPARC -/- mice had markedly increased mortality following myocardial infarction due to cardiac rupture and the development of heart failure [54]. Impaired infarct healing in SPARC null animals was associated with disorganized granulation tissue formation and deposition of immature collagen. TGF-β injection protected SPARC null mice from cardiac rupture. The study suggested that SPARC-mediated actions are essential for matrix organization in the healing infarct.
Periostin is abundantly expressed in the border zone in both mouse and human myocardial infarction and critically modulated fibroblast phenotype and function in the infarcted myocardium [55]. Periostin null mice had impaired healing following myocardial infarction exhibiting increased incidence of cardiac rupture [56], [55] associated with decreased recruitment of myofibroblasts and impaired collagen fiber formation in the infarct. However, surviving mice had less fibrosis and significantly better cardiac performance suggesting attenuated adverse remodeling [56].
Thus, experimental studies using genetically targeted mice suggest essential actions of several matricellular proteins in the healing infarct. Because of the complex effects of matricellular proteins, the molecular basis for their actions remains poorly understood.
5. The matrix during the maturation phase (Figure 4)
Figure 4.

The extracellular matrix during the maturation phase of infarct healing. As the scar matures, lysyl-oxidase upregulation induces collagen cross-linking. Formation of a stable cross-linked extracellular matrix may shield the myofibroblasts (MF) from mechanical tension promoting quiescence. Ultimately, most infarct myofibroblasts undergo apoptosis. Prolonged presence of myofibroblasts in the infarcted myocardium may promote adverse remodeling.
Collagen content in the healing infarct progressively increases with time [57]. As the scar matures increased expression of cross-linking enzymes, such as lysyl-oxidase induces matrix cross-linking in the infarcted myocardium [58]. Formation of a mature scar comprised of dense cross-linked collagen enhances tensile strength of the infarct while increasing passive stiffness and contributing to diastolic dysfunction [59]. During the maturation phase, most myofibroblasts and vascular cells are cleared through a process that may involve activation of apoptotic pathways [60]. Loss of the cellular elements ultimately results in formation of a collagen-based infarct with low metabolic activity.
5.1 The role of the matrix in myofibroblast activity: the concept of “stress-shielding”
As the infarct matures, myofibroblast density decreases [60]. Although the pathways determining the fate of infarct myofibroblasts have not been investigated, descriptive studies have demonstrated that apoptosis may play an important role in elimination of granulation tissue cells from the infarct [61], [62]. Whether the myofibroblasts are deactivated prior to their clearance remains unknown. It has been suggested that formation of a mature cross-linked matrix may “shield” fibroblasts from external loads resulting in their deactivation [41], [63]. Thus, phenotypic alterations of infarct myofibroblasts may reflect the changes in the surrounding matrix. In the early stages of healing matrix degradation exposes the cardiac fibroblasts to mechanical tension contributing to activation and myofibroblast transdifferentiation. In contrast, in the mature scar, deposition of a well-organized scar may result in “stress-shielding” leading to reversal of the myofibroblast phenotype and cell quiescence.
6. Conclusions
The role of the extracellular matrix in the infarcted myocardium extends beyond providing mechanical support. Matrix-related pathways are critically involved in the reparative process and provide essential environmental cues that determine cell survival, phenotype and gene expression. Several key questions need to be answered in order to understand the role of the dynamic alterations in the extracellular matrix of the infarct in the pathogenesis of cardiac remodeling (Table 1). Unfortunately, the complexity of the matrix network and the diverse actions of matrix components on all cell types involved in the reparative response have hampered our efforts to design specific therapeutic strategies aiming at reducing adverse remodeling in patients with myocardial infarction. Extensive experimental evidence has suggested that MMP inhibition may protect the infarcted heart from adverse remodeling [64, 65]; however early clinical experience from the use of a selective MMP inhibitor in myocardial infarction has been disappointing [66]. Because MMPs exert effects beyond matrix degradation by processing cytokines and chemokines, predicting the effects of their inhibition in the clinical context is challenging. Recent evidence suggests that several matricellular proteins attenuate adverse remodeling through effects on the inflammatory and reparative response. Thus, peptides derived from specific matricellular proteins may be promising therapeutic agents in the treatment of myocardial infarction. Although these approaches are of significant interest, it should be emphasized that experimental studies are designed to answer mechanistic questions and cannot predict the success of a specific therapeutic strategy because they cannot simulate the complexity of the clinical context.
Table 1.
Unanswered questions guiding future research
| How do matrix fragments modulate phenotype and function of the various cell types involved in the reparative response following myocardial infarction? |
| What is the role of the provisional matrix in the post-infarction inflammatory response? |
| Which matrix-mediated signals activate fibrosis and angiogenesis in the healing infarct? |
| How does the matrix promote quiescence and apoptosis of myofibroblasts in the mature scar? |
Acknowledgments
Supported by NIH R01 HL-76246 and R01 HL-85440.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–75. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim HE, Dalal SS, Young E, Legato MJ, Weisfeldt ML, D’Armiento J. Disruption of the myocardial extracellular matrix leads to cardiac dysfunction. J Clin Invest. 2000;106:857–66. doi: 10.1172/JCI8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Senzaki H, Paolocci N, Gluzband YA, Lindsey ML, Janicki JS, Crow MT, et al. beta-blockade prevents sustained metalloproteinase activation and diastolic stiffening induced by angiotensin II combined with evolving cardiac dysfunction. Circ Res. 2000;86:807–15. doi: 10.1161/01.res.86.7.807. [DOI] [PubMed] [Google Scholar]
- 4.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–67. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 5.Siwik DA, Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–65. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 6.Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173:57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frangogiannis NG, Mendoza LH, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, et al. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol. 2000;165:2798–808. doi: 10.4049/jimmunol.165.5.2798. [DOI] [PubMed] [Google Scholar]
- 8.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58:88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannon RO, 3rd, Butany JW, McManus BM, Speir E, Kravitz AB, Bolli R, et al. Early degradation of collagen after acute myocardial infarction in the rat. Am J Cardiol. 1983;52:390–5. doi: 10.1016/0002-9149(83)90145-5. [DOI] [PubMed] [Google Scholar]
- 11.Whittaker P, Boughner DR, Kloner RA. Role of collagen in acute myocardial infarct expansion. Circulation. 1991;84:2123–34. doi: 10.1161/01.cir.84.5.2123. [DOI] [PubMed] [Google Scholar]
- 12.Etoh T, Joffs C, Deschamps AM, Davis J, Dowdy K, Hendrick J, et al. Myocardial and interstitial matrix metalloproteinase activity after acute myocardial infarction in pigs. Am J Physiol Heart Circ Physiol. 2001;281:H987–94. doi: 10.1152/ajpheart.2001.281.3.H987. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi S, Barry AC, Factor SM. Collagen degradation in ischaemic rat hearts. Biochem J. 1990;265:233–41. doi: 10.1042/bj2650233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–38. [PMC free article] [PubMed] [Google Scholar]
- 15.Villarreal FJ, Griffin M, Omens J, Dillmann W, Nguyen J, Covell J. Early short-term treatment with doxycycline modulates postinfarction left ventricular remodeling. Circulation. 2003;108:1487–92. doi: 10.1161/01.CIR.0000089090.05757.34. [DOI] [PubMed] [Google Scholar]
- 16.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 17.Cleutjens JP, Kandala JC, Guarda E, Guntaka RV, Weber KT. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27:1281–92. doi: 10.1016/s0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- 18.Villarreal F, Omens J, Dillmann W, Risteli J, Nguyen J, Covell J. Early degradation and serum appearance of type I collagen fragments after myocardial infarction. J Mol Cell Cardiol. 2004;36:597–601. doi: 10.1016/j.yjmcc.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006;324:475–88. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- 20.Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, et al. CD44 Is Critically Involved in Infarct Healing by Regulating the Inflammatory and Fibrotic Response. J Immunol. 2008;180:2625–33. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 21.Gearing AJ, Beckett P, Christodoulou M, Churchill M, Clements JM, Crimmin M, et al. Matrix metalloproteinases and processing of pro-TNF-alpha. J Leukoc Biol. 1995;57:774–7. doi: 10.1002/jlb.57.5.774. [DOI] [PubMed] [Google Scholar]
- 22.Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol. 2007;82:1375–81. doi: 10.1189/jlb.0607338. [DOI] [PubMed] [Google Scholar]
- 23.Adair-Kirk TL, Senior RM. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol. 2008;40:1101–10. doi: 10.1016/j.biocel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J Clin Invest. 1980;66:859–62. doi: 10.1172/JCI109926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12:317–23. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 26.Gaggar A, Jackson PL, Noerager BD, O’Reilly PJ, McQuaid DB, Rowe SM, et al. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180:5662–9. doi: 10.4049/jimmunol.180.8.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–84. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 28.Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, et al. Resolution of lung inflammation by CD44. Science. 2002;296:155–8. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- 29.Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 30.Clark RA. Wound repair. Overview and general considerations. In: RA C, editor. The Molecular and Cellular Biology of Wound Repair. New York: Plenum Press; 1995. pp. 3–50. [Google Scholar]
- 31.Corbett SA, Schwarzbauer JE. Fibronectin-fibrin cross-linking: a regulator of cell behavior. Trends Cardiovasc Med. 1998;8:357–62. doi: 10.1016/s1050-1738(98)00028-0. [DOI] [PubMed] [Google Scholar]
- 32.Petzelbauer P, Zacharowski PA, Miyazaki Y, Friedl P, Wickenhauser G, Castellino FJ, et al. The fibrin-derived peptide Bbeta15-42 protects the myocardium against ischemia-reperfusion injury. Nat Med. 2005;11:298–304. doi: 10.1038/nm1198. [DOI] [PubMed] [Google Scholar]
- 33.Atar D, Petzelbauer P, Schwitter J, Huber K, Rensing B, Kasprzak JD, et al. Effect of intravenous FX06 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction results of the F.I.R.E. (Efficacy of FX06 in the Prevention of Myocardial Reperfusion Injury) trial. J Am Coll Cardiol. 2009;53:720–9. doi: 10.1016/j.jacc.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 34.Drew AF, Liu H, Davidson JM, Daugherty CC, Degen JL. Wound-healing defects in mice lacking fibrinogen. Blood. 2001;97:3691–8. doi: 10.1182/blood.v97.12.3691. [DOI] [PubMed] [Google Scholar]
- 35.Creemers E, Cleutjens J, Smits J, Heymans S, Moons L, Collen D, et al. Disruption of the plasminogen gene in mice abolishes wound healing after myocardial infarction. Am J Pathol. 2000;156:1865–73. doi: 10.1016/S0002-9440(10)65060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown LF, Dubin D, Lavigne L, Logan B, Dvorak HF, Van de Water L. Macrophages and fibroblasts express embryonic fibronectins during cutaneous wound healing. Am J Pathol. 1993;142:793–801. [PMC free article] [PubMed] [Google Scholar]
- 37.Ulrich MM, Janssen AM, Daemen MJ, Rappaport L, Samuel JL, Contard F, et al. Increased expression of fibronectin isoforms after myocardial infarction in rats. J Mol Cell Cardiol. 1997;29:2533–43. doi: 10.1006/jmcc.1997.0486. [DOI] [PubMed] [Google Scholar]
- 38.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–3. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 39.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–16. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142:873–81. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 42.Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002;13:3546–59. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willems IE, Arends JW, Daemen MJ. Tenascin and fibronectin expression in healing human myocardial scars. J Pathol. 1996;179:321–5. doi: 10.1002/(SICI)1096-9896(199607)179:3<321::AID-PATH555>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 44.Imanaka-Yoshida K, Hiroe M, Nishikawa T, Ishiyama S, Shimojo T, Ohta Y, et al. Tenascin-C modulates adhesion of cardiomyocytes to extracellular matrix during tissue remodeling after myocardial infarction. Lab Invest. 2001;81:1015–24. doi: 10.1038/labinvest.3780313. [DOI] [PubMed] [Google Scholar]
- 45.Mackie EJ, Scott-Burden T, Hahn AW, Kern F, Bernhardt J, Regenass S, et al. Expression of tenascin by vascular smooth muscle cells. Alterations in hypertensive rats and stimulation by angiotensin II. Am J Pathol. 1992;141:377–88. [PMC free article] [PubMed] [Google Scholar]
- 46.Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, et al. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol. 2005;167:71–80. doi: 10.1016/S0002-9440(10)62954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, et al. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 48.Schellings MW, Pinto YM, Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res. 2004;64:24–31. doi: 10.1016/j.cardiores.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Murry CE, Giachelli CM, Schwartz SM, Vracko R. Macrophages express osteopontin during repair of myocardial necrosis. Am J Pathol. 1994;145:1450–62. [PMC free article] [PubMed] [Google Scholar]
- 50.Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–77. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hashimoto S, Suzuki T, Dong HY, Yamazaki N, Matsushima K. Serial analysis of gene expression in human monocytes and macrophages. Blood. 1999;94:837–44. [PubMed] [Google Scholar]
- 52.Krishnamurthy P, Peterson JT, Subramanian V, Singh M, Singh K. Inhibition of matrix metalloproteinases improves left ventricular function in mice lacking osteopontin after myocardial infarction. Mol Cell Biochem. 2009;322:53–62. doi: 10.1007/s11010-008-9939-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trueblood NA, Xie Z, Communal C, Sam F, Ngoy S, Liaw L, et al. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res. 2001;88:1080–7. doi: 10.1161/hh1001.090842. [DOI] [PubMed] [Google Scholar]
- 54.Schellings MW, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, van Leeuwen RE, et al. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med. 2009;206:113–23. doi: 10.1084/jem.20081244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, et al. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205:295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res. 2007;101:313–21. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jugdutt BI, Amy RW. Healing after myocardial infarction in the dog: changes in infarct hydroxyproline and topography. J Am Coll Cardiol. 1986;7:91–102. doi: 10.1016/s0735-1097(86)80265-0. [DOI] [PubMed] [Google Scholar]
- 58.Lerman RH, Apstein CS, Kagan HM, Osmers EL, Chichester CO, Vogel WM, et al. Myocardial healing and repair after experimental infarction in the rabbit. Circ Res. 1983;53:378–88. doi: 10.1161/01.res.53.3.378. [DOI] [PubMed] [Google Scholar]
- 59.Holmes JW, Borg TK, Covell JW. Structure and mechanics of healing myocardial infarcts. Annu Rev Biomed Eng. 2005;7:223–53. doi: 10.1146/annurev.bioeng.7.060804.100453. [DOI] [PubMed] [Google Scholar]
- 60.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. 2002;50:71–9. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]
- 61.Zhao W, Lu L, Chen SS, Sun Y. Temporal and spatial characteristics of apoptosis in the infarcted rat heart. Biochem Biophys Res Commun. 2004;325:605–11. doi: 10.1016/j.bbrc.2004.10.064. [DOI] [PubMed] [Google Scholar]
- 62.Takemura G, Ohno M, Hayakawa Y, Misao J, Kanoh M, Ohno A, et al. Role of apoptosis in the disappearance of infiltrated and proliferated interstitial cells after myocardial infarction. Circ Res. 1998;82:1130–8. doi: 10.1161/01.res.82.11.1130. [DOI] [PubMed] [Google Scholar]
- 63.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–37. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 64.Lindsey ML, Gannon J, Aikawa M, Schoen FJ, Rabkin E, Lopresti-Morrow L, et al. Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation. 2002;105:753–8. doi: 10.1161/hc0602.103674. [DOI] [PubMed] [Google Scholar]
- 65.Lindsey ML. MMP induction and inhibition in myocardial infarction. Heart Fail Rev. 2004;9:7–19. doi: 10.1023/B:HREV.0000011390.44039.b7. [DOI] [PubMed] [Google Scholar]
- 66.Hudson MP, Armstrong PW, Ruzyllo W, Brum J, Cusmano L, Krzeski P, et al. Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction: results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J Am Coll Cardiol. 2006;48:15–20. doi: 10.1016/j.jacc.2006.02.055. [DOI] [PubMed] [Google Scholar]