

SUMMARY
AMP activated protein kinase (AMPK) is an evolutionary conserved metabolic sensor that responds to alterations in cellular energy levels to maintain energy balance. While its role in metabolic homeostasis is well documented, its role in mammalian development is less clear. Here we demonstrate that mutant mice lacking the regulatory AMPK β1 subunit have profound brain abnormalities. The β1−/− mice show atrophy of the dentate gyrus and cerebellum, severe loss of neurons, oligodendrocytes and myelination throughout the CNS. These abnormalities stem from reduced AMPK activity with ensuing cell cycle defects in neural stem and progenitor cells (NPC). The β1−/− NPC deficits result from hypophosphorylation of the retinoblastoma protein (Rb), which is directly phosphorylated by AMPK at Ser804. The AMPK-Rb axis is utilized by both growth factors and energy restriction to increase NPC growth. Together, our results reveal that the metabolic sensor AMPK integrates growth factor signaling with cell cycle control to regulate brain development.
INTRODUCTION
AMPK is an integrative metabolic sensor that maintains energy balance both at the cellular and systemic level. It links neuronal functions with energy supply and plays a key role in hypothalamic control of food intake and peripheral energy expenditure (Xue et al., 2006). Systemic AMPK activity is linked to human diseases like diabetes, obesity, stroke, hypertension, myocardial injury and atherosclerosis, and may be involved in the protection afforded by caloric restriction (Clarel et al., 2007; Miller et al., 2008; Dyck 2007). One important neuronal target of AMPK is the GABAB receptor, whose activation helps mediate neuroprotection after ischemia (Kuramoto et al., 2007).
In addition to its metabolic functions, studies in model organisms suggest that AMPK also regulates cell structure and polarity, cell division, as well as normal growth and development (Lee et al., 2007; Baena-González et al., 2007). In particular, AMPK helps maintain genomic integrity in neural precursors as well as the structure and function of mature neurons in Drosophila (Lee et al., 2007). Loss of AMPK activity causes neurodegeneration in Drosophila (Tschäpe et al., 2002; Spasic et al., 2008) and AMPK activation in mice protects hippocampal neurons against metabolic, excitotoxic and oxidative insults (Culmsee et al., 2001). These studies have suggested that AMPK may have additional roles beyond the established metabolic functions both in normal physiology and disease.
AMPK is a heterotrimeric multi-substrate kinase composed of one catalytic (α1 or α2), one regulatory (β1 or β2) and one AMP/ATP binding (γ1, γ2 or γ3) subunit. The C-terminus of the β subunit interacts with both α and γ subunits, and current biochemical and structural evidence indicate that the β subunit is an obligatory component of the active AMPK complex. When intracellular energy levels drop (low ATP:AMP ratio), AMP displaces ATP from the γ subunit causing a conformational change that allows upstream kinases (e.g. LKB1 or CaMKKβ) to phosphorylate and activate the α subunit. In addition to uniting the α and γ subunits, in yeast the β subunits also serve regulatory functions as they direct the AMPK complex to defined substrates in specific subcellular compartments (Vincent et al., 2001).
The analysis of mice lacking AMPKα1 or α2 catalytic subunits demonstrated the widespread and overlapping functions of these proteins and importance of overall AMPK activity (Jørgensen et al., 2005). Human mutations of the γ2 subunit cause cardiomyopathy, characterized by hypertrophy and glycogen accumulation (Blair et al., 2001), where as characterization of mice lacking γ3 subunit demonstrated impaired post-exercise glycogen re-synthesis in skeletal muscle (Barnes et al., 2004). In contrast to studies of these subunits, little is known about the physiologic roles of individual β subunits in mammals. Interestingly, loss of AMPK β subunit in Drosophila causes progressive neurodegeneration, indicating a crucial role in adult neuron maintenance (Spasic et al., 2008). To investigate how β subunits regulate the physiologic functions of AMPK in mammals, we generated AMPKβ1−/− mice. Our results demonstrate that the AMPKβ1 subunit is crucial for proper brain development through its regulation of AMPK phosphorylation of Rb, a pathway that provides for the integration of nutrient and growth factor signaling pathways that influence neural differentiation.
RESULTS
Generation of AMPKβ1 mutant mice
To investigate the biologic roles of the AMPK β1 subunit, we generated mutant mice using ES cells in which the β1 gene was interrupted by the insertion of a βgeo cassette (henceforth called β1−/− mice). The insertion created a β1-βgeo fusion protein containing exons 1–5 of β1. This produces a mutant β1 protein lacking the terminal 46 amino acids (Fig. S1A). This deleted domain is required for generation of the active AMPK heterotrimer through interactions with both α and γ subunits and is highly conserved in the closely related AMPK β2 protein (Iseli et al., 2005). We confirmed the existence of a single βgeo integrant at the predicted site in the β1 locus by Southern blot analysis, PCR genotyping, RT-PCR analysis and nucleotide sequencing (Fig. S1B-D). We detected the β1-βgeo fusion protein by immunoblotting with a β-gal antibody in lysates from multiple tissues, including brain (Fig. S1E). The absence of wildtype β1 in lysates from β1-deficient E14.5 telencephalon (forebrain), P7 cerebellum (Fig. 1A) and MEFs (Fig. S1F) was confirmed using a β1/β2 C-terminal specific antibody. The loss of β1 caused a significant reduction of activated AMPK (phospho-AMPKαThr172) and phosphorylated ACC (phospho-ACCSer79), a downstream target of AMPK, in the brain and other organs (Fig. 1B, C, S1G, H, I, data not shown). A similar reduction in AMPK activity was observed by monitoring phosphorylation of the AMPK artificial substrate (SAMS peptide) (data not shown).
Fig. 1. AMPKβ1-deficient mice show reduced AMPK activity and manifest brain abnormalities.

(A) Immunoblot analysis of wildtype (+/+) and β1−/− brain lysates using β1/β2 C-terminal antibodies or (B) total or phosphorylated AMPK (AMPKThr172) and ACC (ACCSer79) antibodies. (n = 4 independent experiments). (C) Densitometric analysis of bands in A and B. (D) Macroscopic view of wildtype and β1−/−brain at P14. (CB; dotted & lower panel) in β1−/− mice. (E, F) Coronal brain sections showing atrophy of dentate gyrus (E; arrow) and cerebellum (F; IGL inner granule cell layer). NeuN immunohistochemistry of (G) cortex and dentate gyrus (arrow) and (H) cerebellum. (I) Quantification of neuronal losses assessed by counting NeuN+ cells. * p < 0.001 (n = 5).
AMPK β1−/− mice display structural and functional brain abnormalities
The β1−/− mutant mice were born in a proper Mendellian ratio, but failed to gain weight normally and were clearly emaciated by postnatal day 14 (P14) (Fig S2 and Movie1). They displayed severe tremors, ataxic gait and seizure-like activity and died by P21. Most notably, an examination of P14 β1−/− animals revealed a 50% reduction in overall brain size with severe cerebellar atrophy and marked reduction of the cerebral cortex resulting in improper cortical fusion and exposure of the superior (SC) and inferior colliculi (IC) (Fig. 1D; S3A). A histological examination revealed a nearly complete loss of the dentate gyrus (Fig. 1E). The cerebellum was characterized by loss of the inner granule cell layer (IGL), extensive spongiform vacuolation, and disordered laminar organization (Fig. 1F). We did not observe abnormalities in brain structure or behavior in β1+/− animals.
To further characterize the extent of neuronal loss, we used the NeuN antibody to perform immunohistochemistry on brain sections. Significant losses (between 35–65%) of cortical neurons and granule neurons of the dentate gyrus (Fig. 1G) and IGL of the cerebellum were clearly evident in the β1−/− brain (Fig. 1H, I). MAP2 immunohistochemistry and Bielschowsky’s silver staining demonstrated widespread losses of dendritic processes (Fig. 2A) and white matter axonal projections in these mutant mice (Fig 2B), suggesting that absence of β1 causes loss of neurons and neuronal processes.
Fig. 2. AMPKβ1-deficient mice demonstrate both neuronal and glial CNS deficits, seizures and reduced GABA receptor phosphorylation.

MAP2 immunohistochemistry showing dendrites (A), silver staining showing axonal tracts (B), APC immunohistochemistry showing oligodendrocytes (C) and MBP immunohistochemistry showing myelination (D) in the P14 brain. (E) Electron microscopic analysis of P14 β1−/− optic nerves. (F) Quantification of GFAP-positive cells in wildtype and β1−/− brains. (G) Immunohistochemistry of the forebrain of E18.5 wildtype and β1−/− embryos using GFAP antibody. LV, lateral ventricle; arrow heads indicate migrating GFAP+ astroglia. (H) Quantitation of GFAP+ migrating astroglia in E18.5 wildtype and β1−/− brains. (I) EEG showing three seizure episodes recorded for 30 min in P14 β1−/− mice. The trace of one episode is enlarged at the bottom for clarity. (J) Immunohistochemistry of wildtype and β1−/− P14 brain sections and (K) Western blot analysis of brain lysates using phospho-GABABR2Ser783 antibody. (L) Quantification of signal intensities in (K). * p = 0.01; ** p = 0.002; # p = 0.006.
To investigate the effects of β1 loss on CNS glia, we examined oligodendrocytes by immunohistochemistry using the Adenomatous Polyposis Coli (APC) antibody and found a 75–80% loss of oligodendrocytes at P14 (Fig. 2C; S3B). This loss caused severe hypomyelination throughout the brain that was particularly evident in the corpus callosum and striatum (Fig. 2D, Fig S3C). Consistent with the deficit in oligodendrocytes, the β1−/− optic nerve was translucent and ~30% thinner than wildtype nerve (Fig. S3D). Electron microscopic analysis demonstrated severe hypomyelination of mutant P14 optic nerve axons (Fig. 2E).
Astrocyte maturation occurs later in development and was difficult to study due to the early lethality of the β1−/− animals. We performed immunohistochemistry on P14 brains using antibodies to GFAP, an intermediary filament enriched in differentiated astrocytes (Cahoy et al., 2008) and BLBP (brain lipid binding protein), which is expressed in neural progenitors, radial glia and immature/differentiating astrocytes (Domowicz et al., 2008; Hagedus et al., 2008). The results showed that in addition to extensive astrogliosis in β1−/− mice, there were an increased number of astrocytes throughout the brain (Fig. 2F; S4A,). Moreover, astrocytes in P14 dentate gyrus of wildtype animals expressed low levels of GFAP along with BLBP, whereas astrocytes in β1−/− mice expressed similar levels of BLBP but higher levels of GFAP (Fig. S4B). At E18.5, consistent with previous findings (Barnabe-Heider et al., 2005), very few GFAP+ cells were observed in wildtype mice; however β1−/− brain contained many GFAP+ migrating astrocytes (Fig. 2G, H), suggesting premature astrocytic differentiation of glial precursors in these mice. Together, these results suggest that while loss of the AMPKβ1 resulted in fewer neurons and oligodendrocytes, this was accompanied by an increased number of fully differentiated astrocytes.
AMPK directed GABA(B) receptor phosphorylation is reduced in seizure-prone β1−/− mice
The extensive brain hypomyelination in β1−/− mice along with their notable tremor and abnormal behavior prompted us to monitor them for seizure activity. Electroencephalogram recordings revealed spontaneous electrographic seizures in mutant animals that usually recurred within less than 10 min (Fig. 2I). The seizures in these mice could result from the paucity of oligodendrocytes and resulting decreased in myelinated CNS fibers, the severe brain malformations, abnormal energy homeostasis in the brain, or perhaps through decreased inhibitory neuron activity.
GABA is the major inhibitory neurotransmitter in mammalian brain and it exerts its slow, prolonged effects via the GABAB receptor, which is made up of two subunits R1 and R2. Interestingly, phosphorylation of the R2 subunit by AMPK at Ser783, acts to stabilize GABAB activation of inwardly rectifying K+ channels and decrease synaptic activity (Kuramoto et al., 2007). Disturbances of GABA+ neurons, either through selective loss of these neurons or via hypophosphorylation of the GABAB R2 receptor, could facilitate seizure propensity. Immunohistochemistry of P14 brain showed that the loss of GABAergic neurons was similar (~ 40%) to the loss of total neurons in β1−/− P14 brain (not shown). However, immunohistochemical as well as western blot analysis using a GABAB-R2 pSer783-specific antibody revealed that the R2 receptor was hypophosphorylated in β1−/− brain (Fig. 2J–L). Together, these results suggest that loss of AMPK activity could interfere with proper GABAergic signaling, potentially contributing to abnormal electrical activity in the β1−/−postnatal brain.
AMPKβ1−/− NPCs show defective proliferation, differentiation and unregulated apoptosis in vivo
The deficits in multiple cell lineages in the brain suggested that developmental processes were adversely affected by β1 deficiency. We examined mice at a number of embryonic and perinatal ages and found that E18.5 β1−/− embryos were similar in body size to wildtype embryos; however, the β1−/− brain was ~50% smaller (Fig. S5A–D). To determine whether the decreased size and cell number were due to abnormalities in proliferation and/or apoptosis, we first counted the number of cycling cells using Ki67 immunohistochemistry. We found 22.22 ± 4.66% less Ki67-positive cells around the lateral ventricles in β1−/− brain (Fig. 3A). To investigate this cell cycle defect, we pulse-labeled β1−/− E14.5 embryos with BrdU for 1 hr and performed immunohistochemistry with antibodies against BrdU, which marks cells in S-phase, phistone H3 (PH3), which detects cells in M-phase, and Ki67, which detects all actively cycling cells. We observed a similar labeling index (proportion of cells in S-phase [(BrdU+)/total cycling cells (Ki67+)] in wildtype and β1−/− forebrain (wt: 32.83 ± 6.66 vs. β1−/−:33.46 ± 4.25) (Fig. 3B). However, the number of cells in mitosis (PH3+ cells) in mutant E14.5 forebrain (Fig. 3C), as well as in P7 dentate gyrus and cerebellum (Fig. S5E–G) were significantly reduced, suggesting that the cell cycle defect occurs after DNA synthesis.
Fig. 3. Loss of β1 results in neural stem/progenitor cell developmental defects.

Immunohistochemistry of the E14.5 forebrain with antibodies against Ki67 (red) (A), BrdU (green) and Ki67 (red) (B), phosphohistone H3 (red) (C) and cleaved Caspase3 (red) (D). TUNEL staining (red) in conjunction with immunohistochemistry using Sox2 antibodies (green) (E) or nestin antibodies (green) at E14.5 (F). Inset in (F) shows magnified view of TUNEL/Nestin double+ apoptotic neural precursors. (G) Immunohistochemistry using BrdU (green) and cleaved Caspase 3 (red) antibodies 1 hr after BrdU injection. (H) A cartoon illustrating the different layers of the embryonic forebrain. (I) Immunohistochemistry using BrdU (green) and Ki67 (red) antibodies 24 hr after BrdU injection and (J) BrdU (green) and cleaved Caspase 3 (red) antibodies 24 hr after BrdU injection. (K) Immunohistochemistry of E14.5 wild type and β1−/− brain using Tuj1 (green) and cleaved Caspase3 (red), (L) E18.5 brains using Olig2 (green) and cleaved Caspase3 (red) and (M) P7 brains using GFAP (green) and cleaved Caspase3 (red) antibodies to detect apoptotic cells. Inset in (K) shows colocalization of Tuj1 and cleaved Caspase3 in migrating neurons. (N)) Quantitative analysis of apoptotic neural cells. DAPI staining (blue) was used to highlight the nuclei. * p < 0.001.
Increased apoptosis could also be responsible for the decreased numbers of neurons and glia and often occurs in response to abnormalities in cell cycle progression. We found large numbers of apoptotic cells by cleaved Caspase3 immunohistochemistry in β1−/− E14.5 forebrain (Fig. 3D) and P7 cerebellum (Fig. S5H). Although a few apoptotic cells were present in the proliferative Sox2-positive ventricular zone, the majority were found in the Nestin-positive subventricular zone, particularly in the intermediate zone and subplate of the mutant forebrain (Fig. 3E, F). Double immunolabeling with BrdU and cleaved Caspase 3 antibodies confirmed that the majority of apoptotic β1−/− NPCs reside outside the BrdU positive zone (Fig. 3G). As cells that aberrantly exit cell cycle often undergo apoptosis, we examined cell cycle exit of β1−/− NPCs. E14.5 embryos were labeled with BrdU for 24 hr and double immunolabeling with BrdU and Ki67 antibodies was performed. The fraction of BrdU positive cells that were Ki67 negative represents the cells that exited cell cycle during the labeling period. Although, it appeared that a higher number of BrdU positive cells were present outside the subventricular zone in the β1−/− brains (Fig. 3I), the majority of these cell were Ki67-negative but cleaved Caspase 3 positive (Fig. 3J), suggesting that a defect after S phase is responsible for the massive apoptosis of β1−/− NPCs. Moreover, we found increased numbers of apoptotic Tuj1-positive immature neurons and Olig2-positive oligodendrocyte precursors, indicating thatβ1−/− neural precursors undergo apoptosis as they migrate and differentiate into neurons and oligodendrocytes (Fig. 3K–L). In contrast, no cell death was observed in the astrocyte population at P7 (Fig. 3M) or at E18.5 (data not shown).
Although the normal body size of β1−/− embryos suggested that there were minimal cell losses in other tissues, the ubiquitous expression of AMPK prompted us to examine whether defects in proliferation and apoptosis could be detected in other regions of the body. We performed pHistone H3 and cleaved Caspase3 immunohistochemistry on sections of E14.5 embryo body, liver and interdigital junctions. Unlike β1−/− embryonic brain, the number of proliferating and apoptotic cells in these β1−/− tissues was similar to wildtype embryo tissues (Fig. S6A–D). Collectively, these results suggest that loss of the AMPKβ1 subunit leads to proliferative defects and unregulated apoptosis specifically in the progenitors of the developing brain.
Defects in AMPKβ1−/− NPCs are cell autonomous
AMPK is involved in central energy metabolism and the developing brain is sensitive to metabolic imbalances, thus it was important to determine whether the brain anomalies in the β1−/− animals were due to global metabolic abnormalities or whether they were cell autonomous to neural progenitor cells. While global metabolic problems would likely cause deficits in multiple tissues, to clearly address this issue, we isolated mouse embryonic fibroblasts and cultured neurospheres from E12.5 telencephalon from β1−/− and wildtype embryos. In response to energy deprivation, AMPK increases mitochondrial respiration and glucose transporter expression. We examined both glucose transporter expression and basal as well as maximal oxygen consumption and found that they were similar in β1−/− and wildtype NPCs and MEFs (Fig. S7A–C).
To investigate whether the proliferation and apoptosis defects observed in β1−/− NPCs in vivo were cell-autonomous, we cultured neurospheres and found that β1−/− neurospheres were significantly smaller in diameter (Fig. 4A, S7D) and produced fewer numbers of secondary and tertiary neurospheres (Fig. S7E). Direct cell counting revealed that growth and self renewal capacity of β1−/− NPCs was severely impaired (Fig. 4B, C). The proliferative rate of β1−/− NPCs was examined using a CFSE washout experiment (see methods for details). Flow cytometric analysis of mean fluorescence intensities of CFSE-labeled cells at 0 and 96 hr revealed that β1−/− NPCs proliferate at about half the rate of wildtype NPCs (Fig. 4D). We also used propidium iodide (PI) staining to evaluate the extent of cell death in these cultured neurospheres and found that an increased number of β1−/− NPCs were PI-positive (wt: 23.87 ± 3..33% vs. ± β1−/−: 50.95 ± 7.55%) (Fig. 4E, F), indicating that cell intrinsic defects lead to the abnormalities observed in these mutant NPCs.
Fig. 4. AMPKβ1 loss results in cell-autonomous NPC defects.

(A) Photomicrographs of telencephalic neurospheres cultured for 48 hr. Neurospheres were assayed for growth (B) and self renewal (C). 1° & 2° NS = primary & secondary neurospheres. (D) Wildtype and β1−/−NPCs were incubated with CFSE dye and the fluorescence intensity of the cells was measured at 0 and 96 hr. The numbers in parenthesis are mean fluorescence intensities. (E) Propidium iodide staining of unfixed wildtype and β1−/− neurospheres and quantitative analysis (F) of apoptosis of β1+/+, β1+/− and β1−/− NPCs. (G) Quantification of immunohistochemical results obtained from in vitro differentiation of β1+/+, β1+/− and β1−/− neurospheres showing percentage of neurons (Tuj1), oligodendrocytes (O4) and astrocytes (GFAP). * p < 0.001. Data is representative of at least three independent experiments.
To gain insight into how β1-deficiency causes the differential loss of specific CNS cell types, the multilineage differentiation potential of these neurospheres was examined. The β1−/−neurospheres produced fewer Tuj1-positive neurons (55.0 ± 8.25%) and O4-positive oligodendrocytes (60.45 ± 2.5%) but similar numbers of GFAP-positive astrocytes (Fig. 4G; S8A–C). However, β1−/− astrocyte differentiation was not normal. Immunocytochemistry with antibodies to BLBP (immature astrocyte marker), GFAP and Aquaporin4 (mature astrocyte markers) (Bachoo et al., 2004; Cahoy et al., 2008), showed that an increased number of β1−/−astrocytes lost BLBP expression and displayed robust GFAP and Aquaporin4 expression (Fig. S8D–G), indicating that they prematurely differentiate in vitro as they do in vivo.
Finally, the severe cerebellar defects in β1−/− animals prompted us to examine cultured cerebellar granule cell precursors from P2 animals. We found that reaggregate formation as well as neurite projection was severely impaired in the β1−/− precursors (Fig. S9A). We also observed that 60–70% of NeuN+ β1−/− reaggregates were apoptotic (Fig. S9B, C). In sum, these results indicate that cell autonomous defects caused by β1-deficiency result in aberrant proliferation and/or cell fate determination of neural precursors.
AMPK β subunits play functionally distinct roles in NPCs
The abnormalities in β1−/− NPCs are caused by cell intrinsic mechanisms rather than by an altered global metabolic state in these mutant mice. To understand why β1 deficiency results in such devastating NPC defects, we performed western blot analysis on β1−/− neurospheres and found that pAMPK was almost absent, while pACC (a canonical target of AMPK) was reduced by ~50% (Fig. 5A). The low AMPK activity in NPCs prompted us to further investigate why the highly related AMPKβ2 subunit cannot complement the β1 mutation in these cells. We examined their expression and subcellular localization using an antibody that recognizes the C-terminus of both β subunits and found that β1 is expressed at a 6–8-fold higher level than β2 in wildtype NPCs (Fig. S10A). While β2 expression in β1−/− NPCs was increased (Fig. S10A), it was obviously insufficient to compensate for loss of β1 function. Subcellular fractionation studies showed that active AMPK was reduced in both the cytoplasmic and nuclear fractions of β1−/− NPCs (Fig. S10B). Interestingly, immunocytochemistry using β1- or β2-N-terminal specific antibodies (Fig. S10C) as well as Western blot assays using the common β1/β2-C-terminal specific antibody showed that β1 was distributed throughout the cell, whereas the β2 subunit was primarily cytoplasmic (Fig. S10D,E). Moreover, in wildtype NPCs, immunoprecipitated AMPK complexes captured with AMPKα1/2 antibody contained significantly more β1 that β2 (Fig. S10F, G). Interestingly, the absence of the β1 subunit in NPCs resulted in lower levels of α subunits, presumably reflecting decreased stability of the catalytic subunits in cells lacking β1 (Fig. S10B).
Fig. 5. Expression of β1 but not β2 rescues β1−/− NPC phenotypes.
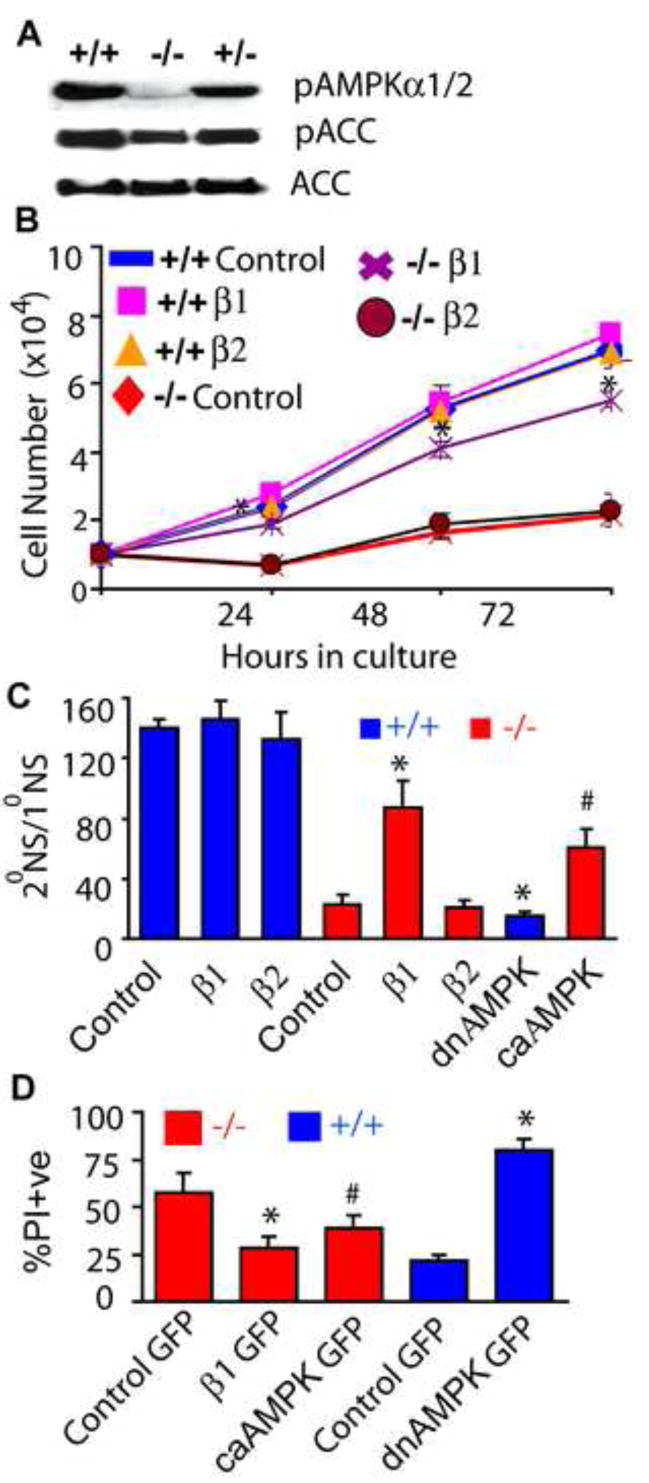
(A) Immunoblot analysis of NPC lysates using phospho-AMPKThr172, total ACC and phospho-ACCSer79 antibodies. AMPKβ1−/− NPCs were infected with lentivirus expressing GFP (control), β1, β2 or constitutively active (ca) AMPKα2, while wildtype NPCs were infected with dominant negative (dn) AMPKα2 and the growth rate (B), self-renewal capacity (C) and number of apoptotic cells (D) were monitored. * p< 0.005; # p <0.05. Data is representative of three independent experiments.
The widespread cellular distribution of the β1 subunit, particularly in the nucleus, may imbue it with functions that are not shared by β2. Expression of His-tagged human β1 or β2 by lentivirus (Fig. S10H) demonstrated that re-introduction of β1 rescued the growth (Fig. 5B), self renewal (Fig. 5C), and apoptosis (Fig. 5D; S10I) defects of β1−/− NPCs, whereas the β2 subunit had no effect. To definitively prove that AMPK activity is necessary for regulated proliferation of NPCs, we expressed constitutively active (ca) and dominant negative (dn) AMPKα2 mutants in β1−/− and wildtype NPCs. We found that while caAMPK partially rescued the self renewal and apoptosis defects of β1−/− NPCs, dnAMPK severely reduced self renewal and caused catastrophic death of wildtype NPCs (Fig. 5C, D). These results indicate that AMPK is a crucial regulator of NPC growth and that its activity is uniquely dependent on the β1 subunit in NPCs.
AMPK directly phosphorylates Rb to regulate NPC cell cycle
The cell autonomous defects in NPC growth caused by loss of β1 prompted us to investigate the MAP kinase (Erk1/2) and PI3 kinase signaling (Akt) pathways, which are crucial for NPC proliferation, survival and differentiation. However we found these pathways were normal in β1−/− NPCs (Fig. S11A, B), suggesting that abnormalities in other effectors are responsible for the deficits in β1−/− NPCs. Other molecules central to NPC health, such as GSK3β, which phosphorylates and destabilizes N-Myc (transcription factor for D-type cyclins) (Galderisi et al., 2003; Kenney et al., 2004), N-Myc, cyclin D1 and D2, and the cell cycle inhibitors p16, p18, p21 and p27 were all expressed at normal levels in β1−/− NPCs (Fig. S11C, D). Another critical regulator of cell growth is p53, which is phosphorylated at Ser15 by AMPK (Jones et al., 2005) was also unaltered in β1−/− NPCs (Fig. S11D). Collectively, these data suggest that other effectors or cell cycle regulators downstream of the cyclins and cell cycle inhibitors must account for the β1−/− NPC deficits.
Despite the seeming normalcy of most cell cycle regulators in β1−/− NPCs, the striking resemblance of the brain abnormalities in β1-deficient mice with those observed in animals lacking N-Myc (Knoepfler et al., 2002), cyclin D1/D2 (Ciemeryc et al., 2002), Rb (Lee et al., 1992) and Rb family proteins (McLear et al., 2006) prompted us to examine the cyclin downstream target Rb. Rb is exquisitely regulated by multiple phosphorylation events and we noticed in searching the Phosphosite database (www.phosphosite.org) that one of the multiple Rb phosphorylation sites, Ser804 (Ser811 in human), resembled the AMPK consensus phosphorylation site (Fig. 6A). Immunoblot analysis using antibodies specific for Rb phosphoSer804 and pan Rb antibodies revealed that phosphorylation of Ser804 is greatly decreased in β1−/− NPCs (Fig. 6B). No change in total Rb or in phosphorylation at another site (Ser780) was observed, suggesting that Ser804 is specifically hypophosphorylated in β1−/− NPCs. The antibody used in our study recognizes both Ser800 and Ser804. However, since only the Ser804 and not the Ser800 conforms to the AMPK consensus site, we believe that Ser804 is the likely residue phosphorylated by AMPK and will be used in the text henceforth. Our mutagenesis studies (see later) further reinforce this view.
Fig. 6. AMPK phosphorylates Rb to regulate Rb-E2F interaction.

(A) AMPK consensus sequence (top), AMPK phosphorylation site in ACC and potential site found in Rb (bottom). (B) Immunoblot analysis of β1−/− NPC lysates using pRb(Ser800/804), pRb(Ser780), pan Rb, and tubulin antibodies. Data is representative of three independent experiments. (C, D) Nonradioactive kinase assay: CDK4/6 complex immunoprecipitated using Cyclin D1/D2 antibodies from wild type and β1−/− NPCs (C) and AMPK holoenzyme immunoprecipitated using AMPKα1/2 antibody from wildtype NPCs, were used to phosphorylate recombinant Rb protein (residues 701–928). Phosphorylation was monitored by immunoblot using phosphoRb800/804 antibody. Data is representative of three independent experiments. (E, F) Immunoprecipitation of NPC lysates with pan Rb antibody followed by Western blot with E2F1 antibody and densitometry showed the increased amount of E2F1-Rb complexes in β1−/−NPCs. (G) Proliferation assay of wildtype and β1−/− NPCs expressing GFP or the indicated Rb proteins. Data is representative of two independent experiments. (H) Flow cytometric analysis of NPCs expressing GFP or Rb proteins displaying the percentage of cells in G2M phase. FACS data is available with Online Supplement (Fig. S12–13; S15–21). (I) Proliferation assay of wildtype NPCs expressing GFP, constitutively active (ca) AMPK, β1, or β2 subunits. Data is representative of three independent experiments. (J) Immunoblot analysis of wildtype NPCs expressing caAMPK using pAMPKαThr172 and pRbSer800/804 and pan AMPKα and Rb antibodies. (K) Immunoprecipitation assay showing the level of E2F1 bound to Rb in NPCs expressing GFP (control), caAMPK, or dnAMPK. * p< 0.005.
The CDK-Cyclin D complex is known to phosphorylate Rb at Ser804, thus the hypophosphorylation at this site in β1−/− NPCs could result from an indirect effect on CDK4/6 activity or a direct effect of AMPK on Rb. CDK4/6, which exist in complexes with cyclinD1/D2, were immunoprecipitated using cyclin D1/2 antibodies and the activity was measured by nonradioactive in vitro kinase assays using bacterially produced Rb fusion protein (residues 701–928) as the substrate. We found that CDK4/6 activity from β1−/− and wildtype NPCs was equivalent (Fig. 6C), indicating that β1-deficiency did not affect CDK activity. To test whether AMPK directly phosphorylates Rb, we immunoprecipitated the AMPK holoenzyme (α/β/γ heterotrimers) using anti-AMPKα1/2 antibody from wildtype NPCs. In vitro kinase assays using this immunoprecipitated AMPK enzyme (or cyclin/CDK4/6 complex, control), and the Rb protein mentioned above, showed that AMPK directly phosphorylated Rb at Ser804 and that this modification could be inhibited by the AMPK inhibitor, compound C (Fig. 6D).
In proliferating cells, growth factor signaling promotes CDK-dependent phosphorylation of Rb to inhibit it from sequestering the E2F transcription factors. As a consequence of the high level of hypophosphorylated Rb in β1−/− NPCs, the amount of E2F1 sequestered by Rb should be higher in these cells. Rb was immunoprecipitated from wildtype and mutant NPCs, and Western analysis using anti-E2F1 antibody showed significantly more Rb-E2F1 complexes in β1−/− NPCs (Fig. 6E, F), suggesting that aberrant regulation of the Rb-E2F1 complex is at least partially responsible for the β1−/− NPC deficiencies.
β1−/− NPCs display defects in the G2M phase of the cell cycle
The Rb-E2F complex plays multiple cellular roles including serving as a gate keeper of the G1-S restriction point, the G2-M phase, cell cycle exit, cellular differentiation, and regulation of apoptotic cell death (Burkhart and Sage, 2008; Rigberg et al., 1999; Naderi et al., 2002; Yen and Sturgill 1998; Niculescu et al., 1998). Many of these defects are present in β1−/− NPCs and our previous analysis demonstrated alterations in cell proliferation. The abnormal regulation of Rb in these cells prompted us to perform flow cytometric analysis to examine cell cycle progression in these cells. In comparing β1−/− and wildtype NPCs, we found comparable numbers of cells in S phase (wt: 16.47 ± 4.7%; β1−/−: 18.06 ± 3.95%), less β1−/− cells in G1 (wt: 71.5 ± 6.5%; β1−/−: 58.25 ± 3.25%, p = 0.005), and almost twice as many β1−/− cells in G2M phase (wt: 12.12 ± 1.7%; β1−/−: 22.56 ± 1.95%, p = 0.001) (Fig S12–13), indicating that β1−/− cells have defects that prevent them properly exiting or re-entering the cell cycle.
To firmly establish a direct connection between these cell cycle defects and the AMPK-Rb signaling axis, we examined the levels of pAMPK and pRb in β1−/− NPCs expressing β1, β2, caAMPK or dnAMPK. β1−/− NPCs expressing caAMPK or β1, but not β2, showed increased levels of pAMPK, pRb and pACC levels (Fig. S14 A–D), while wildtype NPCs expressing dnAMPK showed significantly decreased Rb and ACC phosphorylation (Fig. S14 C, D). Together, these results indicate the importance of the AMPK–Rb signaling axis in NPC growth, a pathway that is largely regulated through the β1 subunit.
The highly orchestrated, cyclical phosphorylation of Rb throughout the cell cycle makes Rb overexpression studies difficult. Nevertheless, we generated lentiviruses expressing wildtype Rb, Rb(S804A), removing the critical phosphorylation site, and Rb(S804E) and Rb(S804D), potentially creating phophomimetics. These lentiviruses were used to examine whether an Rb phosphomimetic mutant could rescue the β1−/− NPC growth defect, and whether a phosphorylation-resistant Rb would cause growth defects in wildtype NPCs. We infected wildtype NPCs with lentivirus expressing GFP (control), wildtype Rb or Rb(S804A) and β1−/−NPCs with lentivirus expressing GFP (control), wildtype Rb, Rb(S804A), or Rb(S804E), Rb(S804D). The cells were counted 24 hr after infection. Both wildtype Rb and Rb(S804A) caused significant growth reduction in wildtype NPCs. β1−/− NPCs expressing GFP, wildtype Rb or Rb(S804A) showed poor growth; however, those expressing the phophomimetic mutants, showed improved growth (Fig. 6G). We continued to observe these cells and found that by 48 hr the neurospheres expressing Rb(S804E) or Rb(S804D) stopped growing and began to look unhealthy, suggesting that constitutive phosphorylation of Rb at this site may prevent cells from re-entering cell cycle.
Previous analysis showed that β1−/− NPC cell cycle progression was blocked at the G2M phase. We therefore investigated whether β1−/− NPCs expressing Rb(S804E) or Rb(S804D) could now transit the G2M stage. Flow cytometric analysis performed on cells 24 hr after lentiviral infection showed that β1−/− NPCs expressing wildtype Rb or Rb(S804A) had 27% cells in G2M, whereas β1−/− NPCs expressing Rb(S804E) or Rb(S804D) had 16% cells in G2M (Fig. 6H, S15–19). Interestingly, wildtype NPCs expressing Rb(S804A) had more cells (~22%) in G2M than those expressing GFP (~10%) (Fig. 6H, S20–21). These results indicate that the hypophosphorylation of Rb at Ser804 is responsible for the G2M defect in β1−/− NPCs, as a phosphomimetic RB mutant can partially restore the ability of these cells to transit G2M. It is interesting that although Rb is phosphorylated at 19 different Ser/Thr residues, many of which could serve overlapping as well as specific functions, the phosphorylation of Rb at Ser804 appears to play an important role in G2M phase and/or cell cycle exit in NPCs.
Stem cell growth factors as well as metabolic perturbations activate AMPK to promote NPC proliferation
We extended our studies to explore whether wildtype NPC growth was enhanced by activation of AMPK via genetic or physiological stimuli. Constitutively active AMPK not only enhanced proliferation of wildtype NPCs (Fig. 6I), but also caused a significant increase in Rb phosphorylation (Fig. 6J). Accordingly, the proportion of E2F1 sequestered by Rb was reduced in wildtype NPCs expressing caAMPK, where as dnAMPK caused significantly more E2F1 sequestration (Fig. 6K). It is intriguing that overexpression of the β1 but not β2 subunit caused a distinct increase in wildtype NPC growth (Fig 5B; 6I), supporting its role in regulating AMPK-mediated NPC proliferation.
The effects of AMPKβ1 deletion on Rb phosphorylation led us to consider whether the highly conserved AMPK signaling pathway is fundamentally important for NPC responses to proliferative signals such as growth factors. We cultured wildtype NPCs in the absence of growth factors for 2 hr (withdrawal phase) and then added back EGF and FGF for 1 hr. Cells treated with growth factors had increased levels of pAMPK and increased phosphorylation of Rb at Ser804. However, when EGF and FGF were added to cells treated with the AMPK inhibitor Compound C, no increase in pAMPK or pRb was observed (Fig. 7A, B). Thus, it appears that the failure of AMPK-directed phosphorylation of Rb in β1−/− NPCs, perhaps in response to endogenous growth factors, results in defects in G2M phase as well as the aberrant differentiation that leads to apoptosis.
Figure 7. Energy restriction and Growth factor signaling utilize the AMPK-Rb axis to enhance NPC proliferation.

Immunoblot (A) and densitometric analysis (B) of wildtype NPCs using indicated antibodies after 2h growth factor stimulation in the presence or absence of AMPK inhibitor Compound C (CC, 5 mM). Proliferation assay of wildtype NPCs (C) and β1−/− NPCs (D) cultured for 48 hr under glucose limiting conditions. (Cont = 25 mM glucose, which is amount in Neurobasal medium). Data is representative of three independent experiments. Immunoblot (E) and densitometric analysis (F–K) using phospho-specific AMPKαThr172, RbSer800/804, ACCSer79, and pan AMPKα, Rb, and ACC antibodies. (L) Immunoprecipitation assay using Rb antibody and Western blot with E2F1 antibody from NPCs grown in the indicated glucose concentration in the absence or presence of Compound C (5 mM). # p= 0.07 * p 0.005. AU = arbitrary units.
AMPK is integrally involved in regulating cellular energy homeostasis and is activated by low cellular ATP levels, such as occurs by limiting oxygen or glucose supplies or exercise, conditions that enhance proliferative capacity of stem cells (Burgers et al., 2008; Fu et al., 2006; Stolzing et al., 2006). To explore whether the proliferative effects of glucose restriction are manifested through activation of the AMPK-Rb axis, we monitored the growth of wildtype NPCs cultured in 2.5 mM to 25 mM (the amount present in Neurobasal medium) glucose for 48 hr. NPC cell numbers were increased when grown in low glucose medium, with 5 mM glucose giving the highest growth rate (Fig. 7C). The NPC growth stimulation by low glucose was not observed in cells treated with Compound C, which severely inhibited neurosphere growth. Furthermore, no effect on growth by low glucose was observed in β1−/− NPCs, consistent with their lack of AMPK signaling (Fig. 7D). Interestingly, there was a small but consistent increase in the phosphorylation of AMPK, Rb and ACC at lower glucose concentrations (Fig. 7E–K). Remarkably, glucose limitation also reduced the proportion of Rb-bound E2F1, whereas Compound C treatment caused higher levels of Rb sequestration of E2F1 (Fig. 7L). Collectively, our results demonstrate that perhaps growth factor signaling as well as other physiological/metabolic stimuli utilize the AMPK-Rb signaling pathway to modulate the growth and differentiation of neural stem and progenitor cells during mammalian brain development.
DISCUSSION
Rb phosphorylation and AMPK in NPC cell cycle progression
In this study we identify AMPK as an important kinase for Rb and show that loss of AMPK activity in AMPKβ1-deficient animals causes Rb hypophosphorylation and multiple NPC defects. AMPK is a ubiquitously expressed kinase with many substrates that influences cellular as well as organismal energy homeostasis. It is likely that the complex phenotype of the β1 mutant mice results from aberrant signaling from multiple AMPK-regulated pathways. Although CDK4/6 can phosphorylate Rb at Ser804 (Zarkowska et al., 1997), the phosphorylation of this site is dramatically decreased in β1−/− NPCs. Data concerning Rb phosphorylation in NPCs is scanty; however, it is noteworthy that most mouse cells lacking two or even three CDKs still proliferate (Malumbres et al., 2004; Berthet et al., 2006; Barriere et al., 2007) and contain residual phosphorylated Rb, providing further evidence that other kinases, such as AMPK, must also phosphorylate Rb.
Despite the importance of Rb phosphorylation as a defining regulatory event in G1, (Sherr 2000; Massague 2004; Oxford and Scadden 2008), β1−/− NPCs with hypophosphorylated Rb appear to enter S-phase normally in vitro as well as in vivo. Rb has 19 Ser/Thr residues that are potentially and transiently phosphorylated during various stages of the cell cycle but information concerning the roles of specific phosphorylation events remains limited. In fact, how different kinases might contribute to differential control of Rb, such as regulating its binding to specific ligands, is an unresolved question (Mittnacht 1998). Rb, which remains hyperphosphorylated from late G1 through the rest of the cell cycle, is now viewed as a transcriptional co-factor (Burkhart and Sage, 2008). Current evidence indicates that Rb hypophosphorylation causes not only G1 arrest but also arrest during the G2M transition (Burkhart and Sage 2008; Rigberg et al., 1999; Naderi et al., 2002; Yen and Sturgill 1998; Niculescu et al., 1998). It is important to note that functional Rb is required specifically for the S and G2 phases in mouse embryonic stem cells (Fluckiger et al., 2006). Furthermore, a number of E2F1 target genes like Mad2 (Sotillo et al., 2007) and SIL (Erez et al., 2008) control the spindle check point during mitosis. Functional complexes containing E2F and Rb are essential for repressing expression of these critical mitotic regulators to maintain the G2M check point (Polager and Ginsberg, 2003). The decreased number of mitotic cells in the ventricular zone of the β1−/− brains where neural stem cells undergo self-renewal, together with the large number of apoptotic cells outside the subventricular zone where progenitor cells migrate and differentiate, suggests that Rb phosphorylation at Ser804 by AMPK is necessary for multiple aspects of NPC biology.
The enhanced growth of β1−/− NPCs expressing phosphomimetic Rb mutants versus the growth suppression of wildtype NPCs expressing phosphorylation resistant Rb is intriguing. Earlier studies demonstrated that introduction of phosphorylation resistant mutant Rb protein in many cell types can efficiently suppress cell proliferation (Jiang and Zacksenhaus 2002; Chang et al., 1995; Roig et al., 2004). It appears from our results that AMPK-dependent phosphorylation of Ser804 is crucial for different phases of NPC cell cycle, particularly the G2M phase.
Caloric restriction, hypoxia and exercise all reduce cellular energy stores and stimulate AMPK activity. The potentiation of NPC proliferation by glucose restriction or constitutively active AMPK is consistent with the AMPK-dependent augmented neurogenesis of adult neural stem cells observed in mice under dietary restriction (Dagon et al., 2005). Interestingly, treatment with the AMPK pharmacological activator AICAR did not increase NPC proliferation (data not shown). While we do not know why AICAR was ineffective, it also exerts many AMPK-independent effects such as inhibition of mitochondrial respiratory chain complex I (Guigas et al., 2007) and regulation of multiple metabolic enzymes (e.g. fructose 1–6, bisphosphatase, glucokinase) (Vincent et al., 1991, Vincent et al., 1992). The potential of AMPK to couple nutrition and stem cell proliferation/differentiation is an important new avenue of exploration that will be enhanced by the identification of more specific AMPK activators and inhibitors.
AMPK -Rb signaling in development and tissue maintenance
AMPK activity is crucial for embryonic development as demonstrated by the stunted growth and reproductive failure in Arabidopsis lacking AMPK activity in (Baena-González et al., 2007), the embryonic lethality in Drosophila lacking either α (Lee at al., 2007) or β subunits (Spasic et al., 2008).. The regulation of Rb by AMPK is particularly intriguing in this regard as the Rb-E2F pathway is involved in fate specification and differentiation of multiple cell types. Appropriate Rb function is necessary for terminal differentiation of progenitor cells undergoing maturation. In C. elegans, developmental defects, rather than changes in cell proliferation, are the most striking consequences of Rb-E2F malfunction (Heuvel and Dyson 2008). In mammals, the Rb-E2F pathway regulates fate and differentiation of a number of cell types including neurons (Lee et al., 1992; Callaghan et al., 1999), cardiac stem cells (Papadimou et al., 2005), adipocytes (Dali-Youcef et al., 2007; Fajas et al., 2002), erythrocytes (Sankaran et al., 2008), and epithelial cells (Wikenheiser-Brokamp, 2004). These cell fate choices depend on the active transcriptional repression mediated by Rb-E2F1 complexes (Weintraub et al., 1995) and, by direct effects of hypophosphorylated Rb on transcriptional regulators like PPARγ (Fajas et al., 2002). Finally, excess E2F activity due to Rb loss results in decreased mitochondrial biogenesis and impaired erythroid differentiation (Sankaran et al., 2008). Thus, the differentiation defects in β1−/− brain are likely to be caused by the wide range of activities disrupted by the altered levels of Rb-E2F complexes in NPCs.
The loss of axonal and dendritic processes in β1−/− postnatal brain is consistent with the neurodegeneration phenotype observed in AMPK γ (Tschäpe et al., 2002) and β subunit deficient (Spasic et al., 2008) Drosophila models. Interestingly, in Drosophila, loss of the entire β subunit gene was functionally equivalent to the loss of β subunit C-terminal exons, consistent with our findings in mice. Thus it appears that besides being required during embryonic differentiation, AMPK is also necessary for maintaining the structural and functional integrity of mammalian neurons. However, unlike the case in epithelial cells where the AMPK-LKB1 axis influences polarity (Lee et al, 2007; Mirouse et al., 2007), we observed no morphological changes reflective of altered neuronal polarity, nor did we observe an altered distribution of the polarity proteins PAR3 or pPKCξ (Fig. S5 I-K). Why AMPKβ1 deficiency causes specific loss of neurons and oligodendrocytes but not astrocytes is unclear; however, it should be noted that astrocyte differentiation can proceed normally in the absence of Rb (Marino et al., 2000). β1−/− astrocytes do not differentiate normally, suggesting that additional AMPK-modulated pathways are important in these cells. On the other hand, it is tempting to speculate that hypophosphorylation of Rb at Ser804 in NPC’s might contribute to the altered ratio of neural cells in vivo.
Finally, the identification of Rb as an AMPK substrate is intriguing because it raises the possibility that cell proliferation and fate choice can be influenced by intracellular energy levels through the actions of AMPK. In fact, AMPK along with the other components of the cellular energy sensing system, namely NaMPT and SIRT1 have been recently shown to influence muscle differentiation based on intracellular energy status (Fulco et al., 2008). Future work will determine whether morphogens and growth factors that orchestrate fate choice also activate AMPK.
Importance of AMPK subunit composition
The inability of the β2 subunit to functionally compensate for loss of β1 is surprising in light of their high homology and the considerable physiologic redundancy observed between the two α subunits (Viollet et al., 2003; Jorgensen et al., 2005). This is highlighted by the increased NPC growth mediated by overexpression of β1 but not β2. One important distinction between these subunits is the failure of β2 to enter the nucleus where it could potentially promote AMPK phosphorylation of Rb. β1 is also heavily phosphorylated, thus it is also possible that differential modifications of the two β subunits directly regulate AMPK substrate choice in a context-dependent manner. For example, it is interesting that AMPK is required for adaptation of nutrient-deprived cancer cells to hypoxia (Yun et al., 2005; Laderoute et al., 2006; Nagata et al., 2003) and that loss of AMPK activation sensitizes cancer cells to apoptosis (Kim et al., 2007; Baumann et al., 2007). If AMPK is required for cancer stem cell self-renewal as it is for NPCs, then AMPK inhibitors could be useful chemotherapeutic agents. Conversely, AMPK activators or dietary manipulation may be useful in promoting adult neurogenesis in disorders where these cells have been damaged such as during radiotherapy or chemotherapy.
EXPERIMENTAL PROCEDURES
Generation of AMPKβ1 mutant mice
Embryonic stem (ES) cells (clone RRR454) containing an AMPKβ1 gene trap allele was obtained from Bay Genomics. Briefly, AMPKβ1 gene trap ES cells were microinjected into blastocysts derived from superovulated pregnant mice, and these blastocysts were injected into pseudopregnant C57BL/6 females. Chimeric males (129/Ola mixed background) were mated with C57BL/6 females and germ line transmission was confirmed by PCR genotyping of tail DNA. All procedures were carried out in the Washington University Animal care facility.
TUNEL staining
Tunel staining was performed on frozen sections as per manufacturer’s instructions (Roche, Indianapolis, IN) and visualized with Cy3-streptavidin and fluorescence microscopy.
In vitro culture of neural progenitors
Neurospheres were dissected from embryonic day 12.5 (E12.5) CNS telencephalic lobes and cultured in Neurobasal medium supplemented with B27 and N2 as described previously (Dasgupta and Gutmann, 2005).. To assess NPC growth, 104 cells from animals of each genotype were seeded in triplicate. At each time point, resulting neurospheres were trypsinized and counted on a hemocytometer. For self-renewal assays, single neurospheres were picked up by pipette and neurosphere diameter was measured individually under microscope. Ten similar-sized neurospheres of each genotype were triturated individually before plating, and the number of resulting secondary neurospheres generated per primary neurosphere was counted after 6 days. Neurosphere diameters were measured using the Metamorph software. For NPC proliferation assay, NPCs were labeled with CFSE {5-(and-6)-Carboxyfluorescein diacetate, succinimidyl ester} as described (Dasgupta and Gutmann, 2005). Briefly, NPCs were pulse labeled with 5 μM CFSE at 37°C for 15 minutes, washed, one-half of the cells were analyzed by FACS while the other half was analyzed after 4 days of growth. Detailed methods are available in Supplemental data.
Non-radioactive in vitro kinase assay
Non-radioactive in vitro kinase assay was performed as described (Lee et al., 2007) with minor modifications. Briefly, wildtype NPCs were lysed with MAPK buffer, AMPK was immunoprecipitated from wild type NPCs with AMPKα1/2 antibody, washed with a buffer containing 10 mM Tris-Hcl, pH 7.5, 0.5 M LiCl and suspended in kinase buffer containing 0.5 mM ATP. CyclinD1/D2 (associated with CDK4/6) was immunoprecipitated with CyclinD1 and CyclinD2 antibodies. Reactions were carried out at 30°C for 1 hr by adding RB-C fusion protein (residues 701–928; Cell Signaling Technology, Beverly, MA) to the reaction mix. Reaction was terminated by adding 6X Laemmli buffer and boiling for 5 min. Samples were resolved in 10% SDS-PAGE and Western blot analysis was performed with phospho-RbSer800/804 (Ser807/811 in human) antibody (Cell Signaling Technology, Beverly, MA).
Quantitative analysis of cell numbers and Statistical methods
Student’s t test was used to calculate statistical significance with p < 0.05 representing a statistically significant difference. For in vivo quantitative analysis of cell numbers, five square regions with identical area were demarcated on each high-power field (20×) image. The regions were demarcated using the Metamorph software and cells were counted in each region. The sections were plane-matched and photographed digitally on a Nikon microscope. Sections from three mice of each genotype were used for manual cell counting. Brain size was measured using the NIH Image J software. Pixels occupying the area of a dorsal view of the brain were quantitated and represented as brain size. Densitometric analysis of immunoblots was performed using GelPro analyzer software. Error bars in all figures indicate SD.
Detail methods for genotyping, RT-PCR and primer sequences, seizure study, electron microscopy, tissue preparation, immunohistochemistry, immunoprecipitation, subcellular fractionation, Western blot analysis, cell culture, AMPK activity assay, cell respiration studies and plasmids and viruses are available with online supplement.
Supplementary Material
Acknowledgments
We thank Eugene Johnson and Shin Imai for fruitful discussions, Craig Press for Oxygraph studies and helpful discussions, Balazs Hegedus and Amy Strickland for immunohistochemical preparations, Michael Wong for EEG analysis, Robert Schmidt for electron microscopy and Zhikang Qian for flow cytometry. This work was supported by National Institutes of Health (NIH) Neuroscience Blueprint Core Grant NS057105 (to Washington University), the HOPE Center for Neurological Disorders, and NIH Grants AG13730 and NS040745 (to J.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baena-González E, Rolland F, Thevelein JM, Sheen J. A central integrator of transcription networks in plant stress and energy signaling. Nature. 2007;448:938–42. doi: 10.1038/nature06069. [DOI] [PubMed] [Google Scholar]
- Bachoo RM, Kim RS, Ligon KL, Maher EA, Brennan C, Billings N, Chan S, Li C, Rowitch DH, Wong WH, DePinho RA. Molecular diversity of astrocytes with implications for neurological disorders. Proc Nat Acad Sci USA. 2004;101:8384–9. doi: 10.1073/pnas.0402140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnabé-Heider F, Wasylnka JA, Karl JL, Fernandes KJL, Porsche C, Sendtner M, Kaplan DR, Miller FD. Evidence that Embryonic Neurons Regulate the Onset of Cortical Gliogenesis via Cardiotrophin-1. Neuron. 2005;48:253–265. doi: 10.1016/j.neuron.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Barnes BR, Marklund S, Steiler TL, Walter M, Hjälm G, Amarger V, Mahlapuu M, Leng Y, Johansson C, Galuska, et al. The 5′-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J Biol Chem. 2004;279:38441–7. doi: 10.1074/jbc.M405533200. [DOI] [PubMed] [Google Scholar]
- Barriere C, Santamaria D, Cerqueira A, Galan J, Martin A, Ortega S, Malumbres M, Dubus P, Barbacid M. Mice thrive without cdk4 and cdk2. Mol Oncology. 2007;1:72–83. doi: 10.1016/j.molonc.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P, Mandl-Weber S, Emmerich B, Straka C, Schmidmaier R. Inhibition of adenosine monophosphate-activated kinase induces apoptosis in multiple myeloma cells. Anticancer Drugs. 2007;18:405–10. doi: 10.1097/CAD.0b013e32801416b6. [DOI] [PubMed] [Google Scholar]
- Berthet C, Klarmann KD, Hilton MB, Suh HC, Keller JR, Kiyokawa H, Kaldis P. Combined loss of Cdk2 and Cdk4 results in embryonic lethality and Rb hypophosphorylation. Dev Cell. 2006;10:563–73. doi: 10.1016/j.devcel.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, Watkins H. Mutations in the 2 subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001;10:1215–1220. doi: 10.1093/hmg/10.11.1215. [DOI] [PubMed] [Google Scholar]
- Bürgers HF, Schelshorn DW, Wagner W, Kuschinsky W, Maurer MH. Acute anoxia stimulates proliferation in adult neural stem cells from the rat brain. Exp Brain Res. 2008;188:33–43. doi: 10.1007/s00221-008-1336-6. [DOI] [PubMed] [Google Scholar]
- Burkhart DL, Sage J. Cellular mechanisms of tumor suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–82. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–78. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan DA, Dong L, Callaghan SM, Hou YX, Dagnino L, Slack RS, et al. Neural precursor cells differentiating in the absence of Rb exhibit delayed terminal mitosis and deregulated E2F 1 and 3 activity. Dev Biol. 1999;207:257–70. doi: 10.1006/dbio.1998.9162. [DOI] [PubMed] [Google Scholar]
- Chang MW, Barr E, Seltzer J, Jiang YQ, Nabel GJ, Nabel EG, Parmacek MS, Leiden JM. Cytostatic gene therapy for vascular proliferative disorders with a constitutively active form of the retinoblastoma gene product. Science. 1995;267:518–22. doi: 10.1126/science.7824950. [DOI] [PubMed] [Google Scholar]
- Ciemeryc MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, Gardner H, Sicinski P. Development of mice expressing a single D-type cyclin. Genes Dev. 2002;24:3277–89. doi: 10.1101/gad.1023602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest. 2007;117:2325–36. doi: 10.1172/JCI31516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- Dagon Y, Avraham Y, Magen I, Gertler A, Ben-Hur T, Berry EM. Nutritional status, cognition, and survival: a new role for leptin and AMP kinase. J Biol Chem. 2005;280:42142–8. doi: 10.1074/jbc.M507607200. [DOI] [PubMed] [Google Scholar]
- Dali-Youcef N, Mataki C, Coste A, Messaddeq N, Giroud S, Blanc S, Koehl C, Champy MF, Chambon P, Fajas L, et al. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc Natl Sci USA. 2007;104:10703–8. doi: 10.1073/pnas.0611568104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Gutmann DH. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J Neurosci. 2005;25:5584–94. doi: 10.1523/JNEUROSCI.4693-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domowicz MS, Sanders TA, Ragsdale CW, Schwartz NB. Aggrecan is expressed by embryonic brain glia and regulates astrocyte development. Dev Biol. 2008;315:114–24. doi: 10.1016/j.ydbio.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyck JR. The ischemic heart: starving to stimulate the adiponectin-AMPK signaling axis. Circulation. 2007;116:2779–81. doi: 10.1161/CIRCULATIONAHA.107.742023. [DOI] [PubMed] [Google Scholar]
- Erez A, Chaussepied M, Castiel A, Colaizzo-Anas T, Aplan PD, Ginsberg D, Izraeli S. The mitotic checkpoint gene, SIL is regulated by E2F1. Int J Cancer. 2008;23:1721–5. doi: 10.1002/ijc.23665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell. 2002;3:903–10. doi: 10.1016/s1534-5807(02)00360-x. [DOI] [PubMed] [Google Scholar]
- Fluckiger AC, Marcy G, Marchand M, Négre D, Cosset FL, Mitalipov S, Wolf D, Savatier P, Dehay C. Cell cycle features of primate embryonic stem cells. Stem Cells. 2006;24:547–56. doi: 10.1634/stemcells.2005-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Tay SS, Ling EA, Dheen ST. High glucose alters the expression of genes involved in proliferation and cell-fate specification of embryonic neural stem cells. Diabetologia. 2006;49:1027–38. doi: 10.1007/s00125-006-0153-3. [DOI] [PubMed] [Google Scholar]
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–73. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galderisi U, Jori FP, Giordano A. Cell cycle regulation and neural differentiation. Oncogene. 2003;22:5208–19. doi: 10.1038/sj.onc.1206558. [DOI] [PubMed] [Google Scholar]
- Guigas B, Taleux N, Foretz M, Detaille D, Andreelli F, Viollet B, Hue L. AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. Biochem J. 2007;404:499–507. doi: 10.1042/BJ20070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus B, Dasgupta B, Shin JE, Emnett RJ, Hart-Mahon EK, Elghazi L, Bernal-Mizrachi E, Gutmann DH. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell. 2008;1:443–57. doi: 10.1016/j.stem.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Heuvel S, Dyson NJ. Conserved function of the pRB and E2F families. Nat Rev Mol Cell Biol. 2008;9:713–24. doi: 10.1038/nrm2469. [DOI] [PubMed] [Google Scholar]
- Iseli TJ, Walter M, van Denderen BJ, Katsis F, Witters LA, Kemp BE, Michell BJ, Stapleton D. AMP-activated protein kinase beta subunit tethers alpha and gamma subunits via its C-terminal sequence (186–270) J Biol Chem. 2005;280:13395–400. doi: 10.1074/jbc.M412993200. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Zacksenhaus E. Activation of retinoblastoma protein in mammary gland leads to ductal growth suppression, precocious differentiation, and adenocarcinoma. J Cell Biol. 2002;156:185–98. doi: 10.1083/jcb.200106084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Jørgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, et al. Effects of alpha-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J. 2005;19:1146–8. doi: 10.1096/fj.04-3144fje. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development. 2004;131:217–28. doi: 10.1242/dev.00891. [DOI] [PubMed] [Google Scholar]
- Kim HS, Hwang JT, Yun H, Chi SG, Lee SJ, KangI, Yoon KS, Choe WJ, Kim SS, et al. Inhibition of AMP-activated protein kinase sensitizes cancer cells to cisplatin-induced apoptosis via hyper-induction of p53. J Biol Chem. 2007;283:3731–42. doi: 10.1074/jbc.M704432200. [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, et al. Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;52:233–47. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–47. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, Lee SH, Shong M, Kim JM, Kim J, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–20. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–94. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Sotillo R, Santamaría D, Galán J, Cerezo A, Ortega S, Dubus P, Barbacid M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504. doi: 10.1016/j.cell.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- McLear JA, Garcia-Fresco G, Bhat MA, Van Dyke TA. In vivo inactivation of pRb, p107 and p130 in murine neuroprogenitor cells leads to major CNS developmental defects and high seizure rates. Mol Cell Neurosci. 2006;33:260–73. doi: 10.1016/j.mcn.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–82. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- Mittnacht S. Control of pRB phosphorylation. Curr Opin Getet Dev. 1998;8:21–27. doi: 10.1016/s0959-437x(98)80057-9. [DOI] [PubMed] [Google Scholar]
- Mirouse V, Swick LL, Kazgan N, St Johnston D, Brenman JE. LKB1 and AMPK maintain epithelial cell polarity under energetic stress. J Cell Biol. 2007;177:387–92. doi: 10.1083/jcb.200702053. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nagata D, Mogi M, Walsh K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem. 2003;278:31000–6. doi: 10.1074/jbc.M300643200. [DOI] [PubMed] [Google Scholar]
- Naderi S, Hunton IC, Wang JYJ. Radiation dose-dependent maintenance of G2 arrest requires retinoblastoma protein. Cell Cycle. 2002;3:193–200. [PubMed] [Google Scholar]
- Niculescu AB, 3rd, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998;18:629–43. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9:115–28. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- Papadimou E, Ménard C, Grey C, Pucéat M. Interplay between the retinoblastoma protein and LEK1 specifies stem cells toward the cardiac lineage. EMBO J. 2005;24:1750–61. doi: 10.1038/sj.emboj.7600652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polager S, Ginsberg D. E2F mediates sustained G2 arrest and down-regulation of Stathmin and AIM-1 expression in response to genotoxic stress. J Biol Chem. 2003;278:1443–9. doi: 10.1074/jbc.M210327200. [DOI] [PubMed] [Google Scholar]
- Rigberg DA, Kim FS, Sebastian JL, Kazanjian KK, McFadden DW. Hypophosphorylated retinoblastoma protein is associated with G2 arrest in esophageal squamous cell carcinoma. J Surg Res. 1999;84:101–5. doi: 10.1006/jsre.1999.5617. [DOI] [PubMed] [Google Scholar]
- Roig JM, Molina MA, Cascante A, Calbó J, Carbó N, Wirtz U, Sreedharan S, Fillat C, Mazo A. Adenovirus-mediated retinoblastoma 94 gene transfer induces human pancreatic tumor regression in a mouse xenograft model. Clin Cancer Res. 2004;10:1454–62. doi: 10.1158/1078-0432.ccr-0442-03. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Orkin SH, Walkley CR. Rb intrinsically promotes erythropoiesis by coupling cell cycle exit with mitochondrial biogenesis. Genes Dev. 2008;22:463–75. doi: 10.1101/gad.1627208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60:3689–95. [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Díaz-Rodríguez E, Teruya-Feldstein J, Cordón-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spasi MR, Callaerts P, Norga KK. Drosophila alicorn is a neuronal maintenance factor protecting against activity-induced retinal degeneration. J Neurosci. 2008;28:6419–29. doi: 10.1523/JNEUROSCI.1646-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolzing A, Coleman N, Scutt A. Glucose-induced replicative senescence in mesenchymal stem cells. Rejuvenation Res. 2006;9:31–5. doi: 10.1089/rej.2006.9.31. [DOI] [PubMed] [Google Scholar]
- Tschäpe JA, Hammerschmied C, Mühlig-Versen M, Athenstaedt K, Daum G, Kretzschmar D. The neurodegeneration mutant löchrig interferes with cholesterol homeostasis and Appl processing. EMBO J. 2002;21:6367–76. doi: 10.1093/emboj/cdf636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent O, Townley R, Kuchin S, Carlson M. Subcellular localization of the Snf1 kinase is regulated by specific beta subunits and a novel glucose signalling mechanism. Genes Dev. 2001;15:1104–14. doi: 10.1101/gad.879301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent MF, Bontemps F, Van den Berghe G. Inhibition of glycolysis by 5-amino-4-imidazolecarboxamide riboside in isolated rat hepatocytes. Biochem, J. 1992;281:267–72. doi: 10.1042/bj2810267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent MF, Marangos PJ, Gruber HE, Van den Berghe G. Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes. 1991;40:1259–66. doi: 10.2337/diab.40.10.1259. [DOI] [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jørgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–8. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub SJ, Chow KN, Luo RX, Zhang SH, He S, Dean DC. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature. 1995;375:812–5. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]
- Wikenheiser-Brokamp KA. Rb family proteins differentially regulate distinct cell lineages during epithelial development. Development. 2004;131:4299–310. doi: 10.1242/dev.01232. [DOI] [PubMed] [Google Scholar]
- Xue B, Kahn BB. AMPK integrates nutrient and hormonal signals to regulate food intake and energy balance through effects in the hypothalamus and peripheral tissues. J Physiol. 2006;574:73–83. doi: 10.1113/jphysiol.2006.113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen A, Sturgill R. Hypophosphorylation of the RB protein in S and G2 as well as G1 during growth arrest. Exp Cell Res. 1998;241:324–31. doi: 10.1006/excr.1998.4007. [DOI] [PubMed] [Google Scholar]
- Yun H, Lee M, Kim SS, Ha J. Glucose deprivation increases mRNA stability of vascular endothelial growth factor through activation of AMP-activated protein kinase in DU145 prostate carcinoma. J Biol Chem. 2005;280:9963–72. doi: 10.1074/jbc.M412994200. [DOI] [PubMed] [Google Scholar]
- Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738–46. doi: 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.