

Summary
Transforming growth factor β1 (TGFβ1) is a multifunctional growth factor involved in wound healing, tissue fibrosis, and in the pathogenesis of many syndromic diseases (e.g., Marfan syndrome, Camurati-Engelmann disease) and muscular, neurological, ophthalmic, cardiovascular and immunological disorders, and cancer. Since the generation of Tgfb1 knockout mice, there has been extraordinary progress in understanding its physiological and pathophysiological function. Here, we report the generation of a conditional knockout allele for Tgfb1 in which its exon 6 is flanked with LoxP sites. As proof of principle, we crossed these mice to LckCre transgenic mice and specifically disrupted Tgfb1 in T cells. The results indicate that T-cell-produced TGFβ1 is required for normal in vivo regulation of peripheral T-cell activation, maintenance of T-cell homeostasis, and suppression of autoimmunity.
Keywords: transforming growth factorβ, conditional knockout mice
Transforming growth factor betas (TGFβs) are regulatory cytokines that function in embryonic development and adults and in the pathogenesis of various diseases and cancer (Derynck and Akhurst, 2007; McLennan and Koishi, 2002; Sporn, 2006). In mammals, there are three TGFβ ligands which express differently in time and space (Azhar et al., 2003; Millan et al., 1991; Pelton et al., 1991). Activated forms of TGFβs usually interact with TGFβR1 and TGFβR2 receptors in conjunction with the TGFβR3 or endoglin and induce phosphorylation of SMADs (Massague and Gomis, 2006). TGFβ signaling can also occur through SMAD-independent pathways (e.g., RAS-MAPK, Ca2+/calcineurin and a non-transcriptional pathway such as platelet aggregation) (Azhar et al., 2003; Bommireddy et al., 2003a).
Tgfb knockout mice display distinct phenotypes (Kaartinen et al., 1995; Kulkarni et al., 1993; Proetzel et al., 1995; Sanford et al., 1997; Shull et al., 1992; Yang et al., 2007). Tgfb1 is expressed in numerous tissues during embryonic development and its expression is dysregulated under pathophysiological conditions in many tissues in adults (Azhar et al., 2003; Kehrl et al., 1986; Pelton et al., 1991; Saxena et al., 2008; Thompson et al., 1989). Tgfb1−/− mice develop severe multifocal inflammatory disease (Kulkarni et al., 1993; Shull et al., 1992), yolk sac defects (Dickson et al., 1995), and colon cancer (Engle et al., 1999). It is thought that TGFβ1 can act in an autocrine or paracrine/juxtacrine manner (Bommireddy and Doetschman, 2007; Longenecker et al., 2002). Therefore, mouse models that could precisely delete Tgfb1 in a cell-type or tissue-specific fashion will be a highly useful tool for determining in vivo functions of TGFβ1.
Here we have generated mice harboring a conditional knockout allele for Tgfb1. Targeting vectors were designed in which exon 6 of Tgfb1 was flanked with LoxP sites in introns 5 and 6 to produce conditional deletion of Tgfb1 in mice using a Cre-LoxP strategy (Fig. 1a). Exon 6 was chosen because we previously showed that its disruption results in TGFβ1-deficient mice (Shull et al., 1992). Tgfb1+/flox-ex 6 mice were intercrossed, and the resulting Tgfb1flox-ex 6/flox-ex 6 mice were born in a normal (1:2:1) Mendelian ratio (Fig. 1e). Both male and female Tgfb1flox-ex 6/flox-ex 6 animals had normal fertility and fecundity, and their longevity was identical to that of wild type mice. Morphological and histological examination of tissues from various organs in age- and sex-matched littermates (n = 6) of Tgfb1flox-ex 6/flox-ex 6 mice indicated normal phenotype.
FIG. 1.
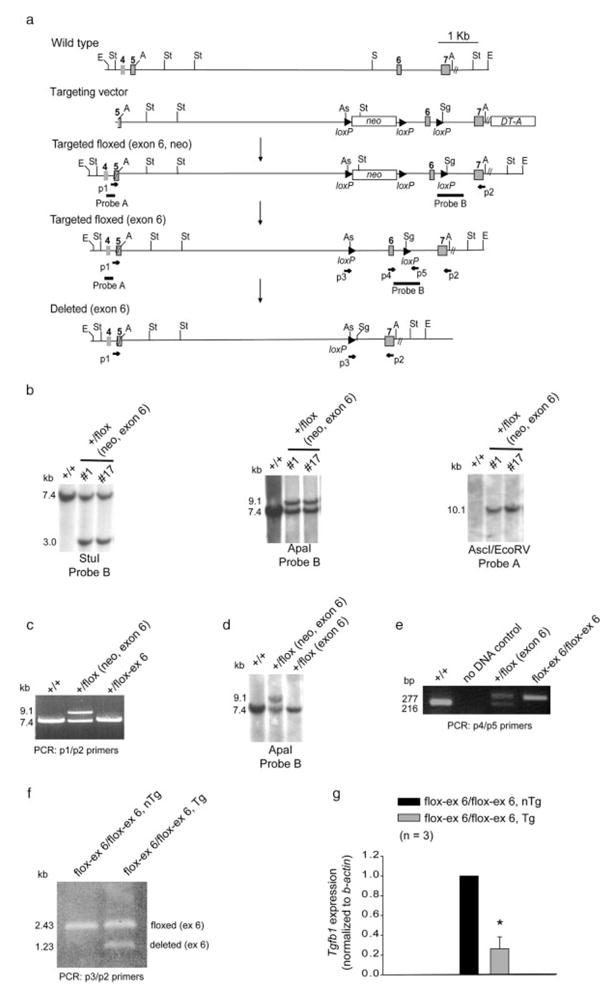
Generation of Tgfb1flox-ex 6 mice. a: A Cre-LoxP based conditional gene targeting scheme depicting the relevant piece of Tgfb1 genomic locus, the Tgfb1flox-ex 6 targeting vector and the Tgfb1flox-ex 6 targeted and deleted locus. PCR primers and southern blot probes are indicated. E, EcoRV; St, StuI; A, ApaI; S, SacI; As, AscI; Sg, SgfI. b: Southern blot screening. StuI cuts both outside and inside and EcoRV cuts only outside at both ends of the targeting vector. AscI and SgfI are only present in the LoxP. Probe A is an outside side probe. Only two (clone #1 and #17) of the five originally targeted clones are shown. StuI, ApaI, and AscI/EcoRV blots confirm the presence of a targeted floxed (exon 6, neo) allele. Positive clones were identified by 3.0 kb (StuI blot), 8.9 kb (ApaI blot), and 10.1 kb (AscI/EcoRV blot). A positive targeted floxed (exon 6, neo) clone (#17) was treated with Cre recombinase and the floxed-neo was deleted in these ES cells. c: Long PCR shows the presence of an expected 8.9-kb band before the Cre-excision and a 7.8-kb band after the Cre-mediated excision in #17 clone, indicating a successful removal of floxed-neo from the targeted floxed (exon 6) allele in ES cells. d: ApaI blot confirms the presence of Tgfb1flox-ex 6 allele in germline chimera. Wild type (+/+) mice and targeted floxed (exon 6, neo) ES cells are used as controls. e: PCR genotyping of wild type, Tgfb1 floxed heterozygous (+/flox (exon 6)) and Tgfb1 floxed homozygous (flox-ex 6/flox-ex 6) mice. f: PCR amplification indicating partial but specific Cre-mediated excision in thymus of Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice. PCR primer p3 is a hybrid primer with sequences from both the Tgfb1 and LoxP and detects specifically Tgfb1flox-ex 6 allele but not the wild-type allele under conditions described in the Materials and Methods. There is no floxed deleted allele (1.23 kb) in control mice which is seen in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice. Note that no deletion product(s) at the Tgfb1 locus in liver, heart, lung, colon and brain tissue of Tgfb1flox-ex 6/flox-ex 6/LckCre nTg or Tgfb1+/+/LckCre Tg mice was found (not shown), indicating that Cre-mediated excision occurred only in the thymus of Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice. g: Q-RT-PCR analysis showing significantly decreased Tgfb1 expression (*P = 3.5747e-3) in T cells from Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice as compared to the control non-transgenic mice. Each control value is normalized to 1.0.
To test the ability of Tgfb1flox-ex 6/flox-ex 6 mice to produce Tgfb1 conditional deletion and gain more specific information about the role of Tgfb1 in T-cell-mediated immune function, these mice were intercrossed to LckCre transgenic (Tg) mice which have been successfully used to conditionally delete genes in T cells. The LckCre transgene first comes on in immature CD4− CD8− T cells in thymus and is later expressed in mature CD4+CD8− and CD4−CD8+ T cells in thymus. It is expressed in T cells that have migrated to the lymphoid organs, including spleen (Orban et al., 1992). PCR analysis of thymic genomic DNA from 1-month-old Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice indicated significant but incomplete conditional deletion of Tgfb1 (Fig. 1f). Since the PCR was done on whole organ which contained both T cells and non-T cells (e.g., thymic epithelial cells), and since Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice are on a mixed genetic background (129/Black-Swiss/C57BL/6), it is possible that these factors have contributed to the partial Cre-mediated excision seen in these mice. Tgfb1flox-ex 6/flox-ex 6 mice are currently being crossed to other Cre transgenic lines to further investigate conditional Tgfb1 ablation in other tissues. Q-RT-PCR data indicated that this partial conditional deletion of Tgfb1 in T cells is sufficient to significantly reduce Tgfb1 expression (Fig. 1g).
FACS analyses using markers of T cells (i.e., CD3, CD4, and CD8) indicated reduced CD3 expression, a substantial increase in percent of CD3+ CD8+ T cells, and a concomitant decrease in the percent of CD3+ CD4+ T cells in 5-month-old Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice (see Fig. 2). These data suggest that TGFβ1 is required for normal T-cell homeostasis. By using CD44 and CD62L as specific markers of T-cell activation and function, this study also revealed significant induction of T-cell activation in 5-month-old Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice (see Fig. 3). This enhanced T-cell activation is seen as early as 1 month of age when these mice were genetically combined with a Tgfb1− allele (Tgfb1flox-ex 6/−/LckCre Tg) (not shown). Together, these data indicate that T-cell-produced TGFβ1 is required for normal activation of peripheral T cells and that juxtacrine production of TGFβ1 by other cell types is not sufficient to rescue the T-cell defect.
FIG. 2.

T cell-specific deletion of Tgfb1 affects T cell homeostasis in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice. Splenocytes are prepared from 5-month-old control and Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice and immunostained for surface expression of CD4, CD8, and CD3 and analyzed by flow cytometry. a: CD3 expression and percent of CD3+ splenocytes. Both the mean fluorescence intensity (MFI) and the percent of CD3+-T cells are decreased in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice. b: Analysis of CD4+ and CD8+ cells. c: CD3+-gated splenocytes indicating distribution of CD4+ and CD8+ T cells. The % of cells positive for each marker is shown in the quadrants in (a, b). The % of CD4+ T cells is reduced and percent of CD8+ T cells is increased in both non-gated and CD3-gated splenocytes. d: CD8 expression and percent of CD8+ splenocytes. The percent of CD8+-T cells is increased but their MFI is slightly reduced in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice.
FIG. 3.

T cell-specific deletion of Tgfb1 results in increased T cell activation in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice. Splenocytes from 5-month-old control and Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice are analyzed for CD4, CD8, CD44 and CD62L by flow cytometry. Expression of CD44 and CD62L is analyzed on CD4+ or CD8+ T cells by gating on these subsets. The percent of cells positive for each marker is shown in the quadrants. a: FACS analyses showing an increased CD44 and reduced CD62L on CD4+ cells in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice. b: CD44 and CD62L expression on CD8+ T cells. CD44 expression is increased, whereas CD62L expression is reduced on CD8+ T cells in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice.
Tgfb1flox-ex 6/flox-ex 6/LckCre Tg adult mice exhibit significant weight loss and decreased cellularity of thymus and spleen as compared with the control mice (see Fig. 4), indicating signs of inflammation and wasting (see Fig. 4). Histological examination of tissues from various organs, including lungs and salivary glands, revealed modest inflammation in 5-month-old Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice (Fig. 5a, b, d, e), which became more severe when Tgfb1 expression was further decreased in Tgfb1flox-ex 6/−/LckCre Tg mice (Fig. 5a–f). Finally, analysis of cytokine profiles of splenic T cells revealed that production of both interferon-γ (IFN-γ) and interleukin-2 (IL-2) was significantly increased in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice (Fig. 5f, g). TGFβ1 has been shown to be required for the maintenance of a diverse and self-tolerant T-cell repertoire and for appropriate T-cell responses necessary for an effective adaptive immune system (Li and Flavell, 2008). Complete absence of TGFβ1 in mice leads to a breakdown in T-cell tolerance, which consequently results in multiorgan inflammation, organ failure, and death at weaning age (Bommireddy and Doetschman, 2007; Shull et al., 1992). Several studies have indicated that reduced TGFβ1 can cause immune disorders such as lupus (Saxena et al., 2008) and inflammation-induced cancer (Engle et al., 1999; Sporn, 2006). Thus, the data presented in this study is novel by indicating that T-cell-produced TGFβ1 is required for suppression of autoimmunity and inflammation.
FIG. 4.

Analysis of body weight and thymus and spleen weight and number of thymocytes and splenocytes in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice. a: Body weight at 5 month. b: Thymic weight at 2 month. c: Spleen weight at 2 month. d: Number of thymocytes at 1 month. e: Number of splenocytes at 5 months. Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice exhibit significant reduction in body weight (n = 4, *P = 8.9415e-4) and thymic (n = 3, *P = 0.0166) and splenic weight (n = 4 for control and n = 5 for Tgfb1flox-ex 6/-/LckCre Tg, *P = 0.0165), and number of total thymocytes (n = 3, *P = 2.4698e-3) and splenocytes (n = 3, *P = 0.0236).
FIG. 5.
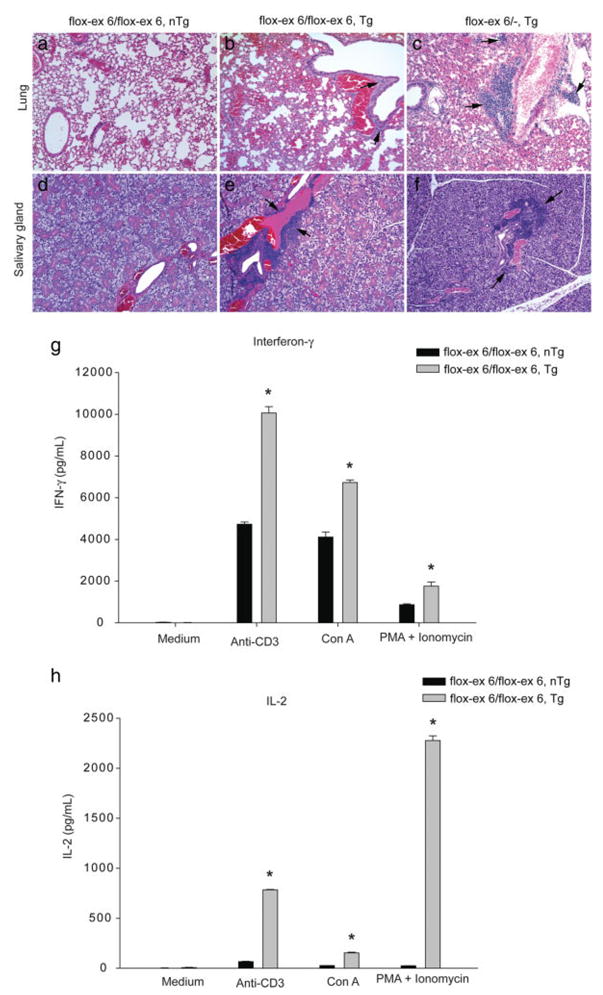
T cell-specific deletion of Tgfb1 results in increased organ inflammation and Th1-effector cytokine production. a–f: Hematoxylin and eosin staining of serial sections of lung (a–c) and salivary gland (d–f) from 5-month-old Tgfb1f-ex 6/f-ex 6 control (a, d), Tgfb1flox-ex 6/flox-ex 6/LckCre Tg (b, e) and Tgfb1f-ex 6/− LckCre Tg (c, f) mice. These histopathology data are representative results of at least three mice per genotype analyzed. All photomicrographs are taken at equal magnification. Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice exhibit moderate inflammation in both the lungs (arrows in b) and the salivary gland (arrows in e) which becomes severe in both organs (arrows in c, f) in Tgfb1flox-ex 6/−/LckCre Tg mice. g: INF-γ production. h: IL-2 production. Splenic T cells (1.5 × 106/ml) from 2-month-old control and Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice are stimulated for 72 h with T cell receptor-dependent (plate-bound anti-CD3 mAb (5 μg/ml) and Con A (1 μg/ml)) and receptor-independent (PMA (25 ng/ml) plus ionomycin (250 ng/ml)) mitogens. The cytokines in the culture supernatants are assayed using ELISA as described in the Materials and Methods section. The mitogen-stimulated T cells of Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice exhibit significant increase in the levels (mean ±SD) of IFNγ (G) (*P < 0.0001 for anti-CD3, *P < 0.0001 for Con A, *P = 0.0017 for PMA+ionomycin; and IL-2 (H) (*P < 0.0001 for anti-CD3, *P < 0.0001 for Con A, *P < 0.0001 (PMA+ionomycin).
Several lines of evidence indicate the importance of the findings presented in this study. First, TGFβ1 is produced by T cells (Thompson et al., 1989) and Tgfb1 knockout mice have activated T cells (Bommireddy et al., 2003b; Robinson and Gorham, 2007), hyper-responsive immature T cells (Bommireddy et al., 2003a; Marie et al., 2005) and TGFβRII-deficient T cells have increased expansion of CD8+ T cells that results in defective T-cell homeostasis (Leveen et al., 2005; Lucas et al., 2000), all of which are consistent with the requirement of an autocrine role of TGFβ1 in T-cell homeostasis and activation. Secondly, endocrine expression of the active form of TGFβ1 in Tgfb1−/− mice fails to ameliorate the lethal inflammatory phenotype (Longenecker et al., 2002). Thirdly, IFN-γ is a proinflammatory cytokine which is produced by Th1-effector cells and that higher levels of INF-γ and IL-2 in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice is in concordance with enhanced Th1-cell differentiation and severe inflammation (Bommireddy et al., 2003b; Saxena et al., 2008). Finally, a mouse model with a floxed Tgfb1 allele has been recently described in which exon 1 of Tgfb1 and exon 4 of the B9 protein domain 2 gene (B9d2 or LOC232987) are both flanked with LoxP sites (Li et al., 2007). The exon 4 of B9d2 resides 340-bp upstream of the transcriptional start site of Tgfb1. The B9d2 gene is highly expressed in thymus, and CD4Cre-mediated excision of floxed Tgfb1/B9d2 alleles results in complete loss of expression of both Tgfb1 and B9d2. Since it is not possible to conditionally delete Tgfb1 using this mouse model without the accidental deletion of one or both alleles of the B9d2 gene, Tgfb1flox-ex 6/flox-ex 6 mice provide a unique mouse model to conditionally delete Tgfb1 in a cell or tissue-specific fashion without the accidental deletion of neighboring genes.
In conclusion, this study reports the generation of an exon 6-specific conditional knockout allele for the Tgfb1 gene in mice. The Tgfb1flox-ex 6/flox-ex 6 mice have been tested as a proof of principle for their ability to produce Tgfb1 conditional deletion in T cells and have also provided initial information which indicates important roles of T-cell-produced Tgfb1 in proper in vivo regulation of peripheral T-cell activation, maintenance of T-cell homeostasis, and suppression of autoimmunity.
METHODS
Conditional Gene Targeting of Tgfb1 Locus and Generation of Mice Carrying Floxed-Tgfb1ex 6 Allele
We used a mouse genomic library clone (129/J) to produce a targeting vector for the Tgfb1flox-ex 6 mice. The targeting strategy was designed to flank exon 6 of Tgfb1 by two LoxP sites to effect the disruption of the mature peptide, while leaving the precursor pro-region and neighboring B9d2 gene (i.e., LOC232987) intact. A similar approach had been successfully used to disrupt Tgfb1 in targeting a Neo gene into exon 6 of the Tgfb1 locus, thereby producing Tgfb1−/− mice (Shull et al., 1992). A 7.4-kb ApaI-digested genomic DNA fragment of Tgfb1 was subcloned in a modified Bluescript plasmid (Stratagene) and used for building the targeting vector (Fig. 1a). A floxed PMC1-Neo gene was placed in intron 5 of Tgfb1. A unique AscI diagnostic restriction enzyme site was introduced into the upstream LoxP sequence of this floxed-Neo gene. A third LoxP site was added in intron 6 of Tgfb1 and this LoxP sequence contained SgfI as a unique diagnostic restriction enzyme site. Diphtheria toxin (PGK-DT-A) gene was used to kill random ES cell recombinants by automatic negative selection. The NotI-linearized targeting vector was used for electroporation of 129-derived KG1 embryonic stem (ES) cells according to the previously published procedure (Azhar et al., 2008). Southern blot analysis revealed that five out of 80 G418-resistant ES cells colonies had a correctly targeted Tgfb1 locus (Fig. 1b). One of these clones (#17) was used to remove the Neo in ES cells by Cre-mediated excision of the floxed-Neo gene. Both southern blotting and PCR screening identified several clones in which Neo was completely removed (Fig. 1c). Two of these clones which contain Tgfb1flox-ex 6 allele were microinjected into C57BL/6 mouse blastocysts. Both southern blotting and PCR screening identified many chimeric mice. Germline transmission of Tgfb1flox-ex 6 allele was established by breeding the male ES cell chimeric mice to Black-Swiss females according to previously described methods (Azhar et al., 2008). PCR analysis on tail clip DNA was used for genotyping mice. Tgfb1+/flox-ex 6 mice were intercrossed, and the resulting Tgfb1flox-ex 6/flox-ex 6 mice were maintained on a mixed genetic background (129 and Black-Swiss, 50:50). Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice (129/J/Black-Swiss/C57BL/6) were generated by crossing Tgfb1flox-ex 6 mice (C57BL/6) (Jax Lab, Bar harbor, ME) to LckCre mice. Tgfb1flox/− mice were produced by crossing Tgfb1flox-ex 6/flox-ex 6 mice to Tgfb1+/− (BALB/c), and breeding of Tgfb1flox/− to Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice resulted in Tgfb1flox-ex 6/−/LckCre Tg mice (129/Black Swiss, C57BL/6, BALB/c). Nontransgenic littermates (Tgfb1flox-ex 6/flox-ex 6/LckCre nTg) mice were used as control animals.
Southern Blot, DNA Sequencing and PCR Analyses
For Southern blotting, genomic DNA from ES cells or mouse tail-clips was digested with StuI, ApaI, and AscI/EcoRV, resolved on 0.8% agarose gels, transferred to nylon membranes (GE Healthcare USA), hybridized to 32P-labeled probes and autoradiographed using Biomax (USA) X-ray films. StuI cuts outside at the 3′ end of the targeting vector. EcoRV cuts outside the targeting vector. AscI was introduced as a unique cutter in the 5′ LoxP sequence of the floxed-Neo gene in the targeting vector. Probe A was used as an outside probe in hybridization with AscI/EcoRV genomic DNA digest to confirm the homologous recombination at both ends of the targeting vector. Probe B was an internal probe and it was used in conjunction with StuI in southern blot hybridization to further confirm the correct integration of the 3′ end of the targeting vector. The Neo primers (forward, CCATT-GAACAAGATGGATTGCACGCAG; reverse, AGAAGAAC TCGTCAAGAAGGCGATAGAAGG) were used in an automated DNA sequencing of a PCR amplified product from the targeted (Neo, exon 6) ES cells to confirm the sequences of both LoxP sites in the floxed-Neo gene. Similarly, Tgfb1 exon 6-specific primer (forward, CCTA TATTTGGAGCCTGGACAC) was used to sequence the LoxP site situated is the intron 6 of the targeted floxed (Neo, exon 6) ES cells.
Southern blotting was also performed to confirm the removal of floxed-Neo gene from the Tgfb1 targeted floxed (Neo, exon 6) allele in the targeted ES cells and to identify germline chimeras. PCR analysis was used to reconfirm the loss of the Neo gene in the targeted (Tgfb1flox-ex 6) ES cells and screen the chimeric mice and their germline offspring using p1 and p2 primers. Primer p1 is located in exon 5 just outside the 5′ end of the targeting vector. PCR analysis was also used for routine genotyping of mice with wild type and Tgfb1flox-ex 6 alleles (primer set p4/p5). Genotyping of Tgfb1+/+, Tgfb1+/− and Tgfb1−/− animals was done as described previously (Shull et al., 1992). In addition, PCR was used for detecting the Cre-mediated excision of the Tgfb1 targeted floxed (Neo, exon 6) allele in ES cells and the Tgfb1flox-ex 6 allele in Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice (primer set p3/p2). Efficiency of Cre-mediated excision of the Tgfb1flox-ex 6 allele was determined by densitometric analysis of floxed (ex 6) and floxed deleted (ex 6) PCR bands in PCR amplified products from the Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice. Densitometry was done by using the program ONE-DScan (Scanalytics, VA). Specific primers that were used for the PCR amplification included (all sequences in 5′-3′ directions): p1 (forward), GCACCATC-CATGACATGAAC; p2 (reverse), GGAGCAGTTGTCCAA-CATGAT; p3 (forward), TACAAAGTGAGCTGGCGCGC-CATAACTT; p4 (forward), AAGAC CTGGGTTGGAAGTG; p5 (reverse), CTTCTCCGTTTC TCTGTCACCCTAT.
Quantitative Real-Time RT-PCR (Q-RT-PCR) Analysis
Total RNA from T cells was extracted by RNeasy Mini kit (Cat # 74704: Qiagen, Valencia, CA). Three Tgfb1flox-ex 6/flox-ex 6/LckCre nTg and Tgfb1flox-ex 6/flox-ex 6/LckCre Tg mice were assessed (each in triplicate) by Q-RT-PCR. Q-RT-PCR was performed using an Opticon 2 real-time PCR machine (Biorad, Hercules, CA) according to the published procedure (Rajan et al., 2006). Primers that were used for the Q-RT-PCR amplification of Tgfb1 included (all sequences in 5′-3′ directions): Tgfb1 forward, ATCCTGTCCAAACTAAGGCTCG; Tgfb1 reverse, ACCTCTTTAGCATAGTAGTCCGC; b-actin forward, GGC TGTATTCCCCTCCATCG; b-actin reverse, CCAGTTGG TAACAATGCCATGT.
Flow Cytometry
Isolation of thymocytes and splenocytes and cell counting, and FACS analysis were done according to the previously described methods (Bommireddy et al., 2003b). Freshly prepared T cells from spleen from at least three animals per genotype were incubated with anti-CD16/32 (2.4G2) to block FcRγII/III, followed by staining with fluorochrome-conjugated mAbs as indicated in figure legends. All antibodies were purchased from BD-Biosciences (USA). Stained cells were analyzed using a Becton Dickinson (Mountain View, CA) LSR or FACSCalibur flow cytometer and CellQuest software as described elsewhere (Bommireddy et al., 2003b). Absolute cell numbers of each cell subset was calculated by percentage of staining cells × total cell number.
Cytokine Assay
T cells from spleen (1.5 × 106/ml) were stimulated with Con A (Sigma, 1 μg/ml), plate-bound anti-CD3 mAb (PharMingen, 5 μg/ml), and PMA (Sigma, 25 ng/ml) plus ionomycin (Sigma, 250 ng/ml) (Bommireddy et al., 2003b; Saxena et al., 2008). The supernatants were collected after 72 h and tested for cytokines by ELISA using mAb pairs and recombinant standards from PharMingen. IFNγ and IL-2 were assayed by sandwich ELISA with PharMingen’s paired rat anti-mouse cytokine mAbs (purified mAb for capture, biotinylated mAb for detection) according to the manufacture’s protocol. These antibodies were R4 6A2 and XMG1.2 for IFN-γ, and JES6-1A12 and JES6-5H4 for IL-2. Alkaline phosphatase-conjugated streptavidin (Jackon Immunoresearch Laboratories, Inc., West Grove, PA) was used in the procedure. Plates were developed with p-nitrophenyl phosphate substrate (Sigma) and OD was determined at 405 nm using Multiskan (Thermo Labsystems, Vantaa, Finland). The cytokines were quantified by reference to the standard curves derived from recombinant cytokines (PharMingen; R&D Systems, Inc.).
Histology
Transverse tissue sections (5 μm) of paraformaldehyde-(4%) fixed and paraffin-embedded organs including lungs and salivary gland from adult Tgfb1+/+, Tgfb1flox-ex 6/flox-ex 6, Tgfb1flox-ex 6/flox-ex 6/LckCre nTg, and Tgfb1flox-ex 6/flox-ex 6/LckCre Tg and Tgfb1flox-ex 6/−/LckCre Tg mice were cut and analyzed for tissue inflammation as described earlier (Bommireddy et al., 2003b). Histological examination was done on hematoxylin and Eosin (H&E)-stained serial sections. All sections were analyzed using bright-field optics with a Zeiss Axio Imager M1 microscope (Carl Zeiss Microimaging, Inc., Thornwood, NY) equipped with a AxioVision 4.6.3 imaging software.
Statistical Analysis
Microsoft Excel was used for managing the data. Findings are reported as means ± SD of the mean, and two-tailed Student’s t-test (SigmaPlot, Systat Software, Inc., Richmond, CA) was used for comparing groups. P-values were calculated, and a P < 0.05 was considered significant.
Acknowledgments
Contract grant sponsor: National Institutes of Health Grant, Contract grant numbers: CA084291, AI067903; Contract grant sponsor: Arizona Biomedical Research Commission, Contract grant number: ABRC #0901; Contract grant sponsors: Steven M. Gootter Foundation; BIO5 Institute of the University of Arizona
We thank Dillihan Gumus, Sandy Schwemberger, Fauzia Nazir, Nabeel Al Moamen, and Dr. Carlos Hidalgo for providing technical assistance.
LITERATURE CITED
- Azhar M, Schultz JE, Grupp I, Dorn GW, Meneton P, Molin DG, Gitten-berger-de Groot AC, Doetschman T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003;14:391–407. doi: 10.1016/s1359-6101(03)00044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azhar M, Yin M, Zhou M, Li H, Mustafa M, Nusayr E, Keenan JB, Chen H, Pawlosky S, Gard C, Grisham C, Sanford LP, Doetschman T. Gene targeted ablation of high molecular weight fibroblast growth factor-2. Dev Dyn. 2008;238:351–357. doi: 10.1002/dvdy.21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommireddy R, Doetschman T. TGFbeta1 and T(reg) cells: Alliance for tolerance. Trends Mol Med. 2007;13:492–501. doi: 10.1016/j.molmed.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommireddy R, Ormsby I, Yin M, Boivin GP, Babcock GF, Doetschman T. TGFbeta1 inhibits Ca2+-Calcineurin-mediated activation in thymocytes. J Immunol. 2003a;170:3645–3652. doi: 10.4049/jimmunol.170.7.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommireddy R, Saxena V, Ormsby I, Yin M, Boivin GP, Babcock GF, Singh RR, Doetschman T. TGF-beta1 regulates lymphocyte homeostasis by preventing activation and subsequent apoptosis of peripheral lymphocytes. J Immunol. 2003b;170:4612–4622. doi: 10.4049/jimmunol.170.9.4612. [DOI] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol. 2007;9:1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999;59:3379–3386. [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Kehrl JH, Wakefield LM, Roberts AB, Jakowlew S, Alvarez-Mon M, Derynck R, Sporn MB, Fauci AS. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. 1986;163:1037–1050. doi: 10.1084/jem.163.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveen P, Carlsen M, White-Makowska A, Oddsson S, Larsson J, Goumans MJ, Cilio CM, Karlsson S. TGF-{beta} type II receptor deficient thymocytes develop normally but demonstrate increased CD81 proliferation in vivo. Blood. 2005;106:4234–4240. doi: 10.1182/blood-2005-05-1871. [DOI] [PubMed] [Google Scholar]
- Li MO, Flavell RA. TGF-beta: A master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Longenecker G, Thyagarajan T, Nagineni CN, Flanders KC, Factor V, Miller G, Ward JM, Nalca A, Rangnekar VM, Thorgeirsson S, Kulkarni AB. Endocrine expression of the active form of TGF-beta1 in the TGF-beta1 null mice fails to ameliorate lethal phenotype. Cytokine. 2002;18:43–50. doi: 10.1006/cyto.2002.1025. [DOI] [PubMed] [Google Scholar]
- Lucas PJ, Kim SJ, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J Exp Med. 2000;191:1187–1196. doi: 10.1084/jem.191.7.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-{beta}1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- McLennan IS, Koishi K. The transforming growth factor-betas: Multifaceted regulators of the development and maintenance of skeletal muscles, motoneurons and Schwann cells. Int J Dev Biol. 2002;46:559–567. [PubMed] [Google Scholar]
- Millan FA, Denhez F, Kondaiah P, Akhurst RJ. Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development. 1991;111:131–143. doi: 10.1242/dev.111.1.131. [DOI] [PubMed] [Google Scholar]
- Orban PC, Chui D, Marth JD. Tissue- and site-specific DNA recombination in transgenic mice. Proc Natl Acad Sci USA. 1992;89:6861–6865. doi: 10.1073/pnas.89.15.6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelton RW, Saxena B, Jones M, Moses HL, Gold LI. Immunohisto-chemical localization of TGF beta 1. TGF beta 2, and TGF beta 3 in the mouse embryo: Expression patterns suggest multiple roles during embryonic development. J Cell Biol. 1991;115:1091–1105. doi: 10.1083/jcb.115.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S, Williams SS, Jagatheesan G, Ahmed RP, Fuller-Bicer G, Schwartz A, Aronow BJ, Wieczorek DF. Microarray analysis of gene expression during early stages of mild and severe cardiac hypertrophy. Physiol Genom. 2006;27:309–317. doi: 10.1152/physiolgenomics.00072.2006. [DOI] [PubMed] [Google Scholar]
- Robinson RT, Gorham JD. TGF-beta 1 regulates antigen-specific CD41 T cell responses in the periphery. J Immunol. 2007;179:71–79. doi: 10.4049/jimmunol.179.1.71. [DOI] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de GA, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena V, Lienesch DW, Zhou M, Bommireddy R, Azhar M, Doetschman T, Singh RR. Dual roles of immunoregulatory cytokine TGF-{beta} in the pathogenesis of autoimmunity-mediated organ damage. J Immunol. 2008;180:1903–1912. doi: 10.4049/jimmunol.180.3.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Doetschman T. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporn MB. The early history of TGF-beta, and a brief glimpse of its future. Cytokine Growth Factor Rev. 2006;17:3–7. doi: 10.1016/j.cytogfr.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Thompson NL, Flanders KC, Smith JM, Ellingsworth LR, Roberts AB, Sporn MB. Expression of transforming growth factor-beta 1 in specific cells and tissues of adult and neonatal mice. J Cell Biol. 1989;108:661–669. doi: 10.1083/jcb.108.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS. Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J Cell Biol. 2007;176:787–793. doi: 10.1083/jcb.200611044. [DOI] [PMC free article] [PubMed] [Google Scholar]