

Abstract
Aims
Several mutations in the ventricular myosin regulatory light chain (RLC) were identified to cause familial hypertrophic cardiomyopathy (FHC). Based on our previous cellular findings showing delayed calcium transients in electrically stimulated intact papillary muscle fibres from transgenic Tg-R58Q and Tg-N47K mice and, in addition, prolonged force transients in Tg-R58Q fibres, we hypothesized that the malignant FHC phenotype associated with the R58Q mutation is most likely related to diastolic dysfunction.
Methods and results
Cardiac morphology and in vivo haemodynamics by echocardiography as well as cardiac function in isolated perfused working hearts were assessed in transgenic (Tg) mutant mice. The ATPase–pCa relationship was determined in myofibrils isolated from Tg mouse hearts. In addition, the effect of both mutations on RLC phosphorylation was examined in rapidly frozen ventricular samples from Tg mice. Significantly, decreased cardiac function was observed in isolated perfused working hearts from both Tg-R58Q and Tg-N47K mice. However, echocardiographic examination showed significant alterations in diastolic transmitral velocities and deceleration time only in Tg-R58Q myocardium. Likewise, changes in Ca2+ sensitivity, cooperativity, and an elevated level of ATPase activity at low [Ca2+] were only observed in myofibrils from Tg-R58Q mice. In addition, the R58Q mutation and not the N47K led to reduced RLC phosphorylation in Tg ventricles.
Conclusion
Our results suggest that the N47K and R58Q mutations may act through similar mechanisms, leading to compensatory hypertrophy of the functionally compromised myocardium, but the malignant R58Q phenotype is most likely associated with more severe alterations in cardiac performance manifested as impaired relaxation and global diastolic dysfunction. At the molecular level, we suggest that by reducing the phosphorylation of RLC, the R58Q mutation decreases the kinetics of myosin cross-bridges, leading to an increased myofilament calcium sensitivity and to overall changes in intracellular Ca2+ homeostasis.
KEYWORDS: Cardiac function, Echocardiography, Myofibrillar ATPase, Myosin regulatory light chain (RLC)
1. Introduction
Familial hypertrophic cardiomyopathy (FHC) is a relatively common autosomal dominant genetic disease characterized by ventricular hypertrophy, myofibrillar disarray, often clinically manifesting as heart failure, and/or sudden cardiac death (SCD).1,2 It originates from mutations in genes encoding the major contractile proteins of the heart,2,3 including mutations in the ventricular myosin regulatory light chain (RLC), which constitute ∼2% of total FHC sarcomeric mutations.4 The RLC-associated disease phenotypes may or may not present as with the classic FHC phenotype and some variants have been associated with SCD without significant hypertrophy (for review see Szczesna5). Interestingly, RLC mutations that affect Ca2+ binding cause variable disease phenotypes in humans, ranging from a relatively mild phenotype in E22K (glutamate to lysine)-mutated patients,6,7 severe mid-ventricular hypertrophy in N47K (asparagine to lysine) individuals,8,9 and to a malignant disease phenotype demonstrated by multiple cases of SCD in R58Q (arginine to glutamine)7,10–12 and D166V (aspartate to valine)8,11 positive patients.
We have recently suggested new potential mechanisms by which calcium-binding mutations of RLC (E22K, N47K, R58Q and D166V) may alter cardiac muscle contraction by changing the function of RLC as a temporary delayed Ca2+ buffer. By changing the properties of the RLC Ca2+–Mg2+-binding site (Figure 1), the FHC mutations may enhance or diminish this intracellular Ca2+-buffering function of RLC. For example, by decreasing the affinity and increasing the specificity of this site for Ca2+, the E22K mutation was shown to decrease the amplitude and duration of [Ca2+] and force transients in transgenic (Tg) intact papillary muscles and led to faster muscle relaxation.13 Consequently, by inactivating this site for Ca2+, the N47K and R58Q mutations resulted in delayed calcium transients in intact muscle fibres and, in addition, the R58Q mutation caused slower muscle relaxation.14 Interestingly, delayed muscle relaxation was recently observed in intact papillary muscle fibres expressing another calcium-binding RLC mutation, D166V.15 Accordingly, both R58Q and D166V mutations were shown to increase the Ca2+-sensitivity of force and ATPase, suggesting a direct mutation-induced effect on the kinetics of myosin cross-bridges and the regulation of cardiac muscle contraction.14,15
Figure 1.

Linear representation of the N-terminal myosin regulatory light chain (RLC) sequence containing the Ca2+–CaM-activated myosin light chain kinase phosphorylation site at Ser 15 and the EF-hand (helix–loop–helix) Ca2+–Mg2+-binding site of RLC. Familial hypertrophic cardiomyopathy mutations are labelled in boxes: alanine to threonine (A13T), phenylalanine to leucine (F18L), glutamate to lysine (E22K), asparagine to lysine (N47K), and arginine to glutamine (R58Q). X, first Ca2+ ligand; Y, second Ca2+ ligand; Z, third Ca2+ ligand; G, glycine; -Y, fourth Ca2+ ligand, provided by a backbone carbonyl; I, isoleucine (although other aliphatic residues are also found at this position); -X, fifth Ca2+ ligand; -Z, sixth and seventh Ca2+ ligands, provided by a bidentate glutamate or aspartate residue.
The objective of this study was to build upon our previous cellular findings assessing the N47K and R58Q mutations of RLC14,16 and investigate whether human phenotypes associated with these mutations can be replicated in Tg mouse models expressing these RLC mutations. Cardiac morphology and ventricular haemodynamics were examined in Tg-N47K and Tg-R58Q mice and the results compared with those obtained with age- and gender-matched Tg-WT (wild type, expressing the human ventricular RLC in mice) and NTg (non-transgenic) mice. The effects of the N47K and R58Q mutations were also assessed in isolated perfused working hearts as previously examined for Tg-E22K mice.13 In addition, based on our solution findings suggesting a correlation between Ca2+ binding to recombinant R58Q mutant protein and its phosphorylation,17 we examined the effects of the N47K and R58Q mutations on RLC phosphorylation in Tg-mice. Interestingly, as we showed recently in rapidly frozen ventricular samples from Tg-D166V mice, the D166V mutation significantly reduced the level of RLC phosphorylation and similar to R58Q mutation caused a series of Ca2+-dependent alterations in cardiac muscle contraction.15 Finally, we also examined the effect of the N47K and R58Q mutations on myofibrillar ATPase activity, cooperativity, and myofilament calcium sensitivity in myofibrils purified from Tg hearts.
2. Methods
2.1. Animals
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All animal studies were conducted in accordance with institutional guidelines (Animal Welfare Assurance Number A3711-01, IACUC Approval Date 06/01/2007). Transgenic mouse models expressing WT or two FHC mutations (N47K and R58Q) of the human ventricular RLC were generated and characterized as described earlier.14 With few exceptions, Tg mutant mice used in the current study were 7 ± 1-month-old males that were age matched with Tg-WT and NTg control littermates. Some of the experiments were repeated with 14 ± 1-month-old mice and if no differences between age groups were found (P > 0.05), the results were combined and averaged. Specific ages of mice are provided in each experimental protocol.
2.2. Protein phosphorylation
After euthanasia, the hearts from ∼6-month-old Tg-N47K, Tg-R58Q, Tg-WT, and NTg mice were excised and the ventricles were immediately isolated and frozen in liquid nitrogen. Prior to the experiment, the tissue was thawed in a buffer consisting of 20 mM phosphate buffer, pH 8.0, 12.5 mM MgCl2, 0.1 M CaCl2, 5 mM ATP, 0.6 mM NaN3, 0.2 mM PMSF, and 1 µL/mL Protease Inhibitor Cocktail (Sigma), homogenized, and dissolved in SDS–PAGE buffer containing 500 mM Tris, pH 6.8, 6 M Urea, 1% SDS, 10% β-mercaptoethanol, and 0.05% Bromophenol Blue and then loaded onto 15% SDS–PAGE. The phosphorylated Tg RLC was detected with +P-human RLC antibodies, specific for the phosphorylated form of the human ventricular RLC (generously provided by Dr Neal Epstein, NIH18) followed by a secondary goat anti-rabbit antibody conjugated with the fluorescent dye, IR red 800. Phosphorylated troponin I (TnI) was detected with Mab14 antibody (MMS-418R, Covance, Berkeley, CA, USA), sensitive to the phosphorylated Ser 24 in the sequence of cardiac TnI, and followed by a secondary goat anti-mouse antibody conjugated with the fluorescent dye, Cy 5.5 (Figure 2). The total RLC protein was detected with a rabbit polyclonal RLC CT-1 antibody (generated in this laboratory), whereas the total TnI was detected with the 6F9 antibody (Research Diagnostics Inc.) both of which served as loading controls.
Figure 2.

The effect of the N47K and R58Q mutations on the phosphorylation status of myosin regulatory light chain (RLC) and troponin I (TnI) in transgenic ventricular extracts blotted with Mab14 MMS-418R antibody recognizing phosphorylated TnI (+P-TnI) (upper panel), and human specific phospho-RLC antibody recognizing transgenic +P-RLChuman (middle panel). Total TnI (TnItotal) and RLC (RLCtotal) proteins were blotted with 6F9 and CT-1 antibodies, respectively (bottom panel). Lane 1, R58Q protein standard (RLCpr.std.); lane 2, Tg-N47K extract; lane 3, Tg-R58Q extract; lanes 4 and 5, NTg extract; lane 6, Tg-WT extract; lane 7, phosphorylated human cardiac RLC-WT protein standard (+P-RLCpr.std.); lane 8, phosphorylated human cardiac TnI-WT protein standard (+P-HCTnIpr.std.).
2.3. Transcript expression, reverse transcriptase–polymerase chain reaction
Mouse ventricles were rapidly harvested from ∼6-month-old NTg, Tg-WT, Tg-R58Q, and Tg-N47K mice, submerged immediately in an appropriate volume of ice cold RNAlater reagent (Qiagen) and stored frozen at −80°C. Total RNA was isolated from adult murine left ventricles using an RNeasy Fibrous Tissue Kit (Qiagen) as described previously.13 First-strand cDNA was synthesized using 2 µg of total RNA and 1 µM oligo-dT primer using Omniscript reverse transcriptase (Qiagen). RT–PCRs were performed using sets of specific sense (S) and antisense (AS) primers designed to amplify the mouse cardiac sarcoplasmic reticulum Ca2+-dependent ATPase (SERCA 2; NCBI accession no. NM_009722), phospholamban (PLB; NM_023129), and α-actin (NM_009608) genes (Table 1). The PCRs were performed using Taq DNA polymerase (Invitrogen) and the products were electrophoresed on 2% agarose gels using ethidium bromide staining. Reactions were documented using a BioRad Gel Imaging System (Gel Doc XR) and Image Quant Software13 (Figure 3).
Table 1.
Primers (S, sense and AS, anti-sense) used for analysis of the expression pattern of mouse cardiac sarcoplasmic reticulum Ca2+-dependent ATPase 2 and phospholamban genes
| Gene/primer | Sequence | Expected size of Amplicon/PCR product (bp) |
|---|---|---|
| SERCA 2 S | 5′-GGGTGTGTGGCAGGAAAGAAATGCT-3′ | 650 |
| SERCA 2 AS | 5′-AAGAGCTAAGCAGGTGGTGATGAC-3′ | |
| PLB S | 5′-CTCACATTTGGCTGCCTGTTGTCA-3′ | 360 |
| PLB AS | 5′-TGACCCTCACAAAGCTGTTCTCAG-3′ | |
| α-Actin S | 5′-AGGTGTCATGGTGGGTATGG-3′ | 200 |
| α-Actin AS | 5′-GCTTCAGTGAGCAGGGTTG-3′ | |
| GAPDH S | 5′-GCTTCACCACCTTCTTG-3′ | 600 |
| GAPDH AS | 5′-TCACCATCTTCCAGGAG-3′ |
Figure 3.

Transcript expression pattern of Ca2+-handling proteins, SERCA 2 and PLB, in transgenic mice. Left ventricular total RNA extracts from Tg-WT, Tg-R58Q, and Tg-N47K mice expressing ≈100% transgene were used. Mouse cardiac α-actin and GAPDH were used as internal loading controls. Lanes 1 and 9, 100 bp and 1 kb plus DNA ladders, respectively; lanes 2 and 3, NTg; lanes 4 and 5, Tg-WT; lane 6, Tg-R58Q; lanes 7 and 8, Tg-N47K.
2.4. Myofibrillar ATPase activity
Cardiac myofibrils were prepared from left and right ventricular walls, septa, and papillary muscles from ∼6-month-old Tg-R58Q, Tg-N47K, Tg-WT, and NTg mice according to Solaro et al.19 Myofibrillar ATPase assays were performed in a solution of 20 mM MOPS, pH 7.0, 40 mM KCl, 2.5 mM MgCl2, 2 mM EGTA, and increasing concentrations of Ca2+ from pCa 9 to pCa 4.5. After 5 min of incubation at 30°C, the reaction was initiated with 2.5 mM ATP and terminated after 10 min with 5% trichloroacetic acid. Released inorganic phosphate was measured according to Fiske and Subbarow.20 Data were analysed with a Hill equation yielding the pCa50 values (50% of the ATPase activity) and the Hill coefficient, nH.21
2.5. Echocardiography
In vivo cardiac morphology and function were assessed non-invasively using a high-frequency, high-resolution echocardiography system consisting of a Vevo 660 ultrasound machine equipped with a 25–50 MHz transducer (Visual Sonics, Toronto, Canada). Six ∼8-month-old- and two ∼15-month-old male Tg-R58Q mice and eight ∼8-month-old- and two ∼14-month-old male Tg-N47K mice were tested and compared with age-matched NTg and Tg-WT controls. Mice were anaesthetized using 3% isoflurane and transferred to an imaging stage equipped with built-in electrocardiography electrodes for continuous heart rate monitoring. The body temperature was maintained at 37°C. Anaesthesia was sustained via a nose cone with 1% isoflurane. High-resolution images were obtained in the parasternal and apical orientations. Standard B-mode (2D) images of the heart and pulsed Doppler images of the mitral valve inflow were acquired. Left ventricular dimensions and wall thickness were measured at the level of the papillary muscles in parasternal short axis, at end-systole, and end-diastole. Left ventricular ejection fraction (LVEF) and mass were determined as described in De Simone et al.22 Global diastolic haemodynamics were evaluated using transmitral Doppler velocities. An apical four-chamber view of the heart was obtained. A pulsed Doppler sample was placed at the tip of the mitral leaflets and transmitral velocities were acquired and stored electronically for offline analysis. Transmitral Doppler data were collected only when the conditions of a stable heart rate, regular rhythm, and consistent velocity profile were met. Early (E) and late (A) transmitral diastolic velocities (cm/s) and the deceleration time (ms) were measured and used as non-invasive indicators of global diastolic function.23
2.6. Isolated mouse working heart perfusion
Experiments with isolated perfused working hearts were performed as described earlier in Szczesna-Cordary et al.13 Hearts were maintained at 37°C during the entire perfusion period. Five 7-month-old male Tg-R58Q and eight 7-month-old male Tg-N47K mice were tested and compared with age-matched NTg and Tg-WT controls. In addition, two ∼15-month-old female Tg-R58Q mice and seven ∼14-month-old male and female Tg-N47K mice were examined. The perfusion protocol included a 30 min aerobic perfusion of spontaneously beating hearts. Functional measurements were acquired for 10 s, three times in 10 min intervals (total 30 min) with an MP100 system from AcqKnowledge (BioPac Systems, Santa Barbara, CA, USA) and data were averaged. Mechanical function (heart rate, peak systolic pressure, and maximum developed pressure), cardiac output, aortic flow, and coronary flow were measured with either in-line pressure transducers or flow probes, as described previously.13 Cardiac work was calculated as the product of peak systolic pressure and cardiac output. Stroke volume was calculated as the cardiac output divided by the heart rate, whereas stroke work was calculated as the stroke volume times the peak systolic pressure. Cardiac power was calculated as the product of developed pressure and cardiac output. A conversion factor of 1.33 × 10–4 was used to convert cardiac power values from millimetres of mercury per millilitre to joules.13
2.7. Statistical analysis
Data are expressed as the average of n experiments ± SEM (standard error of the mean). Multiple comparisons between groups were performed using One-Way ANOVA procedures and an unpaired Student’s t-test (Sigma Plot 11; Systat Software, Inc., San Jose, CA, USA). Statistically significant differences were defined as P < 0.05.
3. Results
3.1. Analysis of protein phosphorylation
Figure 2 demonstrates the effect of R58Q and N47K mutations on the phosphorylation status of the RLC in Tg mouse ventricular extracts blotted with +P-human RLC antibodies, specific for the phosphorylated form of the human ventricular RLC.18 Since both Tg mice express equivalent amounts of transgene which is also similar in Tg-WT,14 the RLC phosphorylation level in Tg-R58Q, Tg-N47K, and Tg-WT ventricular extracts could be directly compared. The +P-RLC and +P-TnI band intensities were corrected for total RLC and TnI content detected with CT-1 and 6F9 antibodies, respectively. As shown, the R58Q mutation reduced phosphorylation of RLC by ∼50% evidenced by the lower +P-RLC band intensity in Tg-R58Q ventricles compared with Tg-WT (Figure 2, middle panel, lane 3 vs. lane 6). Data from three independent experiments showed that compared with Tg-WT hearts, the R58Q myocardium demonstrated only ∼0.48 ± 0.06-fold (n = 3) of RLC phosphorylation. In contrast, the N47K mutation did not change the level of RLC phosphorylation compared with Tg-WT (Figure 2, middle panel, lane 2 vs. lane 6) and there was ∼1.04 ± 0.17-fold (n = 4) of RLC phosphorylation in Tg-N47K vs. Tg-WT hearts. Interestingly, the down-regulation of the phosphorylated form of RLC in Tg-R58Q extracts was accompanied by a slight increase (∼1.24 ± 0.13-fold, n = 3) in TnI phosphorylation compared with Tg-WT (Figure 2, upper panel, lane 3 vs. lane 6). In parallel, a slight decrease (∼0.78 ± 0.09-fold, n = 4) in TnI phosphorylation was observed in Tg-N47K vs. Tg-WT hearts (Figure 2, upper panel, lane 2 vs. lane 6).
3.2. Expression of calcium-handling genes in transgenic mice
To test whether expression of N47K and R58Q mutations in the heart interfered with intracellular Ca2+-handling genes, we examined mRNA expression of SERCA 2 and its regulatory protein, PLB, in the mutated myocardium identified with specific primers designed to amplify the mouse cardiac SERCA 2 and PLB genes (Table 1). As shown in Figure 3, neither mutation, N47K or R58Q, affected transcript expression of SERCA 2 and PLB tested in Tg-R58Q and Tg-N47K ventricular extracts compared with Tg-WT and NTg controls. GAPDH was used as internal loading control for SERCA 2 and α-actin served as control in the reaction for PLB.
3.3. Effects of R58Q and N47K mutations on myofibrillar ATPase activity
In support of our previous experiments on skinned papillary muscle fibres,14 myofibrillar ATPase activity performed under unloaded conditions on myofibrils from ventricles of Tg mice revealed differences in the Ca2+ sensitivity of ATPase for Tg-R58Q mice (Figure 4A). A large increase in Ca2+-sensitivity of ATPase compared with other group of mouse myofibrils was observed in Tg-R58Q myofibrils (Figure 4A) and the respective pCa50 values of the ATPase–pCa dependence are plotted in Figure 4B: 6.48 ± 0.11 (Tg-R58Q), 6.28 ± 0.03 (Tg-N47K), 6.24 ± 0.06 (Tg-WT), and 6.22 ± 0.03 (NTg). The ‘n’ indicates the number of ATPase assays performed on different myofibrillar preparations. Importantly, R58Q-mutated myofibrils demonstrated an impaired inhibition of ATPase activity measured at low [Ca2+] (7.5 < pCa < 8.5) (Figure 4A, P < 0.05). The level of myofibrillar ATPase activity in Tg-R58Q myofibrils was also significantly higher at submaximal Ca2+ concentrations, [Ca2+] ≤ 1 µM, whereas no difference was observed at saturating [Ca2+] (Figure 4A). The Hill coefficient (nH) of the ATPase–pCa relationship was decreased in Tg-R58Q myofibrils compared with Tg-WT or Tg-N47K myofibrils, indicating a poor R58Q-mediated myofibrillar ATPase cooperativity (Figure 4A).
Figure 4.

(A) ATPase–pCa relationships for NTg, Tg-WT, Tg-N47K, and Tg-R58Q mouse cardiac myofibrils. (B) The pCa50 values for the ATPase–pCa dependences of NTg, Tg-WT, Tg-N47K, and Tg-R58Q mouse muscle myofibrils. The differences between the Tg-R58Q myofibrils and myofibrils from other transgenic lines were statistically significant (P < 0.05). Data in (A) and (B) are expressed as mean of n experiments ± SEM.
3.4. Echo-Doppler evaluation
Based on our previous cellular findings, we hypothesized that a malignant FHC phenotype associated with the R58Q mutation could be related to diastolic dysfunction of the R58Q-mutated myocardium. The results summarized in Figures 5 and 6 fully support our hypothesis. However, there were no statistically significant differences in heart rate, LV end-systolic or end-diastolic wall thickness, LV mass, and LVEF between NTg, Tg-WT, Tg-N47K, and Tg-R58Q mice (Table 2). A 30% increase in inter-ventricular septal thickness in diastole [IVS(d)] and posterior wall thickness in diastole [PW(d)] were observed in two 15-month-old Tg-R58Q mice compared with ∼8-month-old Tg-R58Q mice. The same was true for one 14-month-old Tg-N47K mice which showed a 20% increase in IVS(d) and LVPW(d) compared with ∼8-month-old N47K animals (data not shown). Therefore, there might be an age-induced hypertrophy in Tg-R58Q and Tg-N47K hearts, which was not present in control mice. No systolic dysfunction was determined in any of the tested groups of mice (Table 2). In contrast, diastolic dysfunction evidenced by reduced early transmitral diastolic velocity E and increased transmitral velocity A as well as prolonged deceleration time was observed in Tg-R58Q mice compared with Tg-N47K and control of mice (Figures 5 and 6). As a result, the E/A ratio was significantly lower in Tg-R58Q mice (2.2 ± 0.4) compared with all tested groups of mice, NTg (3.9 ± 0.4), Tg-WT (5.3 ± 0.3), and Tg-N47K (4.6 ± 0.9) (P < 0.05) (Figure 6). Deceleration time in Tg-R58Q mice was 53 ± 6 ms, which was 1.71-, 1.36-, and 1.66-fold longer compared with Tg-N47K, Tg-WT, and NTg mice, respectively (Figure 6, P < 0.05). Overall, the results indicate abnormal relaxation and diastolic dysfunction in Tg-R58Q mice.
Figure 5.
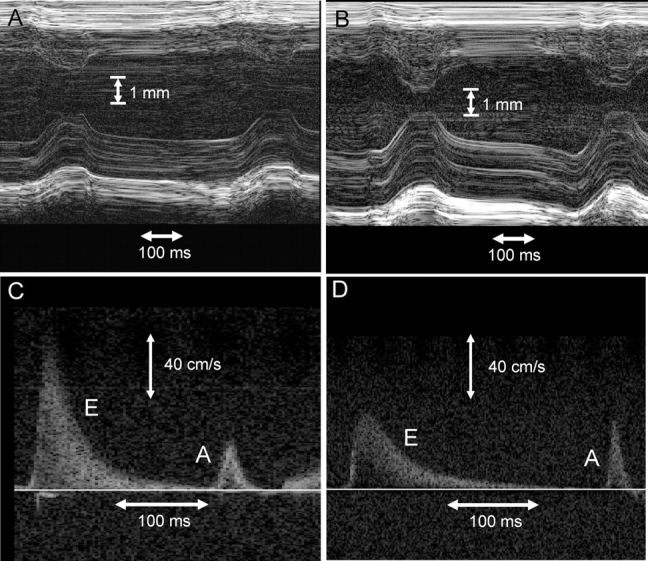
Representative high-resolution echocardiography B-mode images from control (A) and Tg-R58Q mice (B) show no significant difference in chamber dimensions or wall thickness. Representative pulsed Doppler tracings of the mitral valve in controls (C) and Tg-R58Q mice (D) demonstrating reduced E-wave velocity and longer deceleration times in the latter group.
Figure 6.

Assessment of in vivo cardiac function. (A) Early transmitral diastolic velocity ‘E-wave’. (B) Late transmitral diastolic velocity ‘A-wave’. (C) E/A ratio, and (D) deceleration time. Data are expressed as mean of n experiments ± SEM.
Table 2.
In vivo cardiac morphology by echocardiography
| Function | NTg, n = 12 | Tg-WT, n = 6 | Tg-N47K, n = 9 | Tg-R58Q, n = 7 |
|---|---|---|---|---|
| Heart rate (bpm) | 382 ± 58 | 420 ± 62 | 395 ± 72 | 390 ± 60 |
| LVD(d) (mm) | 3.9 ± 0.12 | 4.3 ± 0.15 | 4.0 ± 0.15 | 4.0 ± 0.15 |
| LVD(s) (mm) | 2.6 ± 0.11 | 2.8 ± 0.20 | 2.6 ± 0.11 | 2.5 ± 0.13 |
| IVS(d) (mm) | 0.86 ± 0.03 | 0.91 ± 0.03 | 0.81 ± 0.04 | 0.80 ± 0.13 |
| LVPW(d) (mm) | 0.85 ± 0.03 | 0.93 ± 0.05 | 0.87 ± 0.03 | 0.80 ± 0.15 |
| LVEF (%) | 59 ± 5 | 64 ± 8 | 57 ± 5 | 64 ± 3 |
| LV mass (g) | 0.70 ± 0.004 | 0.73 ± 0.010 | 0.70 ± 0.006 | 0.70 ± 0.012 |
LVD(d), left ventricular dimension at end-diastole; LVD(s), left ventricular dimension at end-systole; IVS(d), inter-ventricular septal thickness in diastole; LVPW(d), left ventricular posterior wall thickness in diastole; LVEF, left ventricular ejection fraction.
3.5. Isolated perfused hearts study
To investigate the relationship between the severity of FHC disease in N47K and R58Q patients and cardiac muscle performance in Tg-N47K and Tg-R58Q mice, we used an isolated perfused working heart model to test the ability of the mutated hearts to perform work. As shown in Tables 3 and 4, cardiac output, cardiac work, and cardiac power were significantly decreased in Tg-N47K and Tg-R58Q aerobically perfused and spontaneously beating hearts compared with controls. Importantly, compared with the N47K mutation, the R58Q mutation resulted in much more severe changes in cardiac function demonstrating even more reduced cardiac work and consequently cardiac power compared with control hearts (Table 3). The changes observed for both mutations indicate compromised cardiac function in both Tg-N47K and Tg-R58Q mice; however, the phenotype associated with Tg-R58Q mice was much more profound (Table 3). No significant differences between female and male mice nor between younger or older animals in any of functional measurements in isolated perfused hearts were observed.
Table 3.
Measurements performed in isolated perfused working hearts from Tg-N47K and Tg-R58Q mice compared with control NTg and Tg-WT mice
| Function | NTg, n = 18 | Tg-WT, n = 14 | Tg-N47K, n = 15 | Tg-R58Q, n = 7 |
|---|---|---|---|---|
| Heart rate (HR) (bpm) | 281 ± 10 | 271 ± 12 | 262 ± 13 | 255 ± 30 |
| Peak systolic pressure (PSP) (mmHg) | 72.9 ± 1.5 | 68.5 ± 1.6 | 68.2 ± 1.0 | 69.9 ± 2.4 |
| Developed pressure (DP) (mmHg) | 31.4 ± 1.3 | 30.9 ± 1.4 | 27.8 ± 1.7 | 27.8 ± 3.0 |
| HR × PSP (bpm × mmHg × 10−3) | 20.4 ± 0.8 | 18.4 ± 0.7 | 17.7 ± 0.8 | 17.4 ± 1.7 |
| HR × DP (bpm × mmHg × 10−3) | 8.6 ± 0.2 | 8.3 ± 0.4 | 7.1 ± 0.4 | 6.5 ± 0.5 |
| Cardiac output (mL/min) | 10.3 ± 0.6 | 9.4 ± 0.9 | 7.1 ± 0.6 | 5.6 ± 0.8 |
| Aortic output (mL/min) | 8.0 ± 0.4 | 7.3 ± 0.8 | 5.3 ± 0.5 | 3.4 ± 0.8 |
| Coronary flow (mL/min) | 2.3 ± 0.3 | 2.1 ± 0.2 | 1.8 ± 0.2 | 2.2 ± 0.4 |
| Cardiac work (ml mmHg/min) | 7.5 ± 0.4 | 6.5 ± 0.6 | 4.8 ± 0.4 | 3.9 ± 0.6 |
| Cardiac power (mJ/min) | 84.4 ± 5.0 | 71.5 ± 6.6 | 53.1 ± 4.3 | 43.5 ± 6.9 |
Table 4.
The P-values resulting from One-Way ANOVA pairwise multiple comparison procedures using the Student–Newman–Keuls method
| Compared mouse groups | Cardiac power | Cardiac work | Cardiac output | Aortic output | HR × DP | P < 0.05 |
|---|---|---|---|---|---|---|
| NTg vs. Tg-R58Q | <0.001 | <0.001 | 0.001 | <0.001 | 0.004 | Yes |
| NTg vs. Tg-N47K | <0.001 | <0.001 | 0.003 | 0.005 | 0.004 | Yes |
| NTg vs. Tg-WT | 0.089 | 0.109 | 0.307 | 0.398 | 0.553 | No |
| Tg-WT vs. Tg-R58Q | 0.014 | 0.012 | 0.008 | 0.002 | 0.013 | Yes |
| Tg-WT vs. Tg-N47K | 0.021 | 0.021 | 0.024 | 0.028 | 0.013 | Yes |
| Tg-N47K vs. Tg-R58Q | 0.314 | 0.291 | 0.203 | 0.066 | 0.371 | No |
Our recent study indicated that the malignant FHC phenotype associated with the R58Q mutation might be related to inefficient energy utilization by the R58Q myocardium.24 We therefore examined the hearts of 7-month-old male Tg-R58Q mice for oxygen consumption while executing work in isolated perfused working hearts and the results were compared with Tg-WT and NTg controls (see Supplementary material online, Figure S1). As expected, cardiac efficiency defined as the ratio of cardiac power to O2 consumption25 was significantly decreased in Tg-R58Q mouse hearts during the initial aerobic perfusion period and also decreased after ischaemia during reperfusion. The difference between Tg-R58Q and control mice during the post-ischaemic reperfusion period was not statistically significant (see Supplementary material online, Figure S1).
4. Discussion
In this report, we present evidence for compromised cardiac function in both Tg-N47K and Tg-R58Q mice that might be responsible for compensatory hypertrophy observed in humans carrying these N47K and R58Q mutations. We also demonstrate that the R58Q-induced changes in cardiac contractility are much more severe than those in the hearts of Tg-N47K mice, and that these differences might explain an observed malignant FHC phenotype and multiple cases of SCD reported for R58Q-mutated patients. Abnormal function of the R58Q myocardium most likely originates from a mutation-mediated decrease in RLC phosphorylation, the R58Q-induced increase in Ca2+-sensitivity of contraction, and impaired relaxation ultimately manifesting as global diastolic dysfunction.
As shown in Figure 1, binding of Ca2+ to cardiac myosin occurs at the RLC Ca2+–Mg2+-binding site that contains a single EF-hand helix–loop–helix motif in the N-terminal domain of RLC and also contains a phosphorylation site at Ser 15, a target for the Ca2+–CaM-activated myosin light chain kinase (MLCK). Interestingly, it is the R58Q and not N47K mutation that results in a decreased level of RLC phosphorylation, as measured in rapidly frozen ventricular samples from mutant Tg mice compared with Tg-WT hearts (Figure 2). As we showed earlier, the R58Q-induced failure of Ca2+ binding to RLC was totally reversed by phosphorylation of recombinant R58Q protein.17 Perhaps, replacing a positively charged arginine with a polar glutamine decreases the affinity of the phosphate group for Ser 15 resulting in lower phosphorylation of RLC in Tg-R58Q hearts (Figure 2). Reduced levels of RLC phosphorylation were paralleled by a slight increase in the PKA-phosphorylation of TnI in Tg-R58Q mice, whereas there was a slight decrease in TnI phosphorylation in Tg-N47K vs. Tg-WT hearts. Possibly, MLCK-dependent RLC phosphorylation is offset by increased or decreased phosphorylation of TnI via sarcomeric protein phosphatases that might be specific for multiple protein targets.
Structurally, ventricular myosin RLC belongs to the superfamily of EF-hand calcium-binding proteins and binds Ca2+ with ∼1.5 µM affinity.17 Calcium is a key regulator of the complex workings of the heart and any alterations in Ca2+ signalling can potentially lead to contractile dysfunction, heart failure, and cardiac death.26,27 As we showed previously, the RLC can function as a molecular regulator of myofilament Ca2+ homeostasis, working in parallel to troponin C (TnC) in maintaining intracellular [Ca2+].13,14 Both FHC mutations, N47K and R58Q, were shown to deactivate RLC for Ca2+ binding and led to Ca2+-dependent changes of muscle contraction at the protein17,24 and cellular13,14,16 levels. Importantly, compared with the N47K mutation, the R58Q substitution resulted in a large increase in myofilament calcium sensitivity.14 The myofilament sensitivity to Ca2+ is a function of two components: (i) the kinetics of force generating myosin cross-bridges and (ii) kon/koff of Ca2+ binding to TnC.28 Therefore, the observed leftward shift in the force/ATPase–pCa relationships in Tg-R58Q myocardium observed earlier14 and in the current study could be due to slower cross-bridge kinetics in Tg-R58Q myocardium or a decreased Ca2+ dissociation from TnC. The latter was fully supported by prolonged force transients measured in intact muscle fibres from Tg-R58Q mice14 and in the current work demonstrating impaired relaxation in vivo. As we confirmed, these RLC Ca2+-dependent changes in contractility seem to be specific for the myofilament network as expression of the key sarcoplasmic reticulum Ca2+ re-uptake genes, SERCA 2, and its regulatory protein PLB was not changed by the N47K or R58Q mutation (Figure 3).
Myofibrillar ATPase activity assays also show a decrease in the cooperativity of the actin–myosin interaction in both Tg-N47K and Tg-R58Q mice with a much more dramatic effect exhibited by the R58Q mutation (Figure 4). Most importantly, compared with Tg-N47K, Tg-WT, or NTg mice, the Tg-R58Q myocardium demonstrated a largely increased ATPase activity at low [Ca2+] (pCa 8), implicating impaired muscle relaxation (Figure 4). The lack of full inhibition of ATPase activity at low [Ca2+] suggests a possibility of an abnormal diastolic filling and our previous findings of delayed calcium and force transients in Tg-R58Q intact muscle fibres14 fully support this notion. But the true evidence of abnormal diastolic function in Tg-R58Q mice comes from our echo-Doppler assessment (Figures 5 and 6) showing significantly altered early and late diastolic transmitral velocities and prolonged deceleration time in Tg-R58Q mice compared with other groups, despite similar ventricular mass and normal systolic function (Table 2). One can conclude that diastolic function is affected earlier in the disease process in this model and may likely be a more sensitive indicator of a malignant FHC phenotype than hypertrophy or systolic dysfunction. Future invasive haemodynamic studies would be helpful to confirm abnormal diastolic function in our Tg FHC mice. Furthermore, diastolic dysfunction as evidenced in Tg-R58Q mice closely parallels significant contractile abnormalities noted in isolated heart preparations. As shown in Table 3, significantly decreased cardiac output, cardiac work, and ultimately cardiac power were observed in isolated perfused working hearts from Tg-N47K and Tg-R58Q mice; however, the changes caused by the R58Q mutation were much more profound (Tables 3 and 4). Additional data attained with Tg-R58Q mice also demonstrated significantly decreased cardiac efficiency in aerobically perfused R58Q hearts compared with controls and poor recovery after an acute ischaemic episode confirming greatly compromised cardiac function in Tg-R58Q mice (see Supplementary material online, Figure S1).
Our results are mirrored by the severity of human phenotypes associated with the N47K and R58Q mutations in RLC showing cardiac hypertrophy with less severe outcomes in N47K patients8,9 and adverse disease progression with multiple cases of SCD in R58Q-positive patients.7,10–12 Both mutations may act through similar mechanisms leading to compensatory hypertrophy of the functionally compromised myocardium but the more severe phenotype associated with the R58Q mutation is most likely due to a series of changes initiated at the molecular level, e.g. mutation-dependent decrease in RLC phosphorylation affecting kinetics of R58Q cross-bridges, compromised Ca2+-dependent regulation of contraction, changes that ultimately alter intracellular Ca2+ homeostasis, and uncouple contraction–relaxation cycle. A large increase in myofilament Ca2+ sensitivity when placed in vivo would contribute to decreased ventricular filling at high heart rates when the tail end of the first [Ca2+] transient begins to fuse with the next. The slow force relaxation could also start to fuse with the next contraction when heart rates are high leading to diastolic dysfunction.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by NIH-HL071778 (to D.S.-C.), NIH-AG22554-01 (to T.P.A.), and a grant from the Canadian Institute for Health Research (to G.D.L.).
Acknowledgements
The authors thankfully acknowledge Neal Epstein at the National Heart, Lung, and Blood Institute, NIH, for the gift of the human phospho-RLC antibodies. The authors thank Zoraida Diaz-Perez and Georgianna Guzman (University of Miami) for their excellent technical assistance.
Conflict of interest: none declared.
References
- 1.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 2.Seidman CE, Seidman JG. Molecular genetic studies of familial hypertrophic cardiomyopathy. Basic Res Cardiol. 1998;93:13–16. doi: 10.1007/s003950050196. [DOI] [PubMed] [Google Scholar]
- 3.Tardiff J. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005;10:237–248. doi: 10.1007/s10741-005-5253-5. [DOI] [PubMed] [Google Scholar]
- 4.Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. 2008;19:104–110. doi: 10.1111/j.1540-8167.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 5.Szczesna D. Regulatory light chains of striated muscle myosin. Structure, function and malfunction. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:187–197. doi: 10.2174/1568006033481474. [DOI] [PubMed] [Google Scholar]
- 6.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, et al. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 7.Kabaeva ZT, Perrot A, Wolter B, Dietz R, Cardim N, Correia JM, et al. Systematic analysis of the regulatory and essential myosin light chain genes: genetic variants and mutations in hypertrophic cardiomyopathy. Eur J Hum Genet. 2002;10:741–748. doi: 10.1038/sj.ejhg.5200872. [DOI] [PubMed] [Google Scholar]
- 8.Andersen PS, Havndrup O, Bundgaard H, Moolman-Smook JC, Larsen LA, Mogensen J, et al. Myosin light chain mutations in familial hypertrophic cardiomyopathy: phenotypic presentation and frequency in Danish and South African populations. J Med Genet. 2001;38:E43. doi: 10.1136/jmg.38.12.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hougs L, Havndrup O, Bundgaard H, Kober L, Vuust J, Larsen LA, et al. One third of Danish hypertrophic cardiomyopathy patients have mutations in MYH7 rod region. Eur J Hum Genet. 2005;13:161–165. doi: 10.1038/sj.ejhg.5201310. [DOI] [PubMed] [Google Scholar]
- 10.Flavigny J, Richard P, Isnard R, Carrier L, Charron P, Bonne G, et al. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J Mol Med. 1998;76:208–214. doi: 10.1007/s001090050210. [DOI] [PubMed] [Google Scholar]
- 11.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 12.Morner S, Richard P, Kazzam E, Hellman U, Hainque B, Schwartz K, et al. Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J Mol Cell Cardiol. 2003;35:841–849. doi: 10.1016/s0022-2828(03)00146-9. [DOI] [PubMed] [Google Scholar]
- 13.Szczesna-Cordary D, Jones M, Moore JR, Watt J, Kerrick WGL, Xu Y, et al. Myosin regulatory light chain E22K mutation results in decreased cardiac intracellular calcium and force transients. FASEB J. 2007;21:3974–3985. doi: 10.1096/fj.07-8630com. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Xu Y, Kerrick WGL, Wang Y, Guzman G, Diaz-Perez Z, et al. Prolonged Ca2+ and force transients in myosin RLC transgenic mouse fibers expressing malignant and benign FHC mutations. J Mol Biol. 2006;361:286–299. doi: 10.1016/j.jmb.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 15.Kerrick WGL, Kazmierczak K, Xu Y, Jones M, Wang Y, Szczesna-Cordary D. Malignant D166V-FHC mutation in the ventricular myosin regulatory light chain causes profound effects in skinned and intact papillary muscle fibers from transgenic mice. FASEB J. 2009;23:000–000. doi: 10.1096/fj.08-118182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szczesna-Cordary D, Guzman G, Ng SS, Zhao J. Familial hypertrophic cardiomyopathy-linked alterations in Ca2+ binding of human cardiac myosin regulatory light chain affect cardiac muscle contraction. J Biol Chem. 2004;279:3535–3542. doi: 10.1074/jbc.M307092200. [DOI] [PubMed] [Google Scholar]
- 17.Szczesna D, Ghosh D, Li Q, Gomes AV, Guzman G, Arana C, et al. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J Biol Chem. 2001;276:7086–7092. doi: 10.1074/jbc.M009823200. [DOI] [PubMed] [Google Scholar]
- 18.Davis JS, Hassanzadeh S, Winitsky S, Lin H, Satorius C, Vemuri R, et al. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001;107:631–641. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- 19.Solaro RJ, Pang DC, Briggs FN. The purification of cardiac myofibrils with Triton X-100. Biochim Biophys Acta. 1971;245:259–262. doi: 10.1016/0005-2728(71)90033-8. [DOI] [PubMed] [Google Scholar]
- 20.Fiske CH, Subbarow Y. The colorimetric determination of phosphorus. J Biol Chem. 1925;66:375–400. [Google Scholar]
- 21.Hill TL. Binding of monovalent and divalent myosin fragments onto sites on actin. Nature. 1978;274:825–826. doi: 10.1038/274825a0. [DOI] [PubMed] [Google Scholar]
- 22.De Simone G, Wallerson DC, Volpe M, Devereux RB. Echocardiographic measurement of left ventricular mass and volume in normotensive and hypertensive rats. Necropsy validation. Am J Hypertens. 1990;3:688–696. doi: 10.1093/ajh/3.9.688. [DOI] [PubMed] [Google Scholar]
- 23.Nishimura RA, Appleton CP, Redfield MM, Ilstrup DM, Holmes DR, Jr, Tajik AJ. Noninvasive doppler echocardiographic evaluation of left ventricular filling pressures in patients with cardiomyopathies: a simultaneous Doppler echocardiographic and cardiac catheterization study. J Am Coll Cardiol. 1996;28:1226–1233. doi: 10.1016/S0735-1097(96)00315-4. [DOI] [PubMed] [Google Scholar]
- 24.Greenberg MJ, Watt JD, Jones M, Kazmierczak K, Szczesna-Cordary D, Moore JR. Regulatory light chain mutations associated with cardiomyopathy affect myosin mechanics and kinetics. J Mol Cell Cardiol. 2009;46:108–115. doi: 10.1016/j.yjmcc.2008.09.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopaschuk GD, Barr R, Thomas PD, Dyck JR. Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ Res. 2003;93:e33–e37. doi: 10.1161/01.RES.0000086964.07404.A5. [DOI] [PubMed] [Google Scholar]
- 26.Bers DM. Calcium and cardiac rhythms: physiological and pathophysiological. Circ Res. 2002;90:14–17. [PubMed] [Google Scholar]
- 27.Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J Clin Invest. 2005;115:556–564. doi: 10.1172/JCI24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson JM, Wang Y, Kerrick WG, Kawai R, Cheung HC. Activation of striated muscle: nearest-neighbor regulatory-unit and cross-bridge influence on myofilament kinetics. J Mol Biol. 2002;322:1065–1088. doi: 10.1016/s0022-2836(02)00855-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.