

Abstract
The mitogen-activated protein kinase (MAPK) pathway allows cells to interpret external signals and respond in an appropriate way. Diverse cellular functions, ranging from differentiation and proliferation to migration and inflammation, are regulated by MAPK signalling. Therefore, cells have developed mechanisms by which this single pathway modulates numerous cellular responses from a wide range of activating factors. This specificity is achieved by several mechanisms, including temporal and spatial control of MAPK signalling components. Key to this control are protein scaffolds, which are multidomain proteins that interact with components of the MAPK cascade in order to assemble signalling complexes. Studies conducted on different scaffolds, in different biological systems, have shown that scaffolds exert substantial control over MAPK signalling, influencing the signal intensity, time course and, importantly, the cellular responses. Protein scaffolds, therefore, are integral elements in the modulation of the MAPK network in fundamental physiological processes.
Keywords: MAPK, Scaffolds, Cancer, Compartmentalisation
1 Introduction
The mitogen activated protein kinase (MAPK) pathway is an intracellular signalling cascade, activated by diverse external cues, that regulates many cellular functions including cell proliferation and differentiation [1]. Several growth factors, such as epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), insulin, neurotrophins, and inflammatory cytokines, activate MAPKs. Cells are stimulated by these factors and respond to changes in their environment through manipulation of MAPK signalling. Five distinct groups of MAPKs have been identified in mammals. These are MAPK/ERK kinase/extracellular regulated kinase (MEK/ERK), c-Jun N-terminal kinase (JNK), p38, ERK5 and ERK3. The MAPKs can control events both in the nucleus, such as gene regulation, and extra-nuclear events, such as cytoskeletal reorganisation, through phosphorylation and activation of targets in the cytosol and nucleus. Several excellent publications have addressed general MAPK functions [1-3]. In this review, we focus on the role of protein scaffolds in the modulation of MAPK signalling.
1.1 The MEK/ERK cascade
The best characterised of the MAPK pathways is the MEK/ERK cascade. This pathway is activated by protein tyrosine kinase receptors, such as EGF receptor (EGFR) or VEGF receptor (VEGFR) [1]. Briefly, when growth factors bind to their cognate receptors, the receptors undergo dimerisation, inducing phosphorylation by intrinsic tyrosine kinases. Proteins that contain SH2 (Src homology 2) domains are recruited to the receptors and bind to specific phosphotyrosine residues. One of these SH2-containing proteins, Grb2, is constitutively bound to the Ras activator Sos, and normally localises to the cytosol. Relocation to the membrane activates Sos, which in turn activates Ras. Ras is a GTPase, which hydrolyses guanosine triphosphate (GTP) to guanosine diphosphate (GDP). Ras is active in the GTP-bound state. GTP-Ras activates downstream effectors (see below), which propagate signalling. Regulation of the GTP/GDP bound state of Ras provides tight control over its activity. This control is provided by two different classes of proteins, GTPase activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs). GAPs reduce the pool of GTP bound Ras by increasing the intrinsic GTPase activity of Ras (and therefore the rate of GTP hydrolysis). Consequently, Ras GAPs act as “off” switches and reduce Ras activity. In contrast to GAPs, GEFs facilitate the exchange of GDP for GTP, increasing the pool of GTP-bound Ras. Consequently, Ras GEFs increase Ras activity. There are three Ras isoforms in mammals, namely H-Ras, K-Ras and N-Ras. At their C-terminal ends, Ras proteins contain a CAAX motif, in which a cysteine (C) is followed by two aliphatic amino acids (A) and any other amino acid (X). This motif is farnesylated by farnesyl transferase, causing Ras to be localised to both the plasma membrane and internal membranes [4].
Ras-GTP recruits Raf kinase to the membrane. There are three known isoforms of Raf, namely A-Raf, B-Raf and C-Raf (also termed Raf-1), each having distinct functions [5]. When localised to the plasma membrane, the Raf kinases become active and catalyse the phosphorylation and activation of MEK1 and MEK2, which in turn activate ERK1 and ERK2. Once active, ERKs dimerise and either translocate to the nucleus, where they phosphorylate transcription factors, or remain in the cytosol where they phosphorylate substrates in multiple cellular compartments (reviewed in [3]). The predominant sequelae of MEK/ERK signalling are proliferation and differentiation.
1.2 JNK Pathway
The c-Jun N-terminal kinase (JNK) family contains three ubiquitously expressed members, JNK1, JNK2 and JNK3 [3]. The major activators of the JNK pathway are cytokines, selected G-protein coupled receptors (GPCR) and cell stress, such as inhibition of DNA or protein synthesis. JNK is phosphorylated by either MEK4 or MEK7, which are themselves phosphorylated by several kinases, including MEKK1-4, MLK2/3 and DLK. Following activation, JNK is translocated to the nucleus where it phosphorylates and activates several transcription factors, including c-Jun, ATF-2, STAT3 and HSF-1 [3]. JNKs control apoptosis and the development of multiple cell types in the immune system.
1.3 p38 Pathway
There are four members of the p38 kinase family, namely α,β,γ and δ. Cytokines, hormones, G-protein coupled receptors and cell stress, for example, heat or osmotic shock, all stimulate these enzymes [1]. p38 kinases, which are targets of both MEK3 and MEK6, have numerous substrates, including MAPK interacting kinases (Mnk) 1 and Mnk 2, and eukaryotic initiation factor 4e (eIF4e). p38 regulates angiogenesis, cell proliferation, inflammation and cytokine production.
2 Protein Scaffolds
Cells have developed a class of proteins, termed scaffolds or adaptor proteins, which confer spatial and temporal regulation of the MAPK pathway. Protein scaffolds bind to multiple components of the MAPK cascade, bringing them into close proximity and thereby facilitating efficient propagation of the signal (Figure 1). Consequently, scaffolds act as signal modules, providing an intricate level of control over MAPK signalling. Originally identified in yeast [6], several scaffolds that modulate MAPK activity in mammalian cells have been recognised [7-9]. While scaffolds have been described in all of the MAPK pathways, in this review we will concentrate on scaffolds that modulate the MEK/ERK pathway.
Figure 1. Model of a molecular scaffold for the MEK/ERK pathway.
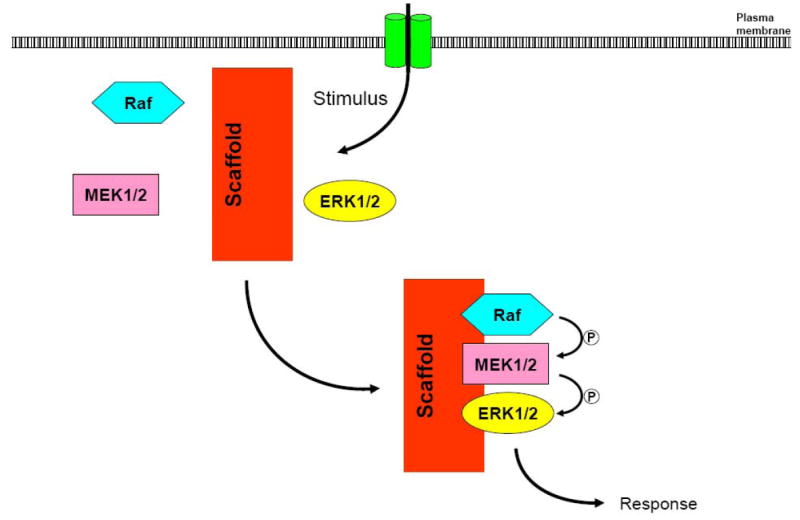
Molecular scaffolds bind to several components of the MAPK cascade, assembling them in a multiprotein complex. The close proximity of the kinases facilitates propagation of the signal.
2.1 Kinase suppressor of Ras
Kinase suppressor of Ras (KSR) was originally isolated from a genetic screen as a positive regulator of MAPK signalling [10]. Although not present in yeast, KSR homologues have been identified in all multicellular organisms examined, including nematodes, Drosophila, C. elegans and mammals [11]. The biological function of KSR remained elusive for some time. Due to a high degree of sequence similarity to C-Raf, KSR was initially thought to be an enzyme, but kinase activity has not been unequivocally demonstrated [11]. Subsequent investigation led to the realisation that KSR is a MAPK scaffold.
KSR is one of the best characterised scaffolds in the MAPK pathway, and binds to C-Raf, MEK1/2 and ERK1/2 [11]. More recent evidence reveals that KSR also binds B-Raf [12], but the physiological relevance of this interaction has not been established. Other proteins known to interact with KSR include 14-3-3, G protein-βγ, heat shock proteins 70 and 90, cdc37 and C-TAK1 [11]. Interestingly, MEK is constitutively associated to KSR, while ERK binds only in response to a stimulus. As is typical for scaffolds, optimal expression levels of KSR are required for maximal responses of MAPK to signalling cues [13].
Loss-of-function analysis has provided some of the best evidence for the in vivo function of KSR in MAPK signalling. There are two ksr genes in C.elegans and between them they are required for most Ras-dependant signalling during development [14]. Although KSR knockout mice are developmentally normal, they have defects in antigen-triggered T cell proliferation [15], and are resistant to antibody-induced arthritis [16]. Furthermore, mouse embryonic fibroblasts from KSR-knockout mice have defective activation of ERK by TNF-α and interleukin-1 [16]. Together, these studies strongly suggest that KSR fulfils an important role in the regulation of MAPK signalling during the immune response and inflammation. Interestingly, KSR null mice are less susceptible to Ras-mediated skin cancer [17], identifying a role for KSR in the regulation of MAPK-mediated cell proliferation.
2.2 IQGAP1
IQGAP1 is a large, widely expressed protein that regulates many signalling pathways and cellular functions (reviewed in [18-20]). With several domains, IQGAP1 is able to bind to a broad spectrum of proteins [18], thereby modulating actin dynamics, microtubule dynamics, cell-cell adhesion and transcriptional regulation. Recent studies have provided strong evidence that IQGAP1 is a scaffold for the MAPK cascade. IQGAP1 binds directly to B-Raf, MEK1, MEK2, ERK1 and ERK2, and regulates their activation in response to EGF [21-23] and CD44 [24]. In addition, fear-conditioning in mice increases the association of IQGAP1 with active ERK2 [25]. Analogous to KSR, both an increase and a decrease in IQGAP1 expression level attenuate EGF-dependant activation of MEK and ERK, suggesting that the correct stoichiometry of IQGAP1 to MAPK components is required for efficient propagation of the cascade [21, 22]. Interestingly, while ERK associates constitutively with IQGAP1 and the binding is not sensitive to EGF, the interaction between IQGAP1 and MEK1 increases, while that with MEK2 decreases, following EGF treatment [22]. This raises the possibility that IQGAP1 preferentially activates the MEK1 signalling pathway. It has been suggested that MEK1 promotes proliferation, while MEK2 promotes differentiation [26], and IQGAP1 may therefore regulate the cellular response to MAPK signalling.
More recent findings reveal that IQGAP1 modulates B-Raf kinase activity [23]. IQGAP1 binds directly to B-Raf and B-Raf isolated from cells in a complex with IQGAP1 has significantly higher kinase than the activity in the absence of IQGAP1 [23]. It is not known whether the interaction with IQGAP1 enhances activation of B-Raf by Ras, or whether IQGAP1 preferentially associates with B-Raf that has already been activated. Importantly, IQGAP1 modulates B-Raf function. For example, knockout of IQGAP1 from cells renders B-Raf insensitive to EGF stimulation [23], suggesting that IQGAP1 acts “upstream” of B-Raf and is necessary for activation of B-Raf by growth factors. Collectively, these findings highlight the importance of IQGAP1 in the B-Raf/MEK/ERK signalling cascade.
Following stimulation with growth factors, the MAPK pathway regulates many cellular responses, one of which is modulation of the cytoskeleton [27]. IQGAP1 is a well-documented regulator of the actin and microtubule cytoskeletons [28, 29], and it is tempting to speculate that IQGAP1 links MAPK signalling to cytoskeletal dynamics. In support of this idea, both IQGAP1 and ERK2 localise to microtubule-associated protein-2 in neuronal cells [30, 31]. In addition, IQGAP1 forms a complex with CD44, Cdc42 and actin in response to hyaluronan, and is also required for hyaluronan-dependant activation of ERK and Elk [24].
2.3 MEK Partner-1
MEK partner-1 (MP-1), a widely expressed scaffold for the MEK/ERK pathway, was originally identified from a yeast-2-hybrid using MEK as bait [32]. In vitro, MP1 promotes the association of MEK with ERK, thereby enhancing phosphorylation of ERK by MEK [32]. In order to augment MAPK signalling in cells, MP1 requires the presence of its interacting protein, p14, which localises MP1 to endosomes [33]. Both MP1 and p14 are essential for EGF-dependant activation of ERK [33].
2.4 β-arrestins
β-arrestins are well-known regulators of GPCRs. Following activation of the receptor, β-arrestin binds to phosphorylated motifs, causing dissociation of the heterotrimeric G-protein. This targets the GPCR to clathrin-coated pits (reviewed in [34]). The first evidence that β-arrestin regulates MAPK signalling came from reports showing that active Src is recruited to β2-adrenergic receptors through a direct interaction with β-arrestin [35]. Here, Src phosphorylates the adaptor protein Shc, resulting in activation of Grb2 and consequently activation of the MAPK cascade. However, β-arrestin also directly regulates the MEK/ERK cascade. In response to activation of protease activated receptor (PAR), β-arrestin recruits Raf, MEK and ERK to the receptor, enhancing activation of ERK [36]. These complexes accompany the receptor to early endosomes to promote efficient MAPK signalling [37]. Importantly, β-arrestin prevents the translocation of active ERK to the nucleus, restricting ERK to cytosolic substrates [36] (discussed in more detail in section 3.2, below).
2.5 Similar Expression to FGF
Similar expression to FGF (Sef) was originally identified as an inhibitor of fibroblast growth factor (FGF)-dependant MEK/ERK signalling in zebrafish [38]. In agreement with this report, overexpression of Sef inhibits both FGF- and NGF-induced differentiation of PC12 cells [39]. These observations appear to be due to Sef capturing active MEK/ERK complexes at the Golgi and inhibiting disassociation of the MEK/ERK complex [40]. Consequently, Sef inhibits nuclear localisation of ERK and restricts ERK to cytosolic substrates [40].
More recently, the subcellular localisation of Sef has been shown to be regulated by phosphorylation of a tyrosine residue at position 330 [41]. Wild type Sef localises to intracellular membranes and vesicles while a Y330F mutant localises to the plasma membrane [41]. Furthermore, SefY330F exhibits greater inhibition of FGF than wildtype Sef. While the kinase that catalyses the phosphorylation has not been identified, these data imply that localisation of Sef is important for its inhibitory activity.
Although an inhibitor of FGF signalling, Sef colocalises and immunoprecipitates with EGF receptor [42]. Interestingly, unlike its role in FGF signalling, Sef enhances the duration of EGF-dependant ERK activation [42]. It is therefore possible that Sef regulates MAPK signalling from factors other than EGF and FGF.
3. Protein scaffolds and MAPK specificity
Despite progress in our comprehension of MAPK signalling, one question that remains poorly understood is how a particular stimulus elicits the correct response. This topic, termed MAPK specificity, seems remarkable when one considers the diverse range of cellular responses induced by many different activators, all of which signal through the MEK/ERK pathway (Figure 2). Spatial and temporal changes to MAPK signalling influence the cellular response to a specific stimulus and are of particular interest when considering MAPK specificity. Protein scaffolds provide one mechanism by which spatiotemporal MAPK signalling is controlled (see below). However, there are additional means by which scaffolds are able to control aspects of MAPK signalling. It has been proposed that scaffolds can provide both positive and negative regulatory mechanisms [43]. By assembling individual components of the MAPK cascade, scaffolds facilitate their interactions and propagation of the signal. However, the assembly of the multiprotein complexes also sequesters these same components away from other signalling pathways. Consequently, protein scaffolds preferentially activate specific cascades, while concomitantly inhibiting others [43].
Figure 2. MAPK specificity.
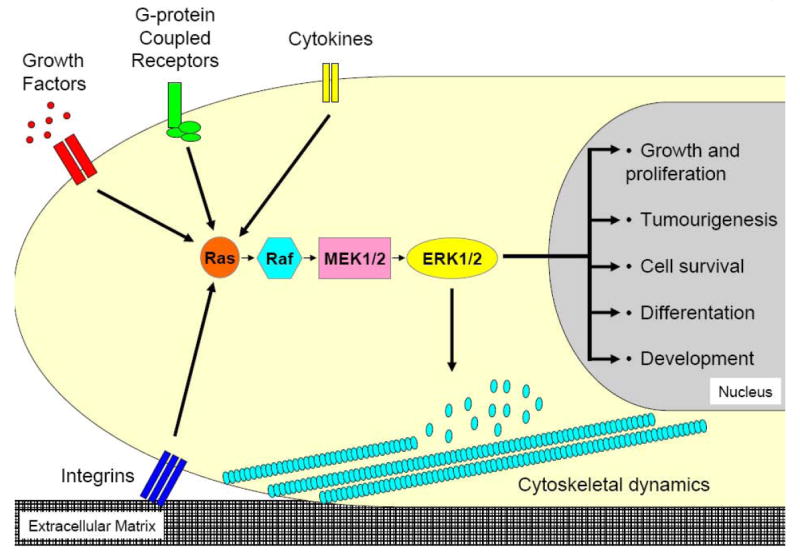
Diverse extracellular inputs stimulate the Ras/Raf/MEK/ERK cascade and each can elicit a distinct cellular outcome.
Another aspect of signalling that scaffolds may bring to the MAPK cascade is that of crosstalk. MAPK scaffolds that interact with, or are regulated by, different signalling pathways, may enable these cascades to impinge on each other. An example of this can be seen with IQGAP1. As described above, IQGAP1 binds to B-Raf, MEK and ERK and promotes EGF-dependant MAPK signalling. In addition, IQGAP1 regulates Rac1 and Cdc42 activity, and is a well-defined modulator of the actin cytoskeleton [18]. Growth factors influence cytoskeletal function by activating MAPK signalling [27], and it is tempting to speculate that IQGAP1 links MAPK signalling to cytoskeletal dynamics. In support of this concept, both IQGAP1 and ERK2 localise to microtubule-associated protein-2 in neuronal cells [30, 31]. Another example can be found with IQGAP1 and B-Raf. Calcium alone enhances the binding of IQGAP1 to B-Raf in vitro, while calcium/calmodulin abrogates this interaction. Interestingly, chelation of intracellular calcium inhibits EGF-dependant B-Raf activation in IQGAP1 knockout cells, but does not affect B-Raf activity when IQGAP1 is present [44]. Consequently, IQGAP1 is required, at least in part, for calcium-dependant modulation of B-Raf activation. It appears, therefore, that IQGAP1 integrates calcium and MAPK signalling.
3.1 Compartmentalised MAPK signalling
The kinase cascade initiated by Ras originates from transmembrane receptors that are activated by external cues. Consequently, it was initially believed that Ras was active only at the plasma membrane and that the MAPK pathway propagated exclusively from the plasma membrane to the nucleus. Subsequent analysis has shown that the signalling mechanism is more sophisticated. It is now recognised that MAPK signalling also propagates from discrete intracellular domains and organelles. This compartmentalisation provides an additional level of control over the MAPK pathway and enables cells to distinguish MAPK signalling from different sources [4]. Protein scaffolds have a fundamental role in this spatial regulation of MAPK. The combination of extracellular signals, compartmentalisation and molecular scaffolds creates an intricate system utilised by cells to regulate a fundamental signalling pathway.
As mentioned above, MAPK signalling frequently originates from the plasma membrane, where transmembrane receptors are activated by factors from the extracellular environment. The signal is transmitted via adaptors proteins and GEFs to Ras. Recent work has identified nanoclusters of active Ras on the plasma membrane, which are important for efficient signalling and also modulation of the MAPK pathway in response to signalling factors [45]. Consequently, the plasma membrane is considered to be vital for both spatial and temporal regulation of MAPK signalling. More detailed analyses have identified the role of specific plasma membrane compartments in MAPK signalling. For example, many signalling receptors, such as EGFR [46], VEGFR2 [47], p75NTR and TrkA, are enriched in caveolae [48]. Interestingly, EGF-dependant ERK phosphorylation is independent of caveolae [49], raising the concept that these membrane compartments provide some degree of signal specification, regulating MAPK activation in response to specific stimuli.
Following ligand binding, receptors such as those for growth factors, are internalised into endosomes, in a process that was originally considered to be exclusively for recycling. However, more recent studies have demonstrated that endosomes also act as signalling platforms, allowing propagation of MAPK cascades [50]. Many MAPK signalling components, including Shc, Grb2, Sos and Ras, have been found on endosomes containing EGFR [51]. Furthermore, EGF does not disassociate from the EGFR following internalisation [52], while tyrosine phosphorylated EGFR colocalises with GTP-Ras in endosomes [53, 54]. Consequently, activation of the MAPK pathway can occur from endosomes, as well as from membrane-associated receptors [55].
Ras proteins localise to both the Golgi and endoplasmic reticulum (ER) [56]. Interestingly, while growth factor-dependant activation of H-Ras at both the plasma membrane and the ER is rapid [57], occurring within 1 minute and reversed after 20-40 minutes, H-Ras activation at the Golgi is delayed, taking 10 minutes, and persisting for 60 minutes [57]. The physiological role of MAPK signalling from the ER or the Golgi has been difficult to elucidate. Nevertheless, some differences in signalling between these two compartments have been demonstrated. When tethered to the Golgi, constitutively active Ras, RasQ61L, is a strong activator of ERK and the Akt protein kinase (also known as protein kinase B), but weakly activates JNK. Conversely, when tethered to the ER, Ras61L strongly activates JNK, while only weakly activating ERK and Akt [57]. In Jurkat T cells, low-grade stimulation of the T cell receptor upregulates the Ras GEF, Ras GRP1, and results in the restriction of active N-Ras to the Golgi [58]. The highly restricted activation suggests N-Ras signalling from the Golgi plays an important role in T cell activation. Finally, an ER-confined Ras effector, ER-associated Ras inhibitor protein 1 (ERI1), was discovered in Saccharomyces cerevisae [59]. ERI1 binds to and inhibits GTP-Ras2p at the ER.
3.2 Scaffold regulation of MAPK compartmentalisation
Many of the protein scaffolds described above have been shown to influence the compartmentalised signalling of MAPK. For example, KSR seems to provide a docking platform at the plasma membrane onto which C-Raf, MEK1/2 and ERK1/2 can form a complex, and allow efficient propagation of the signalling cascade (Figure 3). In support of this notion is the finding that in quiescent cells, KSR is maintained in the cytosol through an interaction with 14-3-3 [60], and in a Triton-insoluble fraction through an interaction with ‘impedes mitogenic signal propagation’ (IMP) [61]. Following stimulation by growth factors, KSR translocates to the plasma membrane, where it facilitates activation of MEK and ERK [60]. Therefore, KSR is able to regulate the spatial activation of MEK/ERK and presumably the cellular response. This concept is bolstered by the observation that overexpression in PC12 cells of B-KSR, a neuronal-specific isoform of KSR, switches EGF signalling from a proliferative signal to a differentiation signal [62].
Figure 3. Spatial regulation of MEK/ERK signalling by protein scaffolds.
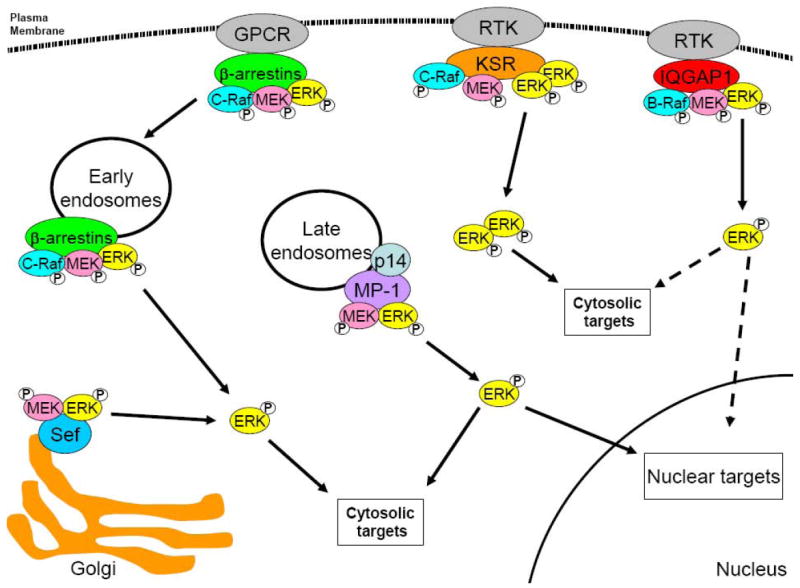
Molecular scaffolds regulate the subcellular distribution of components of the MAPK pathway. Both IQGAP1 and KSR bind to Raf, MEK and ERK and are thought to localise the kinases to the plasma membrane, while MP-1 (through it interaction with p14) localises active MEK and ERK to endosomes. Following activation, ERK is free to phosphorylate cytosolic targets, or translocate to the nucleus and activate nuclear targets. KSR binds to ERK dimers and restricts them to cytosolic targets. Like MP-1, β-arrestins localise Raf, MEK and ERK to endosomes. However, once activated ERK is prevented from translocating to the nucleus and is restricted to cytosolic targets by an unknown mechanism. Sef restricts MEK and ERK to the Golgi and, like β-arrestin, prevents translocation of ERK to the nucleus, limiting it to cytosolic substrates. GPCR, G-protein coupled receptor; RTK, receptor tyrosine kinase.
Phosphorylation of ERK induces dimerisation and this process has been proposed to effect ERK activity [63]. Recent evidence reveals that KSR1 is required for ERK dimerisation [64]. Following EGF activation, KSR1 acts as a platform for the formation of ERK dimers [64] (Figure 3). Importantly, these dimers specifically phosphorylate cytosolic substrates, while ERK monomers, which are not bound to KSR1, are translocated into the nucleus to catalyse phosphorylation of nuclear substrates [64] (Figure 3). Consequently, by regulating dimerisation of ERK, KSR1 can control whether cytosolic or nuclear substrates are activated by MAPKs.
Although no published report has specifically studied the role of IQGAP1 in compartmentalised MAPK signalling, some evidence suggests IQGAP1 assembles the Raf-MEK-ERK complex at the EGFR. In quiescent cells, IQGAP1 is preferentially localised to the cell periphery [65, 66]. Consistent with these findings, IQGAP1 is localised to leading edge of migrating cells [66, 67]. FGF promotes the translocation of IQGAP1 to membrane ruffles of migrating cells [68]. Perhaps most importantly, a mass spectroscopy-based proteomic screen for interactors with activated EGFR in HeLa cells identified IQGAP1 as one of 28 proteins in an EGF-sensitive complex [69]. Moreover, stimulation of MAPK signalling in hippocampal neurones with N-methyl-D-aspartic acid promotes accumulation of IQGAP1 in dendritic spines [25]. Additional studies are required to specifically address whether IQGAP1 provides spatial regulation of MAPK signalling.
β-arrestin prevents the translocation of ERK into the nucleus (Figure 3), thereby reducing phosphorylation of nuclear substrates and consequently MAPK-dependant gene expression [37]. Instead, active ERK is maintained in the cytosol, presumably promoting phosphorylation of cytosolic targets. Like β-arrestin, Sef also prevents translocation of ERK to the nucleus, thereby restricting signalling to cytosolic substrates [40]. Sef captures active MEK/ERK complexes at the Golgi (Figure 3), promoting ERK activation [40]. Because of the distinct roles of MAPK signalling from the Golgi, Sef is likely to influence the cellular response to MAPK activation. Indeed, expression of Sef in PC12 cells prevents FGF and NGF-dependant differentiation [39]. Finally, through an interaction with p14, MP-1 is localised to endosomes (Figure 3). This endosome localisation is crucial for MP-1 activity, as mislocalisation abrogates the ability of MP-1 overexpression to enhance ERK activation [33].
3.3. Temporal regulation of MAPK signalling
Temporal modulation of MAPK signalling elicits distinct cellular responses [2]. The classic example of this mechanism can be found in the neuronal cell line PC12 (pheochromocytoma). PC12 cells proliferate in response to EGF, while nerve growth factor (NGF) induces differentiation [70]. Although very different, both sequelae are mediated via the MEK/ERK pathway [71]. Further investigation has shown that EGF-dependant MAPK signalling is transient, lasting minutes, while NGF-dependant signalling is sustained, continuing for hours [70]. Through stimulation of the TrkA receptor, NGF activates the MAPK cascade by two mechanisms, the Grb2/SOS pathway and the Rap1 pathway [72]. Rap1, a member of the Ras family, modulates integrin-mediated cell adhesion and cadherin-mediated cell junction formation by regulating several effectors, including B-Raf [73]. In response to NGF, Grb2/SOS produces a transient signal due to a negative feedback mechanism, while Rap1-dependant signalling produces sustained activation of B-Raf. However, EGF can activate only the Grb2/SOS pathway, resulting in a transient activation of MAPK [72]. Consequently, these two growth factors induce distinct kinetics of MAPK signalling, culminating in disparate cellular outcomes. While the mechanisms underlying these processes are incompletely understood, there is evidence to suggest that protein scaffolds provide cells with the means to control both the duration of MAPK signalling and the physiological response to activating factors.
3.4 Scaffolds in temporal regulation of MAPK signalling
Analogous to their role in regulating spatial MAPK signalling, protein scaffolds can also control the duration of a MAPK signal. Like the originally identified isoform of KSR, a brain specific isoform of KSR, termed B-KSR, interacts with MEK and ERK [62]. Overexpression of B-KSR in PC12 cells results in increased basal levels of active phosphorylated ERK [62], and increases NGF-induced ERK activation and NGF-dependant differentiation. Interestingly, overexpression of B-KSR also causes a sustained increase in ERK activation following EGF treatment, resulting in differentiation [62]. As described above, PC12 cells usually differentiate or proliferate in response to NGF or EGF, respectively, and these differences are due to the duration of ERK activation [70]. Therefore, it appears B-KSR can alter the time course of MAPK signalling in response to growth factors, and as a consequence, alter the cellular outcome to these growth factors.
Ligand binding induces β2-adrenergic receptors to activate the MEK/ERK cascade [74]. This activation has two separate components: a rapid, transient signal that peaks 2-5 minutes after stimulation, and a slower but more sustained signal peaking 5-10 minutes after stimulation, but lasting for nearly 30 minutes [75]. The early ERK activation is dependant on GPCR and protein kinase A (PKA) but is completely independent of β-arrestin. Conversely, the delayed response is independent of GPCR and PKA, but requires β-arrestin and can be inhibited by siRNA knockdown of β-arrestin [75]. Although it is unclear what the physiological consequences of these two different pathways are, or how they may promote different cellular response, it appears that β-arrestin provides temporal regulation of the MAPK module. Further studies are required to ascertain whether this role played by β-arrestin following adrenergic receptor activation is specific to this receptor, or whether β-arrestin can provide similar temporal controls mediated by other factors.
While MP-1 binds to both MEK and ERK, there are differences in its interactions with the two kinases. MP-1 binds to MEK1 constitutively, but associates with ERK only transiently [76]. Following phosphorylation by MEK1, ERK is released from MP-1. Consequently, it has been proposed that MP-1 is able to translate a brief MEK signal into sustained ERK activation [76]. At present this hypothesis has not be confirmed, but it is an interesting concept that requires further investigation.
3.5 Graded and threshold signalling
A recent characteristic that has been applied to MAPK signalling is that of graded or “all or nothing” signalling. For example, following activation some MAPK pathways reach a critical level of signal strength and behave in a switch-like manner. Therefore individual cells within a population will be either “on” or “off” with respect to the particular outcome [77]. Examples of such cellular responses are proliferation, differentiation and programmed cell death. Other pathways respond in a graded fashion, where all the cells within a population show a uniform “output” increase that is proportional to the activating stimulus [77]. This can be observed in Drosophila embryo development, where graded concentrations of morphogens determine the dorsoventral axis. While still incompletely understood, analysis of the mating MAPK pathway in yeast has shown that protein scaffolds are able to influence these graded and switch-like MAPK signalling [77]. The yeast scaffold, Ste5 is essential for MAPK activation in response to pheromone stimulation [6]. Ste5 interacts with multiple kinases, recruiting them to the plasma membrane. When Ste5 is restricted to the cytosol, the activated kinases are inefficient in signal propagation, and so a strong signal is required in order to produce any significant output [77]. Consequently, pheromone signalling is more switch-like. However, when Ste5 is localised to the membrane, pheromone signalling is more graded, as low levels of active kinases are able to efficiently propagate the signal [77]. While additional studies are required to elucidate how graded and switch-like MAPK signalling is regulated, protein scaffolds do appear to have an important role.
6 MAPK scaffolds as therapeutic targets
The MAPK pathway has been strongly implicated in numerous different cancers (reviewed in [78]). Several MAPK components have been targeted for therapy, but this strategy has failed to yield effective pharmacotherapeutic agents. The lack of efficacy may be due, at least in part, to the lack of selectivity when inhibiting a key signalling kinase, such as MEK, Raf, or p38. Because these kinases regulate myriad cellular functions, inhibiting their activity is likely to affect multiple processes, some not linked to the pathophysiology of the disease being targeted. Therefore, new approaches are being explored to enhance therapeutic specificity.
Protein scaffolds are conceptually appealing therapeutic targets. Multiple scaffolds participate in the MAPK pathway, with each perhaps having a specific, non-overlapping function. Scaffolds are also important regulators of the localisation of many MAPK components. Pharmacologic modulation of scaffolds may enable specific regulation of MAPK function, directing the cellular response towards (or away from) a particular function (e.g. cell proliferation), without attenuating global MAPK activity. Furthermore, targeting scaffolds may allow inhibition of MAPK signalling from a specific cellular compartment, providing further specificity to MAPK modulation. There is little evidence to show effective pharmaceutical targeting of scaffolds. Nevertheless, published data support the concept. For example, KSR appears to be involved in Ras mediated cancer [17] and could, therefore, provide an effective target for tumours displaying hyperactive Ras. In addition, KSR knockout mice are less susceptible to induction of rheumatoid arthritis and exhibit defects in TNF-α and IL-1 signalling [16], suggesting that KSR could also provide a novel target for therapeutic intervention in inflammatory diseases.
Like KSR, IQGAP1 is a scaffold in MAPK signalling and has been implicated in carcinogenesis [18, 29]. For example, IQGAP1 is upregulated by gene amplification in some diffuse types of gastric carcinoma [79], and IQGAP1 protein is overexpressed in colorectal carcinoma, particularly at the invasion front [80]. The translocation of IQGAP1 from the cytoplasm to the cell membrane, which inhibits E-cadherin-mediated cell–cell adhesion [81], correlates with E-cadherin dysfunction and tumour dedifferentiation in gastric carcinoma [82]. In addition, a screen for genes exhibiting altered expression in a mouse model of metastatic melanoma, identified Iqgap1, its regulator, calmodulin, and ERK as 3 of only 32 genes (from ~10,500 arrayed genes) that showed a >2.5-fold increase in expression in metastatic cells [83]. Therefore, IQGAP1 and calmodulin are likely to be important in metastasis. In support of this concept IQGAP1 promotes cell migration and invasion via direct interactions with Cdc42, Rac1, actin and calmodulin [66, 84]. Importantly, overexpression of IQGAP1 in human breast epithelial cells increases formation and invasion of tumours in immunocompromised mice, while siRNA-mediated knockdown of IQGAP1 reduces tumourigenesis [85]. Consistent with these findings, the level of IQGAP1 expression in human breast carcinoma is higher than that in normal breast tissue [85]. Furthermore, the development of hepatocellular carcinoma in IQGAP2 knockout mice is entirely dependant on the presence of IQGAP1 [86]. Collectively, these data suggest IQGAP1 is involved in tumourigenesis and metastasis, and therefore is a conceptually appealing target for therapeutic intervention.
The participation of other MAPK scaffolds in neoplasia is less well documented, but some of it is persuasive. For example, loss of Sef expression correlates with high grade metastatic prostate cancer [87]. In addition, through an interaction with c-Src, β-arrestins modulate transactivation of EGFR by prostaglandin E2 in colorectal carcinoma cell lines [88]. The interaction between β-arrestin and c-Src is crucial for migration of colorectal carcinoma cells in vitro and metastasis in vivo. Thus, disruption of the normal homeostatic regulation mediated by the scaffolds may result in neoplasia. Collectively, the evidence outlined above supports the concept of developing compounds that selectively modulate the interaction of scaffolds with individual targets as a means to specifically modify MAPK activity.
7 Perspectives
Remarkable progress has been made in the last decade in our comprehension of the molecular mechanisms by which MAPK signalling is regulated by protein scaffolds. One of the most intriguing conundrums in understanding MAPK function is how a specific stimulus can induce a specific response, considering the diverse cellular functions that are controlled through MAPKs. This specificity appears to be provided, at least in part, by protein scaffolds. These multidomain proteins control the kinetics of MAPK activation and restrict the localisation of different MAPKs to specific subcellular compartments, such as the ER, endosomes or plasma membrane. Consequently, protein scaffolds are able to provide the specificity to the MAPK pathway that is so important for normal development and cellular homeostasis.
While several scaffolds have been identified in mammals, it is likely that others remain to be discovered. When considering the complexity of the MAPK network, with more than one scaffold probably operating simultaneously within a single cascade, it is clear that considerably more work is required to clearly elucidate how cells utilise this pathway. As we further our knowledge, scaffolds may emerge as appealing targets for novel therapeutics agents for human disease. Targeting scaffolds may allow more subtle and specific modulation of MAPK pathways that have become deregulated in pathological states. We look forward to advances in our understanding of MAPK signalling and the control provided by protein scaffolds.
Acknowledgments
Work in the authors’ laboratory is funded by grants from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Endocr Rev. 2001;22(2):153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 2.Murphy LO, Blenis J. Trends Biochem Sci. 2006;31(5):268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Roux PP, Blenis J. Microbiol Mol Biol Rev. 2004;68(2):320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mor A, Philips MR. Annu Rev Immunol. 2006;24:771–800. doi: 10.1146/annurev.immunol.24.021605.090723. [DOI] [PubMed] [Google Scholar]
- 5.Wellbrock C, Karasarides M, Marais R. Nat Rev Mol Cell Biol. 2004;5(11):875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 6.Elion EA. J Cell Sci. 2001;114(Pt 22):3967–3978. doi: 10.1242/jcs.114.22.3967. [DOI] [PubMed] [Google Scholar]
- 7.Morrison DK, Davis RJ. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 8.Kolch W. Nat Rev Mol Cell Biol. 2005;6(11):827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 9.Sacks DB. Biochem Soc Trans. 2006;34(Pt 5):833–836. doi: 10.1042/BST0340833. [DOI] [PubMed] [Google Scholar]
- 10.Kornfeld K, Hom DB, Horvitz HR. Cell. 1995;83(6):903–913. doi: 10.1016/0092-8674(95)90206-6. [DOI] [PubMed] [Google Scholar]
- 11.Morrison DK. J Cell Sci. 2001;114(Pt 9):1609–1612. doi: 10.1242/jcs.114.9.1609. [DOI] [PubMed] [Google Scholar]
- 12.Ritt DA, Zhou M, Conrads TP, Veenstra TD, Copeland TD, Morrison DK. Curr Biol. 2007;17(2):179–184. doi: 10.1016/j.cub.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 13.Kortum RL, Lewis RE. Mol Cell Biol. 2004;24(10):4407–4416. doi: 10.1128/MCB.24.10.4407-4416.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohmachi M, Rocheleau CE, Church D, Lambie E, Schedl T, Sundaram MV. Curr Biol. 2002;12(5):427–433. doi: 10.1016/s0960-9822(02)00690-5. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen A, Burack WR, Stock JL, Kortum R, Chaika OV, Afkarian M, Muller WJ, Murphy KM, Morrison DK, Lewis RE, McNeish J, Shaw AS. Mol Cell Biol. 2002;22(9):3035–3045. doi: 10.1128/MCB.22.9.3035-3045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fusello AM, Mandik-Nayak L, Shih F, Lewis RE, Allen PM, Shaw AS. J Immunol. 2006;177(9):6152–6158. doi: 10.4049/jimmunol.177.9.6152. [DOI] [PubMed] [Google Scholar]
- 17.Lozano J, Xing R, Cai Z, Jensen HL, Trempus C, Mark W, Cannon R, Kolesnick R. Cancer Res. 2003;63(14):4232–4238. [PubMed] [Google Scholar]
- 18.Brown MD, Sacks DB. Trends Cell Biol. 2006;16:242–249. doi: 10.1016/j.tcb.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Briggs MW, Sacks DB. FEBS Lett. 2003;542(13):7–11. doi: 10.1016/s0014-5793(03)00333-8. [DOI] [PubMed] [Google Scholar]
- 20.Mateer SC, Wang N, Bloom GS. Cell Motil Cytoskeleton. 2003;55(3):147–155. doi: 10.1002/cm.10118. [DOI] [PubMed] [Google Scholar]
- 21.Roy M, Li Z, Sacks DB. J Biol Chem. 2004;279:17329–17337. doi: 10.1074/jbc.M308405200. [DOI] [PubMed] [Google Scholar]
- 22.Roy M, Li Z, Sacks DB. Mol Cell Biol. 2005;25:7940–7952. doi: 10.1128/MCB.25.18.7940-7952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren JG, Li Z, Sacks DB. Proc Natl Acad Sci USA. 2007;104:10465–10469. doi: 10.1073/pnas.0611308104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourguignon LY, Gilad E, Rothman K, Peyrollier K. J Biol Chem. 2005;280(12):11961–11972. doi: 10.1074/jbc.M411985200. [DOI] [PubMed] [Google Scholar]
- 25.Schrick C, Fischer A, Srivastava DP, Tronson NC, Penzes P, Radulovic J. Neuron. 2007;55(5):786–798. doi: 10.1016/j.neuron.2007.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ussar S, Voss T. J Biol Chem. 2004;279(42):43861–43869. doi: 10.1074/jbc.M406240200. [DOI] [PubMed] [Google Scholar]
- 27.Reszka AA, Bulinski JC, Krebs EG, Fischer EH. Mol Biol Cell. 1997;8(7):1219–1232. doi: 10.1091/mbc.8.7.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noritake J, Watanabe T, Sato K, Wang S, Kaibuchi K. J Cell Sci. 2005;118(Pt 10):2085–2092. doi: 10.1242/jcs.02379. [DOI] [PubMed] [Google Scholar]
- 29.Briggs MW, Sacks DB. EMBO reports. 2003;4:571–574. doi: 10.1038/sj.embor.embor867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morishima-Kawashima M, Kosik KS. Mol Biol Cell. 1996;7(6):893–905. doi: 10.1091/mbc.7.6.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, McNulty DE, Marler KJM, Lim L, Hall C, Annan RS, Sacks DB. J Biol Chem. 2005;280:13871–13878. doi: 10.1074/jbc.M413482200. [DOI] [PubMed] [Google Scholar]
- 32.Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ. Science. 1998;281(5383):1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- 33.Teis D, Wunderlich W, Huber LA. Dev Cell. 2002;3(6):803–814. doi: 10.1016/s1534-5807(02)00364-7. [DOI] [PubMed] [Google Scholar]
- 34.Lefkowitz RJ, Whalen EJ. Curr Opin Cell Biol. 2004;16(2):162–168. doi: 10.1016/j.ceb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Science. 1999;283(5402):655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 36.DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW. J Cell Biol. 2000;148(6):1267–1281. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. J Biol Chem. 2003;278(8):6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 38.Furthauer M, Lin W, Ang SL, Thisse B, Thisse C. Nat Cell Biol. 2002;4(2):170–174. doi: 10.1038/ncb750. [DOI] [PubMed] [Google Scholar]
- 39.Xiong S, Zhao Q, Rong Z, Huang G, Huang Y, Chen P, Zhang S, Liu L, Chang Z. J Biol Chem. 2003;278(50):50273–50282. doi: 10.1074/jbc.M306936200. [DOI] [PubMed] [Google Scholar]
- 40.Torii S, Kusakabe M, Yamamoto T, Maekawa M, Nishida E. Dev Cell. 2004;7(1):33–44. doi: 10.1016/j.devcel.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 41.Ren Y, Li Z, Rong Z, Cheng L, Li Y, Wang Z, Chang Z. Biochem Biophys Res Commun. 2007;354(3):741–746. doi: 10.1016/j.bbrc.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 42.Ren Y, Cheng L, Rong Z, Li Z, Li Y, Zhang X, Xiong S, Hu J, Fu XY, Chang Z. Cell Signal. 2008;20(3):518–533. doi: 10.1016/j.cellsig.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garrington TP, Johnson GL. Curr Opin Cell Biol. 1999;11(2):211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 44.Ren JG, Li Z, Sacks DB. J Biol Chem. 2008;283(34):22972–22982. doi: 10.1074/jbc.M804626200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abankwa D, Gorfe AA, Hancock JF. Semin Cell Dev Biol. 2007;18(5):599–607. doi: 10.1016/j.semcdb.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smart EJ, Ying YS, Mineo C, Anderson RG. Proc Natl Acad Sci USA. 1995;92(22):10104–10108. doi: 10.1073/pnas.92.22.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R. Mol Biol Cell. 2003;14(1):334–347. doi: 10.1091/mbc.E02-07-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang CS, Zhou J, Feng AK, Lynch CC, Klumperman J, DeArmond SJ, Mobley WC. J Biol Chem. 1999;274(51):36707–36714. doi: 10.1074/jbc.274.51.36707. [DOI] [PubMed] [Google Scholar]
- 49.Peiro S, Comella JX, Enrich C, Martin-Zanca D, Rocamora N. J Biol Chem. 2000;275(48):37846–37852. doi: 10.1074/jbc.M000487200. [DOI] [PubMed] [Google Scholar]
- 50.Hancock JF. Nat Rev Mol Cell Biol. 2003;4(5):373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 51.Pol A, Calvo M, Enrich C. FEBS Lett. 1998;441(1):34–38. doi: 10.1016/s0014-5793(98)01517-8. [DOI] [PubMed] [Google Scholar]
- 52.Lai WH, Cameron PH, Doherty JJ, 2nd, Posner BI, Bergeron JJ. J Cell Biol. 1989;109(6 Pt 1):2751–2760. doi: 10.1083/jcb.109.6.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burke P, Schooler K, Wiley HS. Mol Biol Cell. 2001;12(6):1897–1910. doi: 10.1091/mbc.12.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang X, Sorkin A. Mol Biol Cell. 2002;13(5):1522–1535. doi: 10.1091/mbc.01-11-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y, Pennock S, Chen X, Wang Z. Mol Cell Biol. 2002;22(20):7279–7290. doi: 10.1128/MCB.22.20.7279-7290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR. Cell. 1999;98(1):69–80. doi: 10.1016/S0092-8674(00)80607-8. [DOI] [PubMed] [Google Scholar]
- 57.Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson RL, 2nd, Cox AD, Philips MR. Nat Cell Biol. 2002;4(5):343–350. doi: 10.1038/ncb783. [DOI] [PubMed] [Google Scholar]
- 58.Perez de Castro I, Bivona TG, Philips MR, Pellicer A. Mol Cell Biol. 2004;24(8):3485–3496. doi: 10.1128/MCB.24.8.3485-3496.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sobering AK, Romeo MJ, Vay HA, Levin DE. Mol Cell Biol. 2003;23(14):4983–4990. doi: 10.1128/MCB.23.14.4983-4990.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK. Mol Cell. 2001;8(5):983–993. doi: 10.1016/s1097-2765(01)00383-5. [DOI] [PubMed] [Google Scholar]
- 61.Matheny SA, Chen C, Kortum RL, Razidlo GL, Lewis RE, White MA. Nature. 2004;427(6971):256–260. doi: 10.1038/nature02237. [DOI] [PubMed] [Google Scholar]
- 62.Muller J, Cacace AM, Lyons WE, McGill CB, Morrison DK. Mol Cell Biol. 2000;20(15):5529–5539. doi: 10.1128/mcb.20.15.5529-5539.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Philipova R, Whitaker M. J Cell Sci. 2005;118(Pt 24):5767–5776. doi: 10.1242/jcs.02683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Casar B, Pinto A, Crespo P. Mol Cell. 2008;31(5):708–721. doi: 10.1016/j.molcel.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 65.Brown MD, Bry L, Li Z, Sacks DB. J Biol Chem. 2007;282:30265–30272. doi: 10.1074/jbc.M702537200. [DOI] [PubMed] [Google Scholar]
- 66.Mataraza JM, Briggs MW, Li Z, Entwistle A, Ridley AJ, Sacks DB. J Biol Chem. 2003;278(42):41237–41245. doi: 10.1074/jbc.M304838200. [DOI] [PubMed] [Google Scholar]
- 67.Fukata M, Watanabe T, Noritake J, Nakagawa M, Yamaga M, Kuroda S, Matsuura Y, Iwamatsu A, Perez F, Kaibuchi K. Cell. 2002;109(7):873–885. doi: 10.1016/s0092-8674(02)00800-0. [DOI] [PubMed] [Google Scholar]
- 68.Bensenor LB, Kan HM, Wang N, Wallrabe H, Davidson LA, Cai Y, Schafer DA, Bloom GS. J Cell Sci. 2007;120(Pt 4):658–669. doi: 10.1242/jcs.03376. [DOI] [PubMed] [Google Scholar]
- 69.Blagoev B, Kratchmarova I, Ong SE, Nielsen M, Foster LJ, Mann M. Nat Biotechnol. 2003;21(3):315–318. doi: 10.1038/nbt790. [DOI] [PubMed] [Google Scholar]
- 70.Marshall CJ. Cell. 1995;80(2):179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 71.Tan PB, Kim SK. Trends Genet. 1999;15(4):145–149. doi: 10.1016/s0168-9525(99)01694-7. [DOI] [PubMed] [Google Scholar]
- 72.York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, Stork PJ. Nature. 1998;392(6676):622–626. doi: 10.1038/33451. [DOI] [PubMed] [Google Scholar]
- 73.Bos JL. Curr Opin Cell Biol. 2005;17(2):123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 74.Ma L, Pei G. J Cell Sci. 2007;120(Pt 2):213–218. doi: 10.1242/jcs.03338. [DOI] [PubMed] [Google Scholar]
- 75.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. J Biol Chem. 2006;281(2):1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 76.Sharma C, Vomastek T, Tarcsafalvi A, Catling AD, Schaeffer HJ, Eblen ST, Weber MJ. J Cell Biochem. 2005;94(4):708–719. doi: 10.1002/jcb.20344. [DOI] [PubMed] [Google Scholar]
- 77.Takahashi S, Pryciak PM. Curr Biol. 2008;18(16):1184–1191. doi: 10.1016/j.cub.2008.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dhillon AS, Hagan S, Rath O, Kolch W. Oncogene. 2007;26(22):3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 79.Sugimoto N, Imoto I, Fukuda Y, Kurihara N, Kuroda S, Tanigami A, Kaibuchi K, Kamiyama R, Inazawa J. J Hum Genet. 2001;46(1):21–25. doi: 10.1007/s100380170119. [DOI] [PubMed] [Google Scholar]
- 80.Nabeshima K, Shimao Y, Inoue T, Koono M. Cancer Lett. 2002;176(1):101–109. doi: 10.1016/s0304-3835(01)00742-x. [DOI] [PubMed] [Google Scholar]
- 81.Li Z, Kim SH, Higgins JM, Brenner MB, Sacks DB. J Biol Chem. 1999;274(53):37885–37892. doi: 10.1074/jbc.274.53.37885. [DOI] [PubMed] [Google Scholar]
- 82.Takemoto H, Doki Y, Shiozaki H, Imamura H, Utsunomiya T, Miyata H, Yano M, Inoue M, Fujiwara Y, Monden M. Int J Cancer. 2001;91(6):783–788. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1121>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 83.Clark EA, Golub TR, Lander ES, Hynes RO. Nature. 2000;406(6795):532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 84.Mataraza JM, Li Z, Jeong HW, Brown MD, Sacks DB. Cell Signal. 2007;19:1857–1865. doi: 10.1016/j.cellsig.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jadeski L, Mataraza JM, Jeong HW, Li Z, Sacks DB. J Biol Chem. 2008;283(2):1008–1017. doi: 10.1074/jbc.M708466200. [DOI] [PubMed] [Google Scholar]
- 86.Schmidt VA, Chiariello CS, Capilla E, Miller F, Bahou WF. Mol Cell Biol. 2008;28(5):1489–1502. doi: 10.1128/MCB.01090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Darby S, Sahadevan K, Khan MM, Robson CN, Leung HY, Gnanapragasam VJ. Oncogene. 2006;25(29):4122–4127. doi: 10.1038/sj.onc.1209428. [DOI] [PubMed] [Google Scholar]
- 88.Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM, DuBois RN. Proc Natl Acad Sci USA. 2006;103(5):1492–1497. doi: 10.1073/pnas.0510562103. [DOI] [PMC free article] [PubMed] [Google Scholar]