

Abstract
Despite numerous candidate gene and linkage studies, the field of type 2 diabetes (T2D) genetics had until recently succeeded in identifying few genuine disease-susceptibility loci. The advent of genome-wide association (GWA) scans has transformed the situation, leading to an expansion in the number of established, robustly replicating T2D loci to almost 20. These novel findings offer unique insights into the pathogenesis of T2D and in the main point towards the etiological importance of disorders of beta-cell development and function. All associated variants have common allele frequencies in the discovery populations, and exert modest to small effects on the risk of disease, characteristics which limit their prognostic and diagnostic potential. However, ongoing studies focussing on the role of copy number variation and targeting low frequency polymorphisms should identify additional T2D-susceptibility loci, some of which may have larger effect sizes and offer better individual prediction of disease risk.
Introduction
Type 2 diabetes (T2D) is a common complex disease, thought to be caused by a complex interplay between environmental and genetic factors. The sibling recurrence risk ratio (λs) for T2D, a measure of the familial aggregation of the disease, has been estimated to be approximately 3 in European populations (1). As with other common complex diseases, the hunt for T2D-susceptibility genes started a few decades ago. Over this period, study designs evolved as a result of progress in technology, in the understanding of the patterns of human genome sequence variation and in the availability of appropriately-ascertained samples. Hundreds of candidate gene studies and more than 30 genome-wide linkage scans for T2D (2,3) were published. Unfortunately, neither approach proved particularly successful in identifying robustly-replicating T2D-susceptibility loci.
This is certainly the case with genome-wide linkage approaches. Such studies typically utilized a few hundred microsatellite markers distributed across the genome to type sib-pairs or families ascertained for diabetes, and relied on available recombination rate information to infer the likely location of disease signals. Although linkage scans took a genome-wide gene-agnostic approach, and might therefore have been expected to have a higher success rate than gene-centric studies, few, if any, regions generated credible statistical evidence for linkage across multiple studies. To date, no susceptibility variant that can reliably account for any of the replicated T2D-linkage peaks has been identified, even though several resource-intensive fine-mapping efforts have been completed. One possible explanation for this may be that the fine-mapping studies have concentrated on evaluating the role of common polymorphisms. It seems increasingly probable (in the light of more recent findings) that most genuine linkage signals reflect the actions of multiple rare variants (copy number or single base pair changes).
Until recently, the candidate-gene approach fared little better in delivering real T2D-susceptibility effects: several factors contributed to this disappointing outcome. Firstly, the majority of studies were limited in terms of power: the small sample sizes deployed were inadequate to detect the kinds of effect sizes that we now know are realistic for most complex traits. Secondly, the candidate genes studied were selected on the basis of limited knowledge of the underlying etiopathology of disease. In addition, poor understanding of the architecture of genetic variation together with the time-consuming, low-throughput genotyping technologies available at the time, meant that it was not possible to undertake comprehensive assessments of the variants within the regions of interest. Finally, irreproducibility of results was further reinforced by liberal thresholds for declaring significance and a widespread tendency to over-interpret results.
The last few years have seen substantial changes in the way genetic association studies are conducted. The availability of large sample collections has led to increased power in both discovery and replication studies. In addition, the development of affordable, high-accuracy, high-throughput genotyping technologies has made large-scale examination of genetic variants (and particularly single nucleotide polymorphisms (SNPs)) possible. International efforts like the Human Genome Sequencing Project and the HapMap (4) have contributed to a much improved understanding of patterns of sequence variation. Genetic association studies have subsequently taken advantage of knowledge about correlation patterns, or linkage disequilibrium (LD), between variants, thereby achieving more streamlined and efficient study designs. These advances have made it possible to undertake genome-wide surveys of common variant association (genome wide association scans or GWAs). Such studies have been facilitated by improved guidelines and tools for study design, analysis and interpretation. The growing number of robustly replicating T2D susceptibility loci is a testament to these recent advances.
Established T2D susceptibility loci
For the purposes of this review, we define T2D-susceptibility loci as signals reaching genome-wide significance levels for T2D, regardless of their primary association. By consensus, genome-wide significance is usually taken as a p value of <5×10-8, which equates to the standard p<0.05 criterion adjusted for ~1M tests (roughly the potential number of independent common variant signals in non-African individuals, after allowing for linkage disequilibrium). Even so, deciding what does and does not constitute a T2D-susceptibility locus is not entirely straightforward. T2D predisposition is causally related to several other phenotypes, most notably body mass index (BMI) and other measures of adiposity, raising a debate as to whether a gene like FTO (which has a primary effect on BMI and obesity risk (5)) should really be counted as a T2D locus. Some would argue not, since the BMI effect is clearly primary: but others would argue that the effect of FTO variants on T2D-risk remains the same whether or not the mechanism responsible is known. Based on the definition cited earlier, there are currently 19 established T2D loci (Table 1) – a number poised to increase substantially.
Table 1.
Established type 2 diabetes susceptibility loci.
| Index SNP | Chr | Position | Region / gene | Identification | λ s * |
|---|---|---|---|---|---|
| rs1001013 1 |
4 | 6343816 | WFS1 | Candidate gene | 1.004 |
| rs1801282 | 3 | 12368125 | PPARG | Candidate gene | 1.005 |
| rs757210 | 17 | 33170628 | HNF1B (TCF2) | Candidate gene | 1.002 |
| rs5219 | 11 | 17366148 | KCNJ11 | Candidate gene | 1.005 |
| rs7901695 | 10 | 114744078 | TCF7L2 | Linkage peak fine- mapping |
1.022 |
| rs1081166 1 |
9 | 22124094 | CDKN2A/B | GWA | 1.003 |
| rs1094639 8 |
6 | 20769013 | CDKAL1 | GWA | 1.002 |
| rs1326663 4 |
8 | 118253964 | SLC30A8 | GWA | 1.003 |
| rs4402960 | 3 | 186994381 | IGF2BP2 | GWA | 1.002 |
| rs5015480 | 10 | 94455539 | HHEX/IDE | GWA | 1.002 |
| rs8050136 | 16 | 52373776 | FTO ** | GWA | 1.009 |
| rs2237892 | 11 | 2796327 | KCNQ1 | GWA | 1.031 |
| rs1083096 3 |
11 | 92348358 | MTNR1B *** | GWA | 1.001 |
| rs1092393 1 |
1 | 120319482 | NOTCH2 | GWA meta-analysis | 1.001 |
| rs1277979 0 |
10 | 12368016 | CDC123/CAMK1D | GWA meta-analysis | 1.002 |
| rs4607103 | 3 | 64686944 | ADAMTS9 | GWA meta-analysis | 1.002 |
| rs7578597 | 2 | 43586327 | THADA | GWA meta-analysis | 1.002 |
| rs7961581 | 12 | 69949369 | TSPAN8/LGR5 | GWA meta-analysis | 1.001 |
| rs864745 | 7 | 28147081 | JAZF1 | GWA meta-analysis | 1.001 |
Calculated in European populations, with the exception of the KCNQ1 locus, which was based on East Asian populations.
The primary association for this locus is with body mass index (BMI).
The primary association for this locus is with fasting glucose levels.
Only four of these 19 T2D-associated variants have been identified through candidate gene studies (Table 1). This represents a rather meagre return given the enormous number of candidate gene studies carried out. The non-synonymous P12A and E23K variants, in PPARG and KCNJ11 respectively, were the first robustly replicating signals to emerge (6,7). The two genes, encoding peroxisome proliferator-activated receptor gamma (PPARG) and Kir6.2 (KCNJ11) were selected for study as their products represent drug targets used in the treatment of diabetes. The more recent findings of WFS1 and HNF1B (previously known as TCF2) SNP associations with T2D arose from in-depth studies of genes in candidate biological pathways (8-10).
The single largest effect size for T2D identified to date resides within the TCF7L2 gene. This locus was unearthed following a linkage peak fine-mapping study on chromosome 10 (11). Although there is evidence for linkage across the region in which the TCF7L2 gene resides, the common polymorphisms driving the T2D association do not account for the linkage signal observed. It therefore remains unclear whether this discovery was pure serendipity, or whether rare causal variants (as yet undiscovered) in TCF7L2 are responsible for the linkage signal.
The 14 remaining T2D-susceptibility loci (mapping near CDKN2A/B, CDKAL1, SLC30A8, IGF2BP2, HHEX/IDE, FTO, KCNQ1, NOTCH2, CDC123/CAMK1D, ADAMTS9, THADA, TSPAN8/LGR5, JAZF1 and MTNR1B; Table 1) have been identified over the course of the last 18 months through individual GWA studies (12-19), through the meta-analysis of GWA scans (20), or in the case of MTNR1B, as follow-up to a signal originally identified through analyses of continuous glycemic traits (21-23).
Published GWAS for T2D
GWA scans have been the most important contributors in finding novel, reproducibly associated T2D variants. For the purposes of this review article, we define GWA scans as studies that have examined over 150,000 SNPs (on the basis that the genome coverage of studies with less than this number is relatively low). Thus far, the literature counts six such GWA scans carried out in European populations (12-18) and one scan carried out in Japanese individuals (19).
The European GWA scans for T2D all used a case-control study design and employed commercially available, fixed-content genotyping platforms. Sample ascertainment, the specific populations examined, sample size and follow-up strategies differed among studies: these factors underlie some of the heterogeneity in the findings. The first published study was conducted in 661 cases and 614 controls from France, using the Illumina platform, and reported the HHEX/IDE and SLC30A8 regions as novel T2D susceptibility loci (16). Four further scans followed shortly afterwards (12-15, 18). Steinthorsdottir et al (18) typed 1399 cases and 5275 controls from Iceland for approximately 300,000 SNPs on the Illumina chip and reported CDKAL1 as a new T2D locus. This same locus was independently found by three further contemporaneous scans performed by the Wellcome Trust Case Control consortium (WTCCC), Diabetes Genetics Initiative (DGI) and Finland-United States Investigation of NIDDM genetics (FUSION) (12-15). The WTCCC (14,15) typed ~500,000 SNPs using the Affymetrix platform in 1924 cases and 2938 controls from the UK. The DGI study (12) employed the same genotyping technology and examined 1464 cases and 1467 controls from Finland and Sweden. The FUSION scan (13) also focused on individuals from Finland (1161 cases, 1174 controls) and used Illumina to type ~300,000 SNPs across the genome. These last three studies collaborated by sharing data and coordinating replication studies, their efforts leading to identification of the CDKN2A/2B, FTO and IGF2BP2 disease loci, as well as confirming other previously-reported loci such as PPARG, KCNJ11 and TCF7L2. The last scan to date published on individuals of European descent (17) examined 500 cases and 497 controls from four populations (Finland, Israel, Germany and UK) but, primarily for reasons of restricted power, failed to reveal any novel T2D susceptibility loci.
The most recent published scan was performed in 194 T2D cases and 1558 controls, all from Japan (19). Perlegen array technology was used to genotype over 200,000 tag SNPs, selected to capture >50% of common variation in the Japanese population. This study confirmed association of the CDKAL1 and IGF2BP2 signals and, through replication efforts, additionally identified KCNQ1 as a novel T2D susceptibility locus. Association at this locus was also identified by a second, smaller scan, again carried out in Japanese individuals (24). The same association signal can be detected in European samples, though differences in allele frequency render the signal much less visible than in East Asians (19,24).
Meta-analysis of GWAS
Although genome-wide association scans have proved successful in identifying T2D susceptibility variants, the power of individual studies to detect small or modest effects at common SNPs has been limited. For example, the WTCCC study, with 1924 cases and 2938 controls, had only 9% power to detect an allelic odds ratio (OR) of 1.2 at a SNP with minor allele frequency (MAF) of 30% at genome-wide significance levels (p=5×10-8).
These limitations have served as an impetus for collaborative meta-analyses of GWA scan data, designed to improve the power to detect genuine susceptibility loci. Power can be increased both through the combination of datasets to achieve a larger sample size, and through the concomitant examination of both directly typed and imputed SNPs. The development and implementation of statistical approaches for the imputation of genotypes at untyped variants on the basis of a common reference (25), such as the HapMap (4), has also facilitated the integration of GWA scans derived from different genotyping platforms.
Investigators from the WTCCC, DGI and FUSION scans joined forces as the DIAGRAM (Diabetes Genetics, Replication And Meta-Analysis) Consortium to provide the first complex disease GWA scan meta-analysis. DIAGRAM (20) combined data across 4549 T2D cases and 5579 controls, all of European descent, from the 3 studies (stage 1). Imputation of genotypes at untyped HapMap SNPs within each scan and subsequent stringent quality control resulted in the final examination of 2,202,892 variants across 10128 samples. These efforts boosted power to detect common susceptibility loci with modest effects, although the power to detect smaller effects remained low (Figure 1), justifying even larger meta-analysis efforts in the future. Following the synthesis of summary results across the genome, 65 independent promising signals were prioritized for replication genotyping in up to 22426 further samples of European descent (stage 2). Then, the 10 strongest signals emerging from stage 2 were selected for a further round of genotyping in an additional, independent set of up to 57366 T2D cases and controls, all of European descent.
Figure 1.
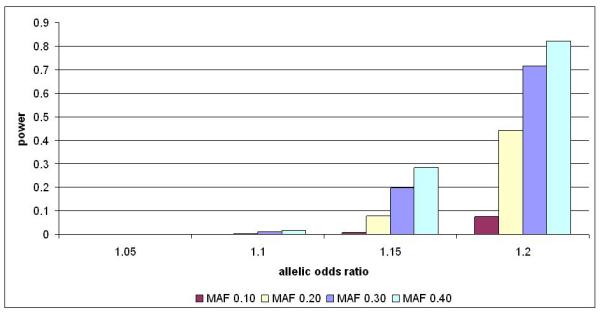
Power of the combined WTCCC, FUSION and DGI data (4549 cases, 5579 controls) to identify an association (alpha of 5×10-8) over a range of different effect sizes and allele frequencies.
When data from all 3 stages were combined using a meta-analysis framework, six novel T2D susceptibility loci reached genome-wide significance levels (mapping near NOTCH2, CDC123/CAMK1D, ADAMTS9, THADA, TSPAN8/LGR5, JAZF1; Table 1). Of these, only NOTCH2 could have been considered a reasonable biological candidate previous to this work. Several other loci (such as DCD and VEGFA) failed to reach stringent association values, but nevertheless were highlighted as interesting candidates. Incorporation of further data will undoubtedly clarify their true relationship to T2D predisposition.
Lessons on genetic architecture of T2D
The GWA studies performed to date have only been designed to detect a particular subset of potential susceptibility effects – those attributable to common SNPs (or copy number variants (CNVs)) well-tagged by the particular variants represented on the various commodity genotyping arrays. Even with the latest wave of high-density arrays, an appreciable minority of common SNPs remains poorly-tagged, even in the European-descent populations which have been the focus of most studies (26). In addition, there is limited coverage of low-frequency variants (27) and, despite the advances in design represented by the new generation of “CNV-aware” chips, the capacity to genotype structural variants (a collective term for polymorphic duplications, deletions and inversions) across the allele frequency spectrum lags well behind that of SNPs (28).
These considerations restrict the allele frequency range of the association signals that can be reliably detected. Since common variants of large effect seem to be few and far between, the consequence has also been to constrain the effect size spectrum of proven susceptibility variants (29). Variants within TCF7L2 remain the basis for the strongest T2D-susceptibility signal in European-descent samples, all other associations displaying per-allele odds ratios between 1.1 and 1.2. Where available, robust estimates of the true effect sizes of many of these proven variants indicate that chance (the “winner’s curse”) often contributed to their initial detection, and that, by inference, many more loci of broadly similar size remain to be found. For some of the confirmed signals, it is probable that fine-mapping efforts will, in time, identify variants causal for the association which have effects larger than those of the tagged variants which led to their discovery (30). In contrast, “winner’s curse” effects may mean that, for others, current measures of effect size overestimate the true state of affairs (31).
How much of the genetic predisposition to T2D is explained by the known variants? Before we benchmark the known T2D-susceptibility variants against some measure of the overall genetic contribution to disease, it is important to acknowledge that estimates of heritability are, by their nature, population- and time-specific. Besides, we have little understanding of the extent to which epigenetic effects (such as the influence of maternal uterine environment on offspring diabetes) (32) are capable of masquerading as apparent genetic predisposition, thereby inflating estimates of heritability. For dichotomous traits, the sibling relative risk (or λs) is often used as a convenient measure of the extent of familial aggregation, though this encapsulates both shared environment and genetic effects. Thus, whilst the frequently-quoted sibling relative risk estimate of T2D in Europeans (~3) (1) can only be considered to provide an upper bound for the total genetic influence, there seems little doubt that the combined effect attributable to known variants (a λs of 1.07) falls well short of any credible assessment of the genetic contribution to T2D. Most probably, the 20 or so known variants explain only 5-10% of the inherited predisposition.
What could explain the missing heritability (or genetic “dark matter” as it has been termed)? Certainly, continued efforts to uncover more common causal variants, through GWA meta-analysis, and fine-mapping, should fill some of the gap. However, given the modest locus effect sizes likely to result from these future discovery efforts, the overall additional contribution to heritability is unlikely to be substantial. At the same time, efforts to use the GWA approach to identify common susceptibility variants in other ethnic groups should, as the recent revelations concerning KCNQ1 demonstrate, improve the proportion of genetic variance explicable in non-European descent populations (19,24,33).
Common CNVs are also likely to be involved in disease-susceptibility (28). In terms of the base pair numbers involved, the sequence diversity attributable to CNVs is roughly equivalent to that due to SNPs, so one might naively expect the overall contribution to disease predisposition made by SNPs and CNVs to be quite similar. However, growing evidence that common CNVs are typically well-tagged by adjacent SNPs (34) suggests that many of the signals for which common CNVs are causal will already have been detected through SNP-based GWA studies. Ongoing efforts to explore global CNV associations for T2D and other common traits should clarify these issues.
Most evaluations of the joint effects of variants assume independence, so non-additivity (in the form of gene-gene and/or gene-environmental interactions) could be another factor contributing to the missing heritability. For the common T2D-susceptibility loci, little evidence of either has been observed (14): this may in part reflect intrinsic low power to detect interaction effects, particularly when the causal variants have not yet been defined. However, it has been argued on population genetics grounds that gene-gene interaction (GGI) is unlikely to have a major effect (35), and, in some scenarios, the variance explained under non-additivity can be LESS than that assumed if the effects are independent (36).
For many researchers in the field, the most probable source of missing heritability lies in the putative contribution of low frequency and/or rare variants (both SNPs and CNVs) of intermediate penetrance. Variants with such characteristics will certainly show marked familial aggregation, though the penetrance would generally be insufficient to allow for detection using classical linkage approaches: at the same time, the risk allele frequency is below the threshold detected reliably through GWAs. As a result, where such variants exist, they are likely to have been refractory to the kinds of systematic genome-wide surveys that have been so successful for rare, penetrant variants (at one extreme) and common variants of modest effect (at the other) (30). The arrival of “next-generation” sequencing technologies brings such variants into play and, in the coming years, it will become possible to examine the hypothesis that susceptibility to T2D (and other common traits) involves the action of multiple low-frequency variants.
Lessons on etiopathogenesis of T2D
The identification of DNA sequence variants shown to have a robust causal relationship to biomedical traits of interest represents one of the most powerful tools available for understanding the mechanisms involved in the pathogenesis of human disease. In principle, definitive statements relating a given association signal to a precise set of molecular and physiological events are generally only possible following identification of the causal variants (most likely through resequencing and fine-mapping) and/or detailed functional studies. In practice, however, it is often possible to make reasonably confident inferences about etiopathological mechanisms before such analyses have been completed.
Analysis of T2D-associated variants in healthy populations has clearly demonstrated that most of the T2D-susceptibility signals so far examined (notably the signals near KCNJ11, TCF7L2, CDKAL1, CDKN2A/B, IGF2BP2, HNF1B, HHEX/IDE, JAZF1 and SLC30A8) have a predominant effect on insulin secretion, with only FTO (which acts primarily through an effect on BMI and risk of obesity) (5) and PPARG shown to influence insulin action (18,37-41). These data reinforce the growing evidence (42) supporting the crucial role of defects in beta-cell function and mass in the development of T2D.
To go beyond these more general physiological observations to inferences about pathways depends on the confidence with which the likely causal genes can be identified. For some signals, the associated interval contains more than one highly credible candidate (e.g. HHEX and IDE in the region on chromosome 10). Nonetheless, some clear stories are emerging. Both HHEX and TCF7L2 are implicated in the regulation of Wnt-signalling, highlighting dysfunction of this pathway in the islet as a key process in T2D development (43). The association signal in SLC30A8 clearly points the finger at disturbances in zinc transport within insulin-secretory granules (16,44).
Perhaps most informative is the evidence incriminating abnormalities in cell cycle regulation. This evidence remains somewhat circumstantial, but at least four of the 18 regions can be tied to genes thought to be involved in this process (these are the signals close to CDKAL1, CDKN2A/B, CDC123 and CDKN1C [KCNQ1]). In rodents, Cdkn2a overexpression compromises maintenance of beta-cell mass and recapitulates the T2D phenotype (45). Further studies at these loci may provide valuable clues to which aspects of the regulation of beta-cell mass (development, regeneration or senescence) are central to the development of diabetes.
Several of the genes implicated in T2D-susceptibility appear to have effects on other common disease traits: these pleiotropic relationships often extend beyond accepted boundaries of co-morbidity. Thus, variation in CDKAL1 has been implicated in inflammatory bowel disease as well as T2D (46), and CDKN2A/B, JAZF1 and HNF1B are each known to play a role in cancer (10,47,48). In the case of HNF1B, alleles at the same variant have opposing effects on predisposition to T2D and prostate cancer, an inverse relationship which is consistent with the epidemiological data (49). The links between diabetes and cancer may betray a variety of shared etiological pathways: these include overlaps in signalling pathways as well as contrasting effects on the regulation of the cell cycle.
Clinical applications of the findings
So far, the modest effect sizes of the common T2D-susceptibility variants limit the extent to which they can be used, individually or in combination, as tools for individual prediction of disease risk (50). Although it is certainly possible to use genetic risk scores to identify individuals who, by virtue of inheriting particularly large or small numbers of risk alleles, differ appreciably in their risk of diabetes (e.g. the top and bottom 1% differ about 4-fold in risk), it is by no means clear that such information is clinically actionable. Indeed, traditional risk factors, such as BMI and age, are much more effective tools for estimating future risk of diabetes, in adults at least, than any available combination of genetic variants. This does not mean that the prospects for individualized genetic profiling will remain of limited value: if, as described above, the coming years provide a bumper harvest of medium-penetrance, low-frequency susceptibility variants, these may provide a much more effective platform for individual prediction than the array of common variants currently available.
In the long-term of course, we can expect novel insights into T2D pathogenesis generated by genetic discoveries to feed forward into drug development. Proof-of-principle is provided by the observation that two of the proven T2D-susceptibility signals lie in genes (PPARG, KCNJ11) whose protein products are already the targets of established and effective therapeutic agents. But what about using genetic information to improve treatment of diabetes at the level of the individual patient? Prescribing on the basis of personal molecular diagnosis is already established practise for those with monogenic forms of diabetes (e.g. sulphonylureas for those with neonatal diabetes due to KCNJ11 mutations) (51,52). And it is possible to show, in cross sectional studies, that TCF7L2 genotype influences the efficiency of the response to sulphonylureas in those with common forms of T2D (53). However, as with disease risk, the modest effect sizes typical of T2D-susceptibility loci mean that the quality of these predictions is far too low for clinical implementation at the level of the individual patient. One potentially powerful use of genetic variant information may be to define groups of individuals which differ appreciably in disease risk as a means to improve the efficiency of therapeutic clinical trials.
Conclusions
So where do we stand? On the one hand, the explosion in confirmed signals, not just for T2D, but also for a range of related traits including BMI and hyperlipidaemia, has been spectacular. This is all the more so, coming, as it does, after so many years of faltering progress. At the same time, recognition that these discoveries have, so far, explained only a small proportion of the inherited predisposition limits our ability to deliver a complete understanding of disease pathogenesis and our capacity to undertake useful disease prediction.
However, the primary stated objective of GWA studies has always been to gain biological insights. This alone warrants detection of confirmed signals, and, as with monogenic diseases, the impact of such discoveries has little bearing on the extent of population variance explained. Substantial work will often be required to define the molecular mechanisms through which these association signals mediate disease risk. However, even before definitive evidence can be produced (through fine-mapping and functional studies), it is possible to make informed and credible inferences about the processes involved and thereby gain important clues to pathogenesis that can initiate translational advances. An obvious example in the T2D field is the relationship between the SLC30A8 association signal and dysfunction of the islet Zn transporter, ZnT-8, which has encouraged efforts to develop pharmaceuticals which target Zn uptake into beta-cell granules (16,44).
Whilst we may well need to look elsewhere to find the bulk of the inherited predisposition to T2D (particularly amongst low frequency variants), there can be little doubt that the implementation of GWA studies has taught us a great deal, not only about diabetes itself, but about the need for rigorous standards of analysis and interpretation, and the value of international collaboration. These lessons will stand us in good stead as we move forward to tackle these new challenges.
Acknowledgement
The authors would like to thank Mandy van Hoek for her contribution in power calculations.
References
- 1.Köbberling J, Tillil H. Empirical risk figures for first degree relatives of non-insulin-dependent diabetics. In: Köbberling J, Tattersall R, editors. The Genetics of Diabetes Mellitus. Academic Press; London: 1982. pp. 201–209. [Google Scholar]
- 2.McCarthy MI. Growing evidence for diabetes susceptibility genes from genome scan data. Current Diabetes Reports. 2003;3:159–167. doi: 10.1007/s11892-003-0040-y. [DOI] [PubMed] [Google Scholar]
- 3.Guan W, Pluzhnikov A, Cox NJ, Boehnke M, for the International Type 2 Diabetes Linkage Analysis Consortium Meta-analysis of 23 Type 2 diabetes linkage studies from the international type 2 diabetes linkage analysis consortium. Human Heredity. 2008;66:35–49. doi: 10.1159/000114164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJ, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CN, Doney AS, Morris AD, Smith GD, Hattersley AT, McCarthy MI. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, Tuomi T, Gaudet D, Hudson TJ, Daly M, Groop L, Lander ES. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nature Genetics. 2000;26:76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 7.Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, McCarthy MI, Hattersley AT, Frayling TM. Large scale association studies of variants in genes encoding the pancreatic beta-cell K-ATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with increased risk of Type 2 Diabetes. Diabetes. 2003;52:568–572. doi: 10.2337/diabetes.52.2.568. [DOI] [PubMed] [Google Scholar]
- 8.Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, Daly A, Lango H, Frayling TM, Neumann RJ, Sherva R, Blech I, Pharoah PD, Palmer CN, Kimber C, Tavendale R, Morris AD, McCarthy MI, Walker M, Hitman G, Glaser B, Permutt MA, Hattersley AT, Wareham NJ, Barroso I. Common variants in WFS1 confer risk of type 2 diabetes. Nature Genetics. 2007;39:951–953. doi: 10.1038/ng2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winckler W, Weedon MN, Graham RR, McCarroll SA, Purcell S, Almgren P, Tuomi T, Gaudet D, Boström KB, Walker M, Hitman G, Hattersley AT, McCarthy MI, Ardlie KG, Hirschhorn JN, Daly MJ, Frayling TM, Groop L, Altshuler D. Evaluation of common variants in the six known MODY genes for association with Type 2 Diabetes. Diabetes. 2007;56:685–693. doi: 10.2337/db06-0202. [DOI] [PubMed] [Google Scholar]
- 10.Gudmundsson J, Sulem P, Steinthorsdottir V, Bergthorsson JT, Thorleifsson G, Manolescu A, Rafnar T, Gudbjartsson D, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, Blondal T, Stacey SN, Helgason A, Gunnarsdottir S, Olafsdottir A, Kristinsson KT, Birgisdottir B, Ghosh S, Thorlacius S, Magnusdottir D, Stefansdottir G, Kristjansson K, Bagger Y, Wilensky RL, Reilly MP, Morris AD, Kimber CH, Adeyemo A, Chen Y, Zhou J, So WY, Tong PC, Ng MC, Hansen T, Andersen G, Borch-Johnsen K, Jorgensen T, Tres A, Fuertes F, Ruiz-Echarri M, Asin L, Saez B, van Boven E, Klaver S, Swinkels DW, Aben KK, Graif T, Cashy J, Suarez BK, van Vierssen Trip O, Frigge ML, Ober C, Hofker MH, Wijmenga C, Christiansen C, Rader DJ, Palmer CN, Rotimi C, Chan JC, Pedersen O, Sigurdsson G, Benediktsson R, Jonsson E, Einarsson GV, Mayordomo JI, Catalona WJ, Kiemeney LA, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nature Genetics. 2007;39:977–983. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 11.Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K. Variant of transcription factor 7-like 2 (TFC7L2) gene confers risk of type 2 diabetes. Nature Genetics. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 12.Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 13.Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS, Wellcome Trust Case Control Consortium (WTCCC) McCarthy MI, Hattersley AT. Replication of genome-wide association signals in U.K. samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:828–830. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 17.Salonen JT, Uimari P, Aalto JM, Pirskanen M, Kaikkonen J, Todorova B, Hyppönen J, Korhonen VP, Asikainen J, Devine C, Tuomainen TP, Luedemann J, Nauck M, Kerner W, Stephens RH, New JP, Ollier WE, Gibson JM, Payton A, Horan MA, Pendleton N, Mahoney W, Meyre D, Delplanque J, Froguel P, Luzzatto O, Yakir B, Darvasi A. Type 2 diabetes whole-genome association study in four populations: the DiaGen consortium. American Journal of Human Genetics. 2007;81:338–345. doi: 10.1086/520599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S, Baker A, Snorradottir S, Bjarnason H, Ng MC, Hansen T, Bagger Y, Wilensky RL, Reilly MP, Adeyemo A, Chen Y, Zhou J, Gudnason V, Chen G, Huang H, Lashley K, Doumatey A, So WY, Ma RC, Andersen G, Borch-Johnsen K, Jorgensen T, van Vliet-Ostaptchouk JV, Hofker MH, Wijmenga C, Christiansen C, Rader DJ, Rotimi C, Gurney M, Chan JC, Pedersen O, Sigurdsson G, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nature Genetics. 2007;39:770–775. doi: 10.1038/ng2043. [DOI] [PubMed] [Google Scholar]
- 19.Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, Ng DP, Holmkvist J, Borch-Johnsen K, Jørgensen T, Sandbæk A, Lauritzen T, Hansen T, Nurbaya S, Tsunoda T, Kubo M, Babazono T, Hirose H, Hayashi M, Iwamoto Y, Kashiwagi A, Kaku K, Kawamori R, Tai ES, Pedersen O, Kamatani N, Kadowaki T, Kikkawa R, Nakamura Y, Maeda S. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nature Genetics. 2008;40:1098–1102. doi: 10.1038/ng.208. [DOI] [PubMed] [Google Scholar]
- *20.Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, Ardlie K, Boström KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P, Ding CJ, Doney AS, Duren WL, Elliott KS, Erdos MR, Frayling TM, Freathy RM, Gianniny L, Grallert H, Grarup N, Groves CJ, Guiducci C, Hansen T, Herder C, Hitman GA, Hughes TE, Isomaa B, Jackson AU, Jørgensen T, Kong A, Kubalanza K, Kuruvilla FG, Kuusisto J, Langenberg C, Lango H, Lauritzen T, Li Y, Lindgren CM, Lyssenko V, Marvelle AF, Meisinger C, Midthjell K, Mohlke KL, Morken MA, Morris AD, Narisu N, Nilsson P, Owen KR, Palmer CN, Payne F, Perry JR, Pettersen E, Platou C, Prokopenko I, Qi L, Qin L, Rayner NW, Rees M, Roix JJ, Sandbaek A, Shields B, Sjögren M, Steinthorsdottir V, Stringham HM, Swift AJ, Thorleifsson G, Thorsteinsdottir U, Timpson NJ, Tuomi T, Tuomilehto J, Walker M, Watanabe RM, Weedon MN, Willer CJ, Wellcome Trust Case Control Consortium. Illig T, Hveem K, Hu FB, Laakso M, Stefansson K, Pedersen O, Wareham NJ, Barroso I, Hattersley AT, Collins FS, Groop L, McCarthy MI, Boehnke M, Altshuler D. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nature Genetics. 2008;40:638–645. doi: 10.1038/ng.120. *Meta-analysis of three type 2 diabetes genome-wide association scans.
- 21.Prokopenko I, Langenberg C, Florez JC, Saxena R, Soranzo N, Thorleifsson G, Loos RJF, Manning AK, Jackson AU, Aulchenko Y, Potter SC, Erdos MR, Sanna S, Hottenga J-J, Wheeler E, Kaakinen M, Lyssenko V, Chen W-M, Ahmadi K, Beckmann JS, Bergman RN, Bochud M, Bonnycastle LL, Buchanan TA, Cao A, Cervino A, Coin L, Collins FS, Crisponi L, De Geus EJC, Dehghan A, Deloukas P, Doney ASF, Elliott P, Freimer N, Gateva V, Herder C, Hofman A, Hughes TE, Hunt S, Illig T, Inouye M, Isomaa B, Johnson T, Kong A, Krestyaninova M, Kuusisto J, Laakso M, Lim N, Lindblad U, Lindgren CM, McCann OT, Mohlke KL, Morris AD, Naitza S, Orru M, Palmer CNA, Pouta A, Randall J, Rathmann W, Saramies J, Scheet P, Scott LJ, Scuteri A, Sharp S, Sijbrands E, Smit JH, Song K, Steinthorsdottir V, Stringham HM, Tuomi T, Tuomilehto J, Uitterlinden AG, Voight BF, Waterworth D, Wichmann H-E, Willemsen G, Witteman JCM, Yuan X, Zao JH, Zeggini E, Schlessinger D, Sandhu M, Boomsma D, Uda M, Spector TD, Penninx BWJH, Altshuler D, Vollenweider P, Jarvelin M-R, Lakatta E, Waeber G, Fox CS, Peltonen L, Groop LC, Mooser V, Cupples LA, Thorsteinsdottir U, Boehnke M, Barroso I, van Duijn C, Dupuis J, Watanabe RM, Stefansson K, McCarthy MI, Wareham NJ, Meigs JB, Abecasis GR. Variants in the melatonin receptor 1B gene (MTNR1B) influence fasting glucose levels. Nature Genetics. doi: 10.1038/ng.290. (Epub Published Dec 7, 2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lyssenko V, Nagorny CLF, Erdos MR, Wierup N, Jonsson A, Spegel P, Buglaini M, Saxena R, Fex M, Pulizzi N, Isomaa B, Tuomi T, Nilsson P, Kussisto J, Tuomilehto J, Boehnke M, Altshuler D, Sundler F, Eriksson JG, Jackson AU, Laakso M, Marchetti P, Watanabe RM, Mulder H, Groop L. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nature Genetics. doi: 10.1038/ng.288. (Epub ahead of print 7th December 2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, Sparso T, Holmkvist J, Marchand M, Deplplanque J, Lobbens S, Rocheleau G, Durand E, DeGraeve F, Chevre J-C, Borch-Johnsen K, Hartikainen A-L, Ruokonen A, Tichet J, Marre M, Weill J, Heude B, Tauber M, Lemaire K, Schuit F, Elliott P, Jorgensen T, Charpentier G, Hadjadj S, Cauchi S, Vaxillaire M, Sladek R, Visvikis-Siest S, Balkau B, Levy-Marchal C, Pattou F, Meyre D, Blakemore AIF, Jarvelin M-R, Walley AJ, Hansen T, Dina C, Pedersen O, Froguel P. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nature Genetics. doi: 10.1038/ng.277. (Epub ahead of print 7th December 2008) [DOI] [PubMed] [Google Scholar]
- 24.Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, Hirota Y, Mori H, Jonsson A, Sato Y, Yamagata K, Hinokio Y, Wang HY, Tanahashi T, Nakamura N, Oka Y, Iwasaki N, Iwamoto Y, Yamada Y, Seino Y, Maegawa H, Kashiwagi A, Takeda J, Maeda E, Shin HD, Cho YM, Park KS, Lee HK, Ng MC, Ma RC, So WY, Chan JC, Lyssenko V, Tuomi T, Nilsson P, Groop L, Kamatani N, Sekine A, Nakamura Y, Yamamoto K, Yoshida T, Tokunaga K, Itakura M, Makino H, Nanjo K, Kadowaki T, Kasuga M. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nature Genetics. 2008;40:1092–1097. doi: 10.1038/ng.207. [DOI] [PubMed] [Google Scholar]
- 25.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nature Genetics. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 26.Fan JB, Chee MS, Gunderson KL. Highly parallel genomic assays. Nature Reviews Genetics. 2006;7:632–644. doi: 10.1038/nrg1901. [DOI] [PubMed] [Google Scholar]
- 27.Zeggini E, Rayner W, Morris AP, Hattersley AT, Walker M, Hitman GA, Deloukas P, Cardon LR, McCarthy MI. HapMap sample size and tagging SNP performance: an evaluation in large-scale empirical and simulated data sets. Nature Genetics. 2005;37:1320–1322. doi: 10.1038/ng1670. [DOI] [PubMed] [Google Scholar]
- 28.McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, Duerr RH, Silverberg MS, Taylor KD, Rioux JD, Altshuler D, Daly MJ, Xavier RJ. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nature Genetics. 2008 Aug 24; doi: 10.1038/ng.215. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007;8:657–662. doi: 10.1038/nrg2178. [DOI] [PubMed] [Google Scholar]
- *30.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome wide association studies for complex traits: consensus, uncertainty and challenges. Nature Reviews Genetics. 2008;9:356–369. doi: 10.1038/nrg2344. *An in-depth review of genome-wide scans for complex disease.
- 31.Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nature Genetics. 2003;33:177–182. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- 32.Dabelea D, Pettitt DJ. Intrauterine diabetic environment confers risks for type 2 diabetes mellitus and obesity in the offspring, in addition to genetic susceptibility. Journal of Pediatric Endocrinology and Metabolism. 2001;14:1085–1091. doi: 10.1515/jpem-2001-0803. [DOI] [PubMed] [Google Scholar]
- 33.McCarthy MI. Casting a wider net for diabetes-susceptibility genes. Nature Genetics. 2008;40:1039–1040. doi: 10.1038/ng0908-1039. [DOI] [PubMed] [Google Scholar]
- 34.McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, Elliott AL, Parkin M, Hubbell E, Webster T, Mei R, Veitch J, Collins PJ, Handsaker R, Lincoln S, Nizzari M, Blume J, Jones KW, Rava R, Daly MJ, Gabriel SB, Altshuler D. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nature Genetics. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 35.Hill WG, Goddard ME, Visscher PM. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 2008;4:e1000008. doi: 10.1371/journal.pgen.1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smyth DJ, Cooper JD, Howson JM, Walker NM, Plagnol V, Stevens H, Clayton DG, Todd JA. PTPN22 Trp620 explains the association of chromosome 1p13 with type 1 diabetes and shows a statistical interaction with HLA class II genotypes. Diabetes. 2008;57:1730–1737. doi: 10.2337/db07-1131. [DOI] [PubMed] [Google Scholar]
- 37.Grarup N, Rose CS, Andersson EA, Andersen G, Nielsen AL, Albrechtsen A, Clausen JO, Rasmussen SS, Jørgensen T, Sandbaek A, Lauritzen T, Schmitz O, Hansen T, Pedersen O. Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects. Diabetes. 2007;56:3105–3111. doi: 10.2337/db07-0856. [DOI] [PubMed] [Google Scholar]
- 38.Pascoe L, Tura A, Patel SK, Ibrahim IM, Ferrannini E, Zeggini E, Weedon MN, Mari A, Hattersley AT, McCarthy MI, Frayling TM, Walker M, RISC Consortium. U.K. Type 2 Diabetes Genetics Consortium Common variants of the novel type 2 diabetes genes, CDKAL1 and HHEX/IDE, are associated with decreased pancreatic beta-cell function. Diabetes. 2007;56:3101–3104. doi: 10.2337/db07-0634. [DOI] [PubMed] [Google Scholar]
- 39.Staiger H, Machicao F, Stefan N, Tschritter O, Thamer C, Kantartzis K, Schäfer SA, Kirchhoff K, Fritsche A, Häring HU. Polymorphisms within novel risk loci for type 2 diabetes determine β-cell function. PLoS One. 2007;9:e832. doi: 10.1371/journal.pone.0000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Staiger H, Stancáková A, Zilinskaite J, Vänttinen M, Hansen T, Marini MA, Hammarstedt A, Jansson PA, Sesti G, Smith U, Pedersen O, Laakso M, Stefan N, Fritsche A, Häring HU. A candidate type 2 diabetes polymorphism near the HHEX locus affects acute glucose-stimulated insulin release in European populations. Diabetes. 2008;57:514–517. doi: 10.2337/db07-1254. [DOI] [PubMed] [Google Scholar]
- 41.Freathy RM, Timpson NJ, Lawlor DA, Pouta A, Ben-Shlomo Y, Ruokonen A, Ebrahim S, Shields B, Zeggini E, Weedon MN, Lindgren CM, Lango H, Melzer D, Ferrucci L, Paolisso G, Neville MJ, Karpe F, Palmer CN, Morris AD, Elliott P, Jarvelin MR, Smith GD, McCarthy MI, Hattersley AT, Frayling TM. Common variation in the FTO gene alters diabetes-related metabolic traits to the extent expected, given its effect on BMI. Diabetes. 2008;57:1419–1426. doi: 10.2337/db07-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leahy JL. Mary, Mary, quite contrary, how do your beta-cells fail? Diabetes. 2008;57:2563–2564. doi: 10.2337/db08-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lyssenko V, Lupi R, Marchetti P, Del Guerra S, Orho-Melander M, Almgren P, Sjögren M, Ling C, Eriksson KF, Lethagen AL, Mancarella R, Berglund G, Tuomi T, Nilsson P, Del Prato S, Groop L. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. The Journal of Clinical Investigation. 2007;117:2155–2163. doi: 10.1172/JCI30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chimienti F, Devergnas S, Pattou F, Schuit F, Garcia-Cuenca R, Vandewalle B, Kerr-Conte J, Van Lommel L, Grunwald D, Favier A, Seve M. In vivo expression and functional characterization of the zinc transporter ZnT8 in glucose-induced insulin secretion. Journal of Cell Science. 2006;119:4199–4206. doi: 10.1242/jcs.03164. [DOI] [PubMed] [Google Scholar]
- 45.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 46.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, NIDDK IBD Genetics Consortium. Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Belgian-French IBD Consortium. Wellcome Trust Case Control Consortium. Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nature Genetics. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z, Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cussenot O, Valeri A, Andriole GL, Crawford ED, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover R, Hayes RB, Hunter DJ, Chanock SJ. Multiple loci identified in a genome-wide association study of prostate cancer. Nature Genetics. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 48.Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–776. doi: 10.1038/nature05985. [DOI] [PubMed] [Google Scholar]
- 49.Kasper JS, Giovannucci E. A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiology, Biomarkers and Prevention. 2006;15:2056–2062. doi: 10.1158/1055-9965.EPI-06-0410. [DOI] [PubMed] [Google Scholar]
- 50.Lango H, UK Type 2 Diabetes Genetics Consortium. Palmer CN, Morris AD, Zeggini E, Hattersley AT, McCarthy MI, Frayling TM, Weedon MN. Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes. 2008;57:3129–3135. doi: 10.2337/db08-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JP, Sumnik Z, van Rhijn A, Wales JK, Clark P, Gorman S, Aisenberg J, Ellard S, Njølstad PR, Ashcroft FM, Hattersley AT. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. New England Journal of Medicine. 2004;350:1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 52.Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, Larkin B, Ashcroft FM, Klimes I, Codner E, Iotova V, Slingerland AS, Shield J, Robert JJ, Holst JJ, Clark PM, Ellard S, Søvik O, Polak M, Hattersley AT, Neonatal Diabetes International Collaborative Group Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. New England Journal of Medicine. 2006;355:467–477. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- 53.Pearson ER, Donnelly LA, Kimber C, Whitley A, Doney AS, McCarthy MI, Hattersley AT, Morris AD, Palmer CN. Variation in TCF7L2 influences therapeutic response to sulfonylureas: A GoDARTs study. Diabetes. 2007;56:2178–2182. doi: 10.2337/db07-0440. [DOI] [PubMed] [Google Scholar]