

Abstract
The current non-redundant protein sequence database contains over seven million entries and the number of individual functional domains is significantly larger than this value. The vast quantity of data associated with these proteins poses enormous challenges to any attempt at function annotation. Classification of proteins into sequence and structural groups has been widely used as an approach to simplifying the problem. In this article we question such strategies. We describe how the multi-functionality and structural diversity of even closely related proteins confounds efforts to assign function based on overall sequence or structural similarity. Rather, we suggest that strategies that avoid classification may offer a more robust approach to protein function annotation.
Keywords: protein function annotation, protein classification, fold space
Introduction
Protein classification schemes codify various relationships between groups of proteins that are often based on global sequence or structural similarities. However, the complex nature of evolutionary relationships between proteins raises questions about the possibility of generating a reliable classification [1]. The function of a protein is not easy to define and difficulties in describing it can occur at all levels of a classification hierarchy, even when an unambiguous sequence relationship is evident. The problem is exemplified by the so-called “moonlighting” proteins [2,3], which can play multiple roles in the cell. Such proteins have been able to acquire new functions with only minimal changes in either sequence or structure. Conversely, it has become increasingly apparent that proteins can undergo significant changes in sequence and/or structure while still maintaining the same or a similar function [4●,5●]. The picture that is emerging of protein sequence/structure/function space is one of multiple and complex relationships that in many ways defy classification.
One example of the problem involves the organization of proteins into discrete categories based on their “fold”. Structural alignments have revealed numerous geometric relationships between local fragments of proteins that have been classified, globally, as belonging to different folds. Such observations have led to the view that protein structure space is continuous rather than discrete [6–9●]. The importance of this issue is amplified by observations that proteins can have different global topologies, and hence be assigned different fold classifications, but can still share a biological function [10●]. Indeed that evolutionary relationships might exist between proteins that have been classified differently can be inferred from the very existence of so many different folds and topologies (over 1000 topologies in CATH and folds in SCOP). As has been recently pointed out, it is unlikely that each of these folds appeared independently but, rather they probably evolved from a smaller, less diverse set of ancestral proteins [11●]. This in turn suggests that there are numerous functional relationships between proteins with seemingly unrelated structure. The potential existence of such relationships implies that there is a richness of diverse information in structural databases that has yet to be uncovered. This article provides specific examples and suggests a general strategy for mining this information.
Mechanisms for the evolution of structurally diverse proteins with common functions
A conceptual problem that arises in understanding the origins of structural diversity is that most mutations are neutral in the sense that they do not usually cause structural changes while those that do would generally be expected to result in disordered structures (see [12], for example) and hence would be expected to also abrogate function. However, evidence has recently accumulated implying that the standard mechanisms of evolution, ranging from point mutations, to large deletions, insertions or rearrangements of secondary structure elements (SSEs) can result in structural diversity while maintaining function. That small changes in sequence can lead to large changes in structure has been known for some time and there have been recent reports of this phenomenon using both designed [13] and naturally occurring proteins [14,15]. At the extreme are “chameleon” sequences which can adopt multiple conformations depending on factors such as oligomeric state [16] or temperature [17].
The conservation of the sequence and structure of a functional site combined with structural diversity in other regions provides a simple mechanism to conserve function but not global structure. The α/β and all-α ferrodoxins offer an apparent example of this mechanism. Despite a structural similarity that only involves a pair of helices surrounding the functional Fe-S cluster, an evolutionary relationship between these two groups of proteins [18] is suggested both by their sequence similarity and by the existence of both types of ferredoxins as individual domains in the protein dihydropyrimidine dehydrogenase.
Insertion, deletion and rearrangement of individual domains are other mechanisms for the evolution of novel functions [19,20]. A related mechanism can occur within domains, for example the rearrangement of structural fragments that consist of a number of SSEs that play a functional role [4●,5●,9●,10●] and would also account for proteins with different global topologies and related functions. A possible objection to the idea is that significant changes within a domain would be expected to be highly destabilizing. However, recent experimental evidence suggests that this is not an issue. Graziano et al. [21] created a library of DNA elements representing random SSEs from proteins with known structure. Members of the library were randomly combined, producing several stably folded proteins one of which was highly homologous to the protein aspartate racemase from Polaromonas sp. In another example, individual members of the family of cobaltochelatases still preserve function even after significant deletions, insertions, duplications and substitutions of multiple SSE’s [22]. Finally, Peisajovich et al. [23] proposed and experimentally verified a mechanism involving circular permutation in a DNA methyltransferases which preserves function even after significant rearrangement.
Analysis of protein structures has also suggested evolutionary mechanisms by which different structures can share a common function. For example, it has been shown that proteins with different topologies can evolve from a conserved structural core [4●,5●,10●,24●]. Moreover, analyses of individual functional families [5●] and of homologous superfamilies in CATH [4●] have shown that structural changes associated with the evolutionary mechanisms summarized above are in fact quite common and often occur while still preserving overall function. An important example is provided by the evolution of heteromeric protein-protein interactions via the duplication of genes that are involved in homodimeric interactions [25]. It was found that in the evolution of heteromeric proteins, the general location of the interface seen in homodimers was conserved. This suggests that the prediction of protein-protein interactions sites may benefit from the exploitation of apparently remote structural relationships.
Classification can obscure functional relationships
The observation of functional relationships between proteins that have been classified as structurally unrelated provides some of the most striking consequences of the evolutionary mechanisms summarized in the previous section. A recent example is the identification of an evolutionary path relating the P22 and phage λ Cro proteins. Both proteins are transcription factors and both contain a DNA-binding helix-turn-helix motif but they have very different global topologies; P22 Cro is an all α protein and λ Cro is an α/β protein. The two proteins have a low level of sequence identity (25%) and could not be unambiguously related based on sequence alone. However, a relationship has been established through the identification of a series of proteins with sequence similarities (>40%) using transitive sequence searches [26●,27●]. Classifications that would place the two Cro proteins in different categories would clearly obscure the relationship between them. Related studies has been reported by Lupas and coworkers [28,29] who demonstrated an evolutionary connection between proteins in different folds. As in the Cro protein example, they identified functional relationships that involve only a few SSEs, in one case a small βαβ fragment containing conserved residues [24●].
We have recently discussed other cases where common structural fragments in proteins with different overall topologies play a similar functional role [9●]. For example, Figure 1 represents the SSE’s of three sugar-binding proteins. Each is classified as a different fold in SCOP and a different architecture in CATH (a “jelly roll”, a “β-prism” and a “β-propeller”). Nevertheless, they all contain a common substructure consisting of a 4-stranded and 3-stranded sheet with identical connectivity. Moreover, the overall locations of sugar binding sites on the surface of this substructure are also conserved [9●]. An evolutionary relationship between these proteins is also suggested by their more general biological function shown in Figure 1B. Each protein plays a similar role in distinct but related pathways: the jelly-roll facilitates viral entry into bacterial cells and the β-propeller facilitates bacterial entry into eukaryotic cells. Moreover, both of these activities are mediated through interactions with sugar-modified proteins on the cell surface. Although the specific function of the β-prism proteins has not been determined, it is known that they are involved in apoptosis [30]. Based on the relationships identified here, it seems likely that they play a role in autophagy, a particular type of apoptotic mechanism where sugar-binding is a component of the fusion of the phagosome with the lysozome.
Figure 1.
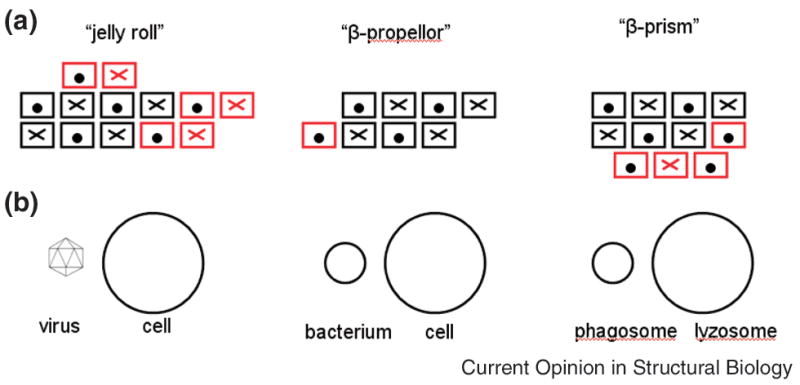
Relationships between three all-β proteins that have been classified as belonging to different folds/topologies. A) Secondary structure elements. Boxes represent β-strands that are perpendicular to the plane of the page. Strands marked with an ‘X’ are directed into the page and those marked with a ‘●’ are directed out of the page. The substructure conserved between all three proteins is shown in black and the nonconserved strands are shown in red. Except for a β-hairpin in the jelly-roll like proteins, all non-conserved strands occur at the N and C-termini of the conserved region and are easily accommodated structurally. B) Similarities between the biological roles of the three proteins. The jelly-roll and β-propeller are known experimentally to be involved in membrane binding associated with viral and bacterial entry into the cell, respectively. As summarized in the text, we suggest that the function of β-prism like proteins might involve the fusion of the phagosome with the lysozome.
Observations such as these suggest that the identification of remote similarities between differently classified proteins can have important practical applications, even when the structural similarity between two proteins is highly remote. For example, Xie and Bourne have identified relationships in the ligand binding properties of highly divergent proteins based on similarities in the sequence profiles of residues surrounding the binding site [31] and have used this information to identify off-targets for pharmaceuticals [32●]. What general strategies can be used to uncover the type of apparently remote relationships described in this section?
Enhanced function annotation and prediction
Structure-based function annotation often begins with a protein of unknown function but known structure, either determined experimentally or from a model. A set of structurally similar proteins is then identified and this often constitutes the end-point for protein function annotation servers, i.e. the identification of a set of possible functions derived from structural neighbors. But how are these neighbors found? If we restrict our definition of structural neighbors to proteins that have already been classified as belonging to the same fold or superfamily, we will be missing a large number of relationships that are not covered by such classifications [9●]. An alternate strategy is to identify structural neighbors, independent of classification, for example to accept the alignment of as few as three SSEs as “hits” [9●]. This will clearly significantly increase the number of possible relationships but many of these will constitute false positives so that list has to be paired down with other information.
Figure 2 provides a schematic of how this can be accomplished. It assumes that a researcher interested in a particular protein could, if interactive tools were available, pose a series of detailed queries that would enable the assignment of function based on a structural similarity. Relevant questions would include determining the “important” parts of each protein on the list and their specific functions and, conversely, given a particular function that may be known from experiment, identifying those regions of the sequence and structure that actually participate in this function. Such information can be crucial in the decision of whether homology transfer to the protein of unknown function is appropriate.
Figure 2.
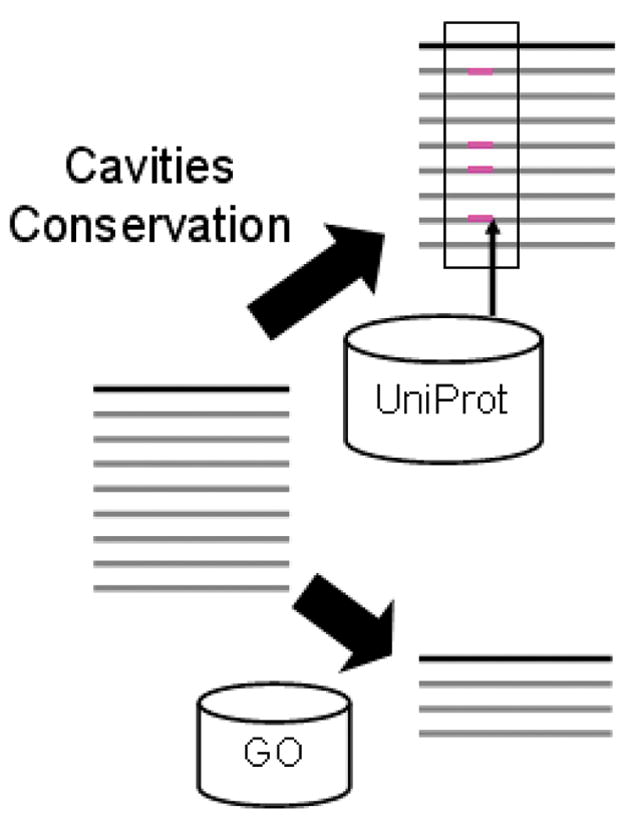
Schematic of an interactive approach to function annotation. On the left of the figure a query protein and structurally similar proteins are represented in a manner similar to BLAST output. Interactive tools can then be used to address two types of biological questions. In the upper right, a functionally important region (enclosed by the black rectangle) is identified, based on properties such as sequence conservation, electrostatic conservation, and/or the presence of a surface cavity. UniProt features that represent known functions that occur in structural neighbors in similar locations are “mapped” to the alignment. (magenta rectangles). In the lower right, the initial set of proteins that are structurally similar to the query is filtered based on a known function of interest. The resulting reduced set can then be further analyzed to identify the structural determinants of that function.
As indicated schematically in Figure 2, biophysical information combined with bioinformatics analyses of an entire set of structurally related proteins can address such questions, especially when facilitated by the availability of interactive query and visualization tools directed at widely used databases such as UniProt, the Gene Ontology (GO) and the PDB. For example in cases where there are no original functional hypotheses, sequence conservation patterns and the identification of cavities can be used in combination with UniProt sequence “features” to locate functionally important regions on a protein surface. The presence of such regions in a subset of structural neighbors should then enable a plausible function annotation. In cases where a researcher already has a functional hypothesis, the initial set of structural neighbors can be filtered based on the GO term representing that function. The reduced set of proteins generated in this way can then be further analyzed to identify the sequence and structural determinants of that function. We have implemented a server, called MarkUs (http://luna.bioc.columbia.edu/honiglab/mark-us/cgi-bin/submit.pl), that enables this type of analysis.
Although we have focused here on structure, categorizing proteins into families based on global sequence similarity is also problematic, as highlighted by the example shown in Figure 3. The figure shows a sequence alignment of a functionally important region for three members of the thioredoxin superfamily. Sequence differences unambiguously place thioredoxin and glutaredoxin-4 in different families. However, although the protein Q8L5D4 is included in the thioredoxin family (see the Pfam classification, for example), it has a CXFS motif that is characteristic of the glutaredoxin and not the thioredoxin family. In fact, it has been suggested that thioredoxin family members with the CXFS motif be reclassified as glutaredoxins [33]). This then is a case where global and local sequence similarities lead to different classifications.
Figure 3.
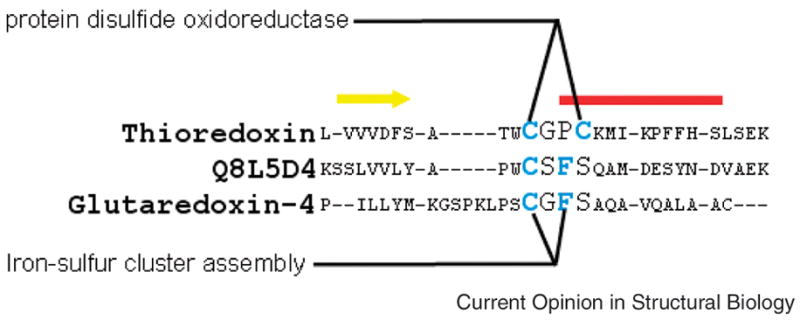
Functional motifs in thioredoxins. The multiple sequence alignment shows the primary functional regions, a strand-loop-helix containing the catalytic cysteines, of three members of the thioredoxin superfamily (strand=yellow bar, helix=red bar). As illustrated, the GO term “protein disulfide oxidoreductase” is associated with the two cysteines (shown in blue) that determine this functionality specific to thioredoxins while the term “iron-sulfur cluster assembly” is associated with that functionality in the monothiol glutaredoxin family.
The examples presented above highlight the difficulties associated with describing protein function based on the classification of proteins into specific groups. The alternative that we propose begins with the identification of possible evolutionary relationships by using sequence and/or structural alignment tools to identify related proteins independent of their classification, but then filtering the list based on more local criteria. The successful implementation of such a strategy, which should be maximally flexible and adaptable to the needs of a particular problem, will require the development of tools necessary both to generate hypotheses and to eliminate obvious false positive that are generated in the first stage.
An important step in this direction would be a more fine-grained description of function. For example, significant improvements in the accuracy of annotation transfer have been observed when GO terms are associated with specific residues [34,35] or specific structural features such as similar loop conformations [36,37], and where the validity of annotation transfer was based on the conservation of such specific features. While, as in these studies, the association between GO term and structure/sequence features was done automatically, a combination of expert-identified associations and computational tools would probably be optimal. For the proteins shown in Figure 3, a GO/residue association would both prevent the misclassification of the protein Q8L5D4 as a thioredoxin and facilitate an alternate functional hypothesis based on its local similarity to the glutaredoxins.
Summary
The difficulties associated with classification based on structural similarity exist in other areas of biology as well. For example, ambiguities in Linnaean classification of biological organisms equivalent to the type of classification used in SCOP and CATH with “fold”, “superfamily”, “family” replaced by “kingdom”, “phylum”, etc. continue to come to light as more complete genomes become available. Indeed, whole kingdoms have been removed or replaced [38]. The possibility of actually generating an unambiguous classification that represents the “tree of life” has been questioned [39] as alternatives to the traditional descent with modification for the transfer of genes between organisms have been discovered. Such complexities have led to the proposition that evolutionary relationships are more appropriately represented as a “web of life” [40].
In full analogy, we have argued here that the complex web of relationships that characterize protein structure/function space cannot be properly represented by placing proteins into discrete categories, especially when based on global structural similarity. On this basis, we are suggesting a number of changes in how function annotation is addressed. Specifically, we propose a more dynamical approach to annotation which involves the use of tools based on sequence and structure alignments that identify many possible relationships that can then be used as a starting point for more detailed analysis. This analysis should be based on local, residue-level information offered by as the integration of biophysical tools, evolutionary conservation patterns, and expanded versions of databases such as GO and UniProt. It is our opinion that the development of computational technologies that enable this overall approach will lead to a far more productive exploitation of the information contained in sequence/structure/function databases than is currently possible. The implications of such an approach, for example on the impact of structural genomics initiatives, could be enormous. Every new structure that is solved and every new homology model that is constructed would then have the potential of uncovering multiple unanticipated relationships thus playing an important role in the development of a new description of the web of relationships that characterizes “protein space”.
Acknowledgments
We acknowledge many helpful conversations with Markus Fisher and many other members of our lab. This work was supported by NIH grants R01-GM030518 and U54-CA121852.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Taylor WR. Evolutionary transitions in protein fold space. Current opinion in structural biology. 2007;17:354–361. doi: 10.1016/j.sbi.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Gancedo C, Flores C-L. Moonlighting proteins in yeasts. Microbiology and molecular biology reviews: MMBR. 2008;72:197–210. doi: 10.1128/MMBR.00036-07. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Commichau FM, Stulke J. Trigger enzymes: bifunctional proteins active in metabolism and in controlling gene expression. Molecular microbiology. 2008;67:692–702. doi: 10.1111/j.1365-2958.2007.06071.x. [DOI] [PubMed] [Google Scholar]
- 4●.Reeves GA, Dallman TJ, Redfern OC, Akpor A, Orengo CA. Structural Diversity of Domain Superfamilies in the CATH Database. Journal of Molecular Biology. 2006;360:725–741. doi: 10.1016/j.jmb.2006.05.035. This paper, along with reference [5], describe the surprising amount of structural diversity that can arise in proteins that are related evolutionarily as a result of variations functional necessities, such as novel oligomeric states or binding of ligands with different moieties. [DOI] [PubMed] [Google Scholar]
- 5●.Andreeva A, Murzin AG. Evolution of protein fold in the presence of functional constraints. Current opinion in structural biology. 2006;16:399–408. doi: 10.1016/j.sbi.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Yang AS, Honig B. An integrated approach to the analysis and modeling of protein sequences and structures. I. Protein structural alignment and a quantitative measure for protein structural distance. J Mol Biol. 2000;301:665–678. doi: 10.1006/jmbi.2000.3973. [DOI] [PubMed] [Google Scholar]
- 7.Shindyalov IN, Bourne PE. An alternative view of protein fold space. Proteins: Structure, Function, and Genetics. 2000;38:247–260. [PubMed] [Google Scholar]
- 8.Kolodny R, Petrey D, Honig B. Protein structure comparison: implications for the nature of 'fold space', and structure and function prediction. Current Opinion in Structural Biology. 2006;16:393–398. doi: 10.1016/j.sbi.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 9●.Petrey D, Fischer M, Honig B. Functional relationships between apparently unrelated proteins: implications for the nature of protein structure space. Proceedings of the National Academy of Sciences of the United States of America; 2009. Submitted. This paper along with [10,24] discuss an array of functional relationship between proteins that would normally be considered structurally dissimilar and evolutionarily unrelated, and highlights the potential for remote structural relationships to reveal useful functional information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10●.Krishna SS, Grishin NV. Structural drift: a possible path to protein fold change. Bioinformatics. 2005;21:1308–1310. doi: 10.1093/bioinformatics/bti227. [DOI] [PubMed] [Google Scholar]
- 11●.Davidson AR. A folding space odyssey. Proceedings of the National Academy of Sciences of the United States of America; 2008. pp. 2759–2760. This paper discusses the implications of the results described in references [26–27] which demonstrate an unambiguous evolutionary link between proteins that would normally be classified differently in terms of their overall structure, i.e., an all-α protein and an α/β protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanco FJ, Angrand I, Serrano L. Exploring the conformational properties of the sequence space between two proteins with different folds: an experimental study. Journal of molecular biology. 1999;285:741–753. doi: 10.1006/jmbi.1998.2333. [DOI] [PubMed] [Google Scholar]
- 13.He Y, Chen Y, Alexander P, Bryan PN, Orban J. NMR structures of two designed proteins with high sequence identity but different fold and function. Proceedings of the National Academy of Sciences of the United States of America; 2008. pp. 14412–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meier S, Jensen PR, David CN, Chapman J, Holstein TW, Grzesiek S, Ozbek S. Continuous molecular evolution of protein-domain structures by single amino acid changes. Current biology: CB. 2007;17:173–178. doi: 10.1016/j.cub.2006.10.063. [DOI] [PubMed] [Google Scholar]
- 15.Meier S, Ozbek S. A biological cosmos of parallel universes: does protein structural plasticity facilitate evolution? BioEssays: news and reviews in molecular, cellular and developmental biology. 2007;29:1095–1104. doi: 10.1002/bies.20661. [DOI] [PubMed] [Google Scholar]
- 16.Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nature structural & molecular biology. 2004;11:338–345. doi: 10.1038/nsmb748. [DOI] [PubMed] [Google Scholar]
- 17.Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF. Interconversion between two unrelated protein folds in the lymphotactin native state. Proceedings of the National Academy of Sciences of the United States of America; 2008. pp. 5057–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishna SS, Sadreyev RI, Grishin NV. A tale of two ferredoxins: sequence similarity and structural differences. BMC structural biology. 2006;6:8. doi: 10.1186/1472-6807-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bashton M, Chothia C. Structure. Vol. 15. London, England: 2007. The generation of new protein functions by the combination of domains; pp. 85–99. 1993. [DOI] [PubMed] [Google Scholar]
- 20.Moore AD, Bjorklund AK, Ekman D, Bornberg-Bauer E, Elofsson A. Arrangements in the modular evolution of proteins. Trends in Biochemical Sciences. 2008;33:444–451. doi: 10.1016/j.tibs.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 21.Graziano JJ, Liu W, Perera R, Geierstanger BH, Lesley SA, Schultz PG. Selecting folded proteins from a library of secondary structural elements. Journal of the American Chemical Society. 2008;130:176–185. doi: 10.1021/ja074405w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pisarchik A, Petri R, Schmidt-Dannert C. Probing the structural plasticity of an archaeal primordial cobaltochelatase CbiX(S) Protein engineering, design & selection: PEDS. 2007;20:257–265. doi: 10.1093/protein/gzm018. [DOI] [PubMed] [Google Scholar]
- 23.Peisajovich SG, Rockah L, Tawfik DS. Evolution of new protein topologies through multistep gene rearrangements. Nature genetics. 2006;38:168–174. doi: 10.1038/ng1717. [DOI] [PubMed] [Google Scholar]
- 24●.Coles M, Hulko M, Djuranovic S, Truffault V, Koretke K, Martin J, Lupas AN. Structure. Vol. 14. London, England: 2006. Common evolutionary origin of swapped-hairpin and double-psi beta barrels; pp. 1489–1498. 1993. [DOI] [PubMed] [Google Scholar]
- 25.Pereira-Leal JB, Levy ED, Kamp C, Teichmann SA. Evolution of protein complexes by duplication of homomeric interactions. Genome biology. 2007;8:R51. doi: 10.1186/gb-2007-8-4-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26●.Roessler CG, Hall BM, Anderson WJ, Ingram WM, Roberts SA, Montfort WR, Cordes MHJ. Transitive homology-guided structural studies lead to discovery of Cro proteins with 40% sequence identity but different folds. Proceedings of the National Academy of Sciences of the United States of America; 2008. pp. 2343–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27●.Van Dorn LO, Newlove T, Chang S, Ingram WM, Cordes MH. Relationship between sequence determinants of stability for two natural homologous proteins with different folds. Biochemistry. 2006;45:10542–10553. doi: 10.1021/bi060853p. [DOI] [PubMed] [Google Scholar]
- 28.Alva V, Koretke KK, Coles M, Lupas AN. Cradle-loop barrels and the concept of metafolds in protein classification by natural descent. Current opinion in structural biology. 2008;18:358–365. doi: 10.1016/j.sbi.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Ammelburg M, Hartmann MD, Djuranovic S, Alva V, Koretke KK, Martin J, Sauer G, Truffault V, Zeth K, Lupas AN, et al. A CTP-dependent archaeal riboflavin kinase forms a bridge in the evolution of cradle-loop barrelsStructure (London, England: 1993) 2007151577–1590. [DOI] [PubMed] [Google Scholar]
- 30.Karasaki Y, Tsukamoto S, Mizusaki K, Sugiura T, Gotoh S. A garlic lectin exerted an antitumor activity and induced apoptosis in human tumor cells. Food Research International. 2001;34:7–13. [Google Scholar]
- 31.Xie L, Bourne PE. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments. Proceedings of the National Academy of Sciences of the United States of America; 2008. pp. 5441–5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32●.Xie L, Wang J, Bourne PE. In silico elucidation of the molecular mechanism defining the adverse effect of selective estrogen receptor modulators. PLoS computational biology. 2007;3:e217. doi: 10.1371/journal.pcbi.0030217. This paper is an illustration of how useful biological information with important clinical applications can be derived from structures whose global similarity is remote but which have local similarities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pace NR. Time for a change. Nature. 2006;441:289. doi: 10.1038/441289a. [DOI] [PubMed] [Google Scholar]
- 34.Doolittle WF, Bapteste E. Pattern pluralism and the Tree of Life hypothesis. Proceedings of the National Academy of Sciences of the United States of America; 2007. pp. 2043–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koonin EV, Wolf YI. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic acids research. 2008;36:6688–6719. doi: 10.1093/nar/gkn668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rouhier N, Gelhaye E, Jacquot JP. Plant glutaredoxins: still mysterious reducing systems. Cellular and molecular life sciences: CMLS. 2004;61:1266–1277. doi: 10.1007/s00018-004-3410-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian W, Arakaki AK, Skolnick J. EFICAz: a comprehensive approach for accurate genome-scale enzyme function inference. Nucleic Acids Research. 2004;32:6226–6239. doi: 10.1093/nar/gkh956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pazos F, Sternberg MJ. Automated prediction of protein function and detection of functional sites from structure. Proceedings of the National Academy of Sciences of the United States of America; 2004. pp. 14754–14759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manikandan K, Pal D, Ramakumar S, Brener NE, Iyengar SS, Seetharaman G. Functionally important segments in proteins dissected using Gene Ontology and geometric clustering of peptide fragments. Genome biology. 2008;9:R52. doi: 10.1186/gb-2008-9-3-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Espadaler J, Querol E, Aviles FX, Oliva B. Identification of function-associated loop motifs and application to protein function prediction. Bioinformatics (Oxford, England) 2006;22:2237–2243. doi: 10.1093/bioinformatics/btl382. [DOI] [PubMed] [Google Scholar]