

Abstract
The type of immune response induced by a vaccine is a critical factor that determines its effectiveness in preventing infection or disease. Inactivated and live rabies virus (RV) vaccine strains elicit an IgG1-biased and IgG1/IgG2a-balanced antibody response, respectively. However, IgG2a antibodies are potent inducers of anti-viral effector functions, and therefore, a viral vaccine vector that can elicit an IgG2a-biased antibody response may be more effective against RV infection. Here we describe the humoral immune response of a live replication-deficient phosphoprotein (P)-deleted RV vector (SPBN-ΔP), or a recombinant P-deleted virus that expresses two copies of the RV glycoprotein (G) gene (SPBN-ΔP-RVG), and compare it to a UV-inactivated RV. Mice inoculated with UV-inactivated RV induced predominantly an IgG1-specific antibody response, while live recombinant SPBN-ΔP exhibited a mixed IgG1/IgG2a antibody response, which is consistent with the isotype profiles from the replication-competent parental viruses. Survivorship in mice after pathogenic RV challenge indicates a ten-fold higher efficiency of live SPBN-ΔP compared to UV-inactivated SPBN-ΔP. In addition, SPBN-ΔP-RVG induced a more rapid and robust IgG2a response that protected mice more effectively than SPBN-ΔP. Of note, 103 ffu of SPBN-ΔP-RVG induced anti-RV antibodies that were 100% protective in mice against pathogenic RV challenge. The increased immune response was directed not only against RV G but also against the ribonucleoprotein (RNP), indicating that the expression of two RV G genes from SPBN-ΔP-RVG enhances the immune response to other RV antigens as well. In addition, Rag2 mice inoculated intramuscularly with 105 ffu/mouse of SPBN-ΔP showed no clinical signs of rabies, and no viral RNA was detected in the spinal cord or brain of inoculated mice. Therefore, the safety of the P-deleted vectors along with the onset and magnitude of the IgG2a-induced immune response by SPBN-ΔP-RVG indicate that this vector holds great promise as either a therapeutic or preventative vaccine against RV or other infectious diseases.
Keywords: rabies virus, replication-deficient, viral vector, isotypes, antibody subclass, vaccine, phosphoprotein, post-exposure prophylaxis
Introduction
The development of vaccines against a wide variety of infectious diseases is one of the greatest accomplishments of the scientific community. However, the World Health Organization (WHO) and the Global Alliance for Vaccines and Immunizations (GAVI) report that almost 27 million children worldwide do not receive vaccines. Due to cost, complicated vaccine strategies and lack of availability, almost two million deaths occur annually from otherwise preventable diseases (WHO Fact Sheet, No 169). For example, current pre- or post-exposure rabies virus (RV) vaccine regimens are highly effective in the prevention of human rabies infections, if administered in a timely and appropriate manner. Nonetheless, WHO estimates that the annual number of deaths worldwide caused by RV is between 40,000 to 70,000, and an estimated 10 million people receive post-exposure prophylaxis (PEP) after exposure to potentially infected animals. In addition, the financial cost of rabies prevention is prohibitively expensive for much of the world; the cost of rabies prevention in Africa and Asia alone is almost $600 million dollars per year (1). Therefore, alternative RV vaccine strategies are needed that are affordable, effective, safe and simple to administer,.
We have shown that live, highly attenuated recombinant RV-based vectors are safe and immunogenic in mice (2) and non-human primates [(3), and WHO (Report of the Fourth W.H.O. Consultation on Oral Immunization of Dogs Against Rabies [W.H.O./Rab.Res./93.42], 1993)], which might indicate their potential use as human RV vaccines. However, as with any viral vector used as a vaccine, including those currently licensed for use in humans (4), residual vector-associated pathogenicity is a concern. For RV, a wide array of variants exist, ranging from highly pathogenic strains to attenuated RV vaccine strains such as the molecular clone SAD B19 (5). However, since even SAD B19 is pathogenic when inoculated directly into mouse brains, further efforts to attenuate the virus are necessary. One promising alternative is the use of replication-deficient viral vectors that lack an essential gene(s), which renders the vector unable to complete its viral life cycle. However, there is often a trade-off between diminished immunogenicity for increased safety, and the development of replication-deficient viral vectors that are safe and yet retain potent and protective immune responses is desirable and would greatly enhance their utility as vaccine vectors.
To that end, we have developed replication-deficient RV-based recombinant vaccine vectors in which the P gene has been deleted (SPBN-ΔP). We have also produced SPBN-ΔP that expresses two copies of the RV glycoprotein (G) gene (SPBN-ΔP-RVG). The RV P serves as a nonenzymatic cofactor and regulator protein for the RV polymerase protein (L), and interacts with viral and cellular proteins to aid in viral replication (6, 7). It also serves as a type-1 interferon (IFN) antagonist (8). A P-deleted RV was previously shown to be immunogenic in mice and provided protection against pathogenic RV challenge in a pre-exposure setting (9, 10); however, the type of immunity induced by the P-deleted replication-deficient RV vectors has not be previously characterized, nor has the issue been raised as to whether there might be alternative antibody responses that provide immunity that is superior to the current RV vaccines. Our goal is to develop a new and improved vaccine strategy that can be used in both pre- and post-exposure settings against RV. In order for a vaccine vector to be considered a viable alternative to conventional vaccines for post-exposure treatment (PET), it must induce a rapid immune response to limit infection and/or prevent disease. Therefore, we sought to quantitatively increase the onset and magnitude of the induced immune response and/or qualitatively modulate the type of immune response that is induced.
Several other rabies vaccine strategies are being tested to improve immunizations of humans in both pre- and post exposure settings. These involve heterologous recombinant viruses (e.g., vaccinia virus and adenoviruses) (11-17) that express RV glycoprotein (G), or DNA or subunit protein vaccines containing RV G or nucleoprotein (N) (18-24). Due to safety concerns, replication-deficient vectors are being tested as potential RV vaccines and replacements for replication-competent vaccine vectors. For example, capripoxvirus, which is replication-deficient in non-ruminants was found to protect mice from an intracranial RV challenge (25). Replication-deficient adenoviruses are also being tested for their ability to produce RV G-specific virus neutralization capabilities and protection against pathogenic virus challenge in mice. Positive results have been shown with human adenovirus (HuAd5) (17), although pre-existing immunity to the vector itself may limit its usefulness. However, an adenovirus that was isolated from a chimpanzee and made replication-deficient by deleteing the E1 gene was developed as a vaccine to circuvent preexisting immunity to huAd5 in humans. The chimpanzee adenovirus, which expresses RV G has also been shown to protect mice against RV challenge (26).
The promising results regarding immunogenicity described above for other replication-deficient vectors support the use of replication-deficient RV-based vectors as potentially immunogenic RV vaccines. Here we also tested a mechanism that could potentially increase both the speed of onset and magnitude of the anti-RV immune response induced by a replication-deficient RV-based vaccine vector. It was previously shown that the expression of two copies of the RV G gene from a replication-competent RV vector significantly increases anti-RV-specific immune responses (27). Our results here show that the expression of two copies of the RV G gene from replication-deficient RV vaccine vector increases the speed of onset, quantity and quality of the induced humoral immune response compared to the replication-deficient vector that expresses only a single copy of the RV G gene, and provides for a highly effective replication-deficient vaccine vector that requires only minimal vaccines doses for protection against pathogenic RV challenge.
Materials and Methods
Plasmid construction
Using pSPBN, which was derived from the SAD-B19 vaccine strain (28), as our template, a 432 bp fragment corresponding to the C-terminus of N to the stop codon in the N gene was amplified (PCR#1) using plus primer RP300 5’-TTTCACGTGTTCAA TCTCATTCAC-3’ (PmlI, underlined) and minus primer RP301 5’AAAACGCGTTTATG AGTCACTCGAATATGTC-3’ (MluI, underlined; RV N gene stop codon bold). A 900 base pair fragment corresponding to the transcriptional stop/start sequence between RV P and matrix protein (M), including sequences from RV M was amplified (PCR #2) using plus primer RP302 5’-TTTACGCGTCCGAACC TCTCCCCTCAG-3’ (MluI, underlined) and minus primer RP303 5’-AAACCCGGGGTCTTTTGAGGG-3’ (XmaI, underlined). The PCR products from PCR #1 and #2 were each digested with MluI and ligated; the ligation product was used as the template for an additional PCR (PCR #3) using plus primer RP300 and minus primer RP303. PCR #3 product (1.3 kb) was digested with PmlI and XmaI and inserted into pSPBN also digested with PmlI and XmaI. The resulting plasmid was named pSPBN-ΔP. The integrity of the gene-deleted status was verified by direct sequencing. pSPBN-ΔP-RVG was constructed by digesting pSPBN-RVG, which contains one copy of the wild-type RV G gene and one copy of the RV G-333 gene (2, 29) and flanked by the two restriction sites PacI and NheI, with PacI and NheI and inserting the two RV G genes into pSPBN-ΔP also digested with PacI and NheI, resulting in pSPBN-ΔP-RVG. The sequences encoding the RV G genes were confirmed by direct sequence analysis.
Generation of a stable cell line expressing RV P
BSR cells (a baby hamster cell line) stably expressing RV P were developed using the inducible Tet-off system as described by the manufacturer (Clonetech). Briefly, BSR cells were transfected with the pTet-Off regulator plasmid along with pTRE2-RV-P by the calcium phosphate method. pTRE2-RV-P was constructed by PCR amplification of the RV P gene from pSPBN using plus primer 5’-TTTGCTAGCAACATGAGCAAGATCTTTGTC-3’ (NheI, underlined) and minus primer 5’-AAAGCGGCCGCTTAGCAAGA TGTATAGCGATTC-3’ (NotI, underlined). The PCR product was digested with NheI/NotI and ligated to pTRE2 also digested with NheI/NotI. After transfection, cells were selected with 400mg/ml G418 and treated with 1 mg/ml tetracycline to keep RV P transcription turned off. Single cell clones were isolated and immunostained with an anti-RV P antibody for the verification of expression after the removal of tetracycline. The new cell line was termed BSR-RVP.
Recovery of infectious virus
Due to the replication-deficient nature of the P-deleted viruses, the standard protocol for the recovery of infectious recombinant virus (30) needed to be modified. Briefly, BSR cells stably expressing bacteriophage T7 RNA polymerase were transfected with four different plasmids, three plasmids encoding the RV viral proteins (N, P, and L) and one encoding the anti-genomic RNA of SPBN-ΔP or SPBN-ΔP-RVG. Three days post-transfection, supernatant from transfected cells were transferred to BSR-RVP cells. Recovered virus was confirmed by immunostaining with a FITC-conjugated anti RV N antibody (31, 32). RNA from virus stock was isolated and converted to DNA by RT-PCR. The deletion within SPBN-ΔP and SPBN-ΔP-RVG was confirmed by direct sequencing.
Flow cytometry
Expression of RV G on the surface of cells infected with SPBN, SPBN-ΔP and SPBN-ΔP-RVG was analyzed by FACS analysis as described previously (27). Briefly, NA cells were infected with recombinant RVs at a MOI of 3 and incubated for 24 or 48 h at 37°C. Cells were suspended in Cell Stripper (Mediatech), pelleted at 300 × g for 5 min, resuspended in 100 μl of blocking solution (1%BSA, 10 mM glycine in PBS), and fixed in suspension by addition of 100 μl of Cytofix solution (BD Bioscience). After 20 minutes, cells were washed twice with blocking solution and incubated with rabbit anti-RV G antiserum (1:2000) followed by a Cy-2-conjugated affinity-purified goat anti-rabbit antibody (1:500; Jackson ImmunoResearch Laboratories Inc., West Grove, Pa.). Flow cytometry was performed on an EPICS profile analyzer.
Immunization of mice and pathogenic challenge
Groups of 6- to 8-week-old female BALB/c mice were inoculated intramuscularly (i.m.) with different concentrations of the P-deleted vectors, as described in the Figures and Figure Legends. For the UV inactivated vaccine, a single lot of SPBN-ΔP was divided into two parts. One part was subjected to UV irradiation for ten minutes to inactivate the virus, and one part was not UV-inactivated. This helped to ensure virus input, and glycoprotein quantity, were similar. Virus inactivation was confirmed by inoculating an aliquot of UV-treated virus on BSR cells and immunostaining for the RV nucleoprotein 48 hours post-inoculation. Treated and non-treated viruses were diluted in PBS to the appropriate concentrations for immunization. Four to six weeks post-immunization, mice were challenged i.m. with 100 LD50 pathogenic Challenge Virus Strain (CVS)-N2c, which is a mouse-adapted subclone of CVS-24 RV (33), and observed for at least three weeks for clinical signs of rabies. Mice were euthanized at the onset of neurological symptoms.
Antibody ELISAs
ELISA plates (96-well) were coated with 100 ng/well RV G or RNP in coating buffer (5 mM Na2CO3, pH 9.6) overnight at 4 °C. Plates were washed four times in PBS-tween and blocked with 5% low-fat milk in PBS for 1 h at room temperature. Serum samples (100 μl) diluted in PBS (1:50) were added to wells, serially diluted 1:3, and incubated for 1 h at room temperature. After washing the plates 4 times in PBS-tween, 100 μl HRP-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch, Inc.) was added per well and incubation continued at 37 °C for 30 min. Plates were washed four times with PBS-tween before o-phenylenediamine dihydrochloride (OPD) substrate, prepared according to the manufacturer’s instructions (Sigma, Inc.), was added. Incubation was continued for 30 min at room temperature in the dark and the reaction was stopped by the addition of 2 M H2SO4. Optical density at 490 nm was determined. Gamma-globulin subclass-specific ELISAs were performed as described above using secondary antibodies IgG1 (1:2000) or IgG2a (1:2000) (Southern Biotechnology, Inc.) (34).
Virus Neutralization Assays
Sera collected from inoculated mice were heat inactivated at 56° C for 30 minutes to inactivate complement. The Rapid Fluorescent Focus Inhibition Test (RFFIT) was used to determine RV antibody neutralization titers. Serial three-fold dilutions of the serum were made in 96-well plates and 100 FFD50 (focus forming dose) CVS-11 was added to each well. Serial dilutions of the World Health Organization reference standard serum were analyzed in parallel. Plates were incubated for one hour at 37° C. Virus plus serum mixture (100 μl) was transferred to a 96-well plate previously seeded with NA cells (neuroblastoma cells) and incubated 34°C for 48 hours. Infectious virus was determined by immunostaining with a FITC-conjugated anti-RV N antibody. Neutralization titers, defined as the inverse of the highest serum dilution that neutralizes 50% of the challenge virus, were normalized to international units/ml (IU/ml) using the WHO anti-RV antibody reference standard.
Safety and spread of P-deleted RV in Rag2 KO mice
We selected B- and T-cell deficient mice (Rag2) as our model for pathogenicity and viral spread since this would be the most stringent test for the ability of the vector to spread to the CNS. Groups of five 6- to 8-week-old RAG2 KO mice (Taconic Farms, Germantown, NY) were infected i.m. with 105 focus-forming units (ffu) of SPBN, SPBN-333 or SPBN-ΔP in the gastrocnemius muscle. SPBN, the parental vaccine strain, has been shown to migrate to and be pathogenic in the CNS of Rag2 mice and served as a positive control. SPBN-333 (2) is a highly attenuated vaccine strain that contains a single amino acid exchange at position 333 of RV G. SPBN-333 is apathogenic after direct intracranial inoculation of immune-competent mice, yet it retains residual pathogenicity in Rag2 mice after a peripheral inoculation and served as a positive control. Clinical signs of rabies from mice inoculated with SPBN-333 were used as an indicator for when to collect samples from SPBN-ΔP-inoculated mice since the possibility existed that this vector would not cause disease even in severely immuno-deficient mice. Mice were monitored daily for weight changes and signs of rabies. At either 12 days post-infection (dpi) (SPBN) or 24 dpi (SPBN-333 and SPBN-ΔP), mice were euthanized and the muscle, brain, and spinal cord were harvested. Total RNA was isolated from the tissues, and cDNA synthesis and quantitative real-time PCR were performed as previously described (35).
Results
Construction of SPBN-ΔP
SPBN-ΔP was constructed using a PCR strategy and standard cloning techniques (Figure 1), and infectious virus was recovered, as described in the Materials and Methods section. Supernatant from SPBN-ΔP recoveries were transferred to BSR cells stably expressing RV P (BSR-RVP). Recovered virus was confirmed by immunostaining with a FITC-conjugated anti RV N antibody (31, 32). Virus titers greater than 107 ffu/ml were obtained (data not shown). RNA from viral stocks was isolated, and the gene-deletion status was confirmed by direct sequencing of the RT-PCR product.
Figure 1.
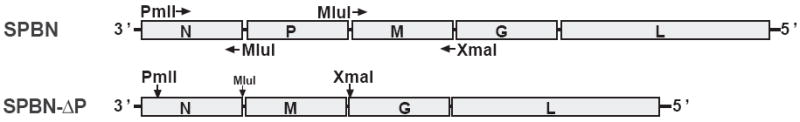
Construction of SPBN-ΔP. The P gene was deleted from SPBN (28) (top), which is based on the attenuated SAD-B19 RV vaccine strain, using a PCR strategy and standard cloning techniques. Infectious virus was recovered and propagated on BSR cells stably expressing RV P, as described in Materials and Methods
Immunogenicity and protective capacity of SPBN-ΔP virus
Mice immunized with live SPBN-ΔP induced higher antibody responses against RVG and RNP than the UV-inactivated virus (36-38) at doses of 105 (RVG p=0.0176; RNP p=0.0007) and 104 (RVG p=0.0007; RNP p<0.0001) ffu/mouse (Figure 2A). At a dose of 106 ffu/mouse, the response was similar (RV G) or higher (RNP p=<0.0001) in the case of the UV-inactivated virus. However, mice immunized with 105 or 104 ffu of live SPBN-ΔP showed statistically higher VNA responses than from mice immunized with the same dose of UV-inactivated virus (105 p<0.0001; 104 p<0.0001) (Figure 2B). No VNA responses were detected in mice immunized with 103 ffu/mouse of either live or killed vaccine. Consistent with the VNA titers, all mice that received 105 ffu/mouse of live SPBN-ΔP survived after pathogenic virus challenge with 100 LD50 of CVS-N2c 4 weeks post-immunization (Figure 2C). Ninety percent (90%) of those mice that received 104 ffu/mouse also survived pathogenic RV challenge. This compares with only a 70% or 30% survival rate after receiving 105 or 104 ffu/mouse of the UV-inactivated vaccine, respectively. The calculated ED50 indicates a ten-fold higher efficiency of live versus UV-inactivated SPBN-ΔP (Figure 2D).
Figure 2.
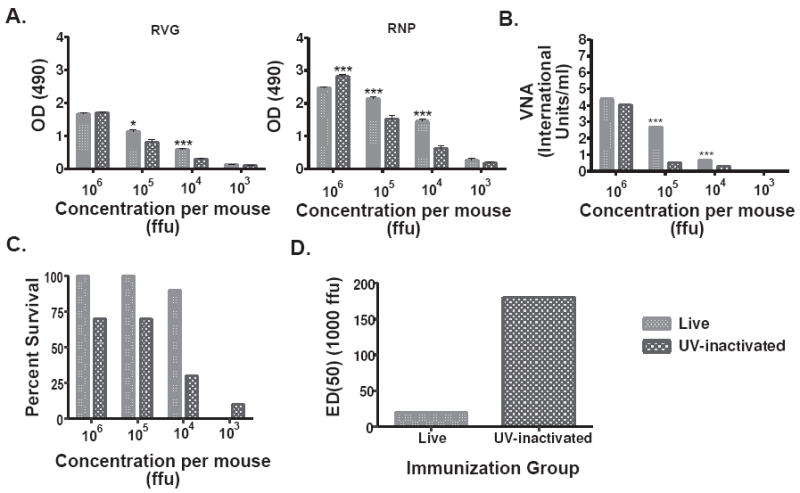
Live SPBN-ΔP induces high levels of anti-RV G and anti-RNP antibodies, and protects mice more effectively against pathogenic RV challenge than UV-inactivated RV. Groups of 10 Balb/c mice were immunized intramuscularly (i.m.) with the indicated doses of live or UV-inactivated SPBN-ΔP and sera from mice 14 days post-immunization were tested for total IgG antibodies against RV G or RNP by ELISA at a 1:50 sera dilution (A), or virus neutralizing antibodies (VNA) expressed in international units/ml (IU/ml) (B). The data represents the average and standard error of the mean (SEM) of n=10 completed in duplicate and analyzed using the unpaired t-test, 95% confidence (see the text for p values). Four weeks post-immunization, the mice were challenged i.m. with 100 LD50 of pathogenic Challenge Virus Strain (CVS)-N2c and observed for three weeks and their survivorship recorded (C). The ED50 was calculated using the survivorship rates between the two groups of immunized animals (D) and indicates a 10-fold higher efficiency of live SPBN-ΔP versus UV-inactivated SPBN-ΔP. (***p<0.001; **p=0.001-0.01; *p=0.01-0.05)
We suspected that the increased protection, however, was not solely explained by the magnitude of the induced immune response and that other factors were involved. This was based partly on the finding that mice inoculated with 106 ffu/mouse of live or UV-inactivated SPBN-ΔP induced similar antibody responses (Figure 2A and 2B) but showed differences in their ability to protect mice against pathogenic RV challenge (Figure 2C and 2D). Inasmuch as inactivated vaccines generally induce an IgG1-biased antibody response, live viral vectors can induce both IgG1 and IgG2a antibodies (39). Mice immunized with the 104 ffu/mouse, which was the dose that induced the largest difference in antibody titers and protection between live and UV-inactivated SPBN-ΔP, showed significantly higher anti-RV G IgG2a antibodies in the case of live SPBN-ΔP (Figure 3A). The subclass profile was antigen-independent since it was observed for both RV G (p<0.0001) and RNP (p<0.0001). In addition, mice immunized with 106 ffu/mouse of live or inactivated SPBN-ΔP, which was the dose that induced similar antibody responses between the two groups but different levels of protection, also showed a significantly higher IgG2a antibody titers against both RVG (p<0.0001) and RNP (p<0.0001) (Figure 3B). Of note, except for mice that received 104 ffu/mouse of the inactivated vaccine that showed a significant increase in the IgG1 antibody titers compared with the live vaccine (p=0.0032), the IgG1 antibody responses were largely unaffected by the type of vaccine used. Taken together, these data suggest that live SPBN-ΔP induces an immune response that provides the benefits of a live virus vaccine and that the induction of higher IgG2a antibodies, or a mixed IgG2a/IgG1 response, is likely more beneficial in RV vaccine-induced protection.
Figure 3.
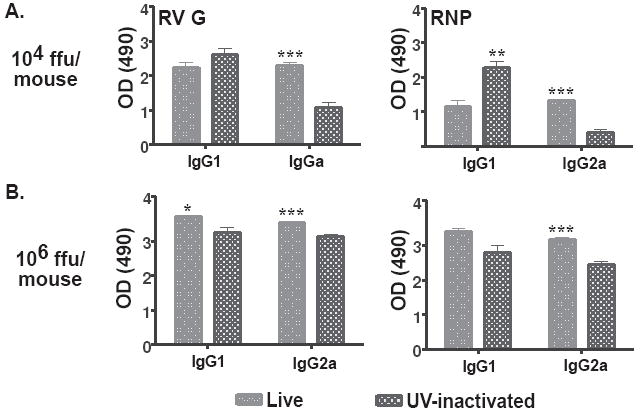
Live SPBN-ΔP induces a mixed IgG1/IgG2a antibody titer compared with an IgG1-biased response from UV-inactivated RV. Fourteen days post-immunization, sera from mice that received either 104 (A) or 106 ffu (B) of live SPBN-ΔP or UV-inactivated RV, as described in Figure 2, were tested by ELISA for anti-RV G (left) or anti-RNP (right) antibodies at a 1:50 dilution. The data shows an increase in IgG2a-specific antibody titers against both RV G and RNP at both doses used of live SPBN-ΔP compared with UV-inactivated RV. The data represents the average and SEM of n=10 completed in duplicate and analyzed using the unpaired t-test, 95% confidence (p values provided in the text; ***p<0.001; **p=0.001-0.01; *p=0.01-0.05).
Construction, immunogenicity and protective capacity of SPBN-ΔP-RVG
The expression of two RV G genes from a replication-competent RV genome induces apoptosis that results in increased anti-RV immunity (27, 40). To test whether this is a viable strategy to increase immune responses from replication-deficient RVs, we constructed and recovered a P-deleted virus that expresses two RV G genes (Figure 4 and Material and Methods). Higher levels of anti-RV G (Figure 5A) and anti-RNP (Figure 5B) antibodies were detected as early as five days post-infection from mice immunized with SPBN-ΔP-RVG when compared with SPBN-ΔP at doses of 105 (RVG p<0.0001; RNP p=0.0008) and 104 (RVG p<0.0001; RNP p=0.0132) ffu/mouse. By day 7 or 14, mice that received only 103 ffu/mouse of SPBN-ΔP-RVG showed higher levels of anti-RV G or anti-RNP antibodies, respectively, compared with mice that received SPBN-ΔP (RVG p=0.0002; RNP p<0.0001). This finding continues through the last time point of 28 days post-inoculation, when almost a 10-fold increase in anti-RVG antibodies was observed, or a 5-fold increase in anti-RNP antibodies, in mice immunized with 103 ffu/mouse of SPBN-ΔP-RVG compared with those immunized with SPBN-ΔP (RVG p<0.0001; RNP p<0.0001). Since an increase in anti-RNP antibodies was also observed, the increase in immunogenicity induced by SPBN-ΔP-RVG was not presumably a result solely of the increase in RV G expression, and it could have been due to other factors (please see Discussion).
Figure 4.
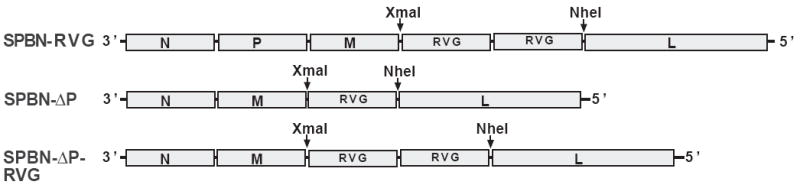
Construction of SPBN-ΔP-RVG. Two copies of the RV G gene were digested from SPBN-RVG (top) using the two unique restriction sites, XmaI and NheI. The two RV G genes were inserted into SPBN-ΔP (middle) also digested with XmaI and NheI, resulting in SPBN-ΔP-RVG (bottom).
Figure 5.
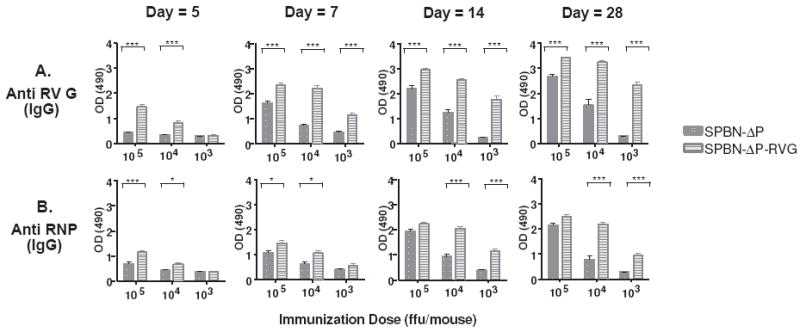
SPBN-ΔP-RVG induces rapid and potent anti-RV G and anti-RNP antibodies. Groups of five Balb/c mice were immunized i.m. with 105, 104,, or 103 ffu of SPBN-ΔP or SPBN-ΔP-RVG and sera was collected on days 5, 7, and 14 post-immunization (Experiment #1). The experiment was repeated with an additional five mice, except in this experiment, sera were collected on days 5, 7, 14 and 28 post-immunization. At each time point, sera were tested for total IgG by ELISA for anti-RV G (A) or anti-RV RNP (B) antibodies. The data for days 5, 7, and 14 represent the average and SEM for both Experiments #1 and #2 (n=10) completed in duplicate and for day 28 from Experiment #2 (n=5) also completed in duplicate. The data were analyzed using the unpaired t-test, 95% confidence (see the text for p values; ***p<0.001; **p=0.001-0.01; *p=0.01-0.05).
Consistent with the antibodies detected above by ELISA, the kinetics of the VNA responses showed that mice immunized with SPBN-ΔP-RVG induced VNA titers as high as 0.8 IU/ml within 5 days post-inoculation with 105 ffu/mouse (Figure 6A). This exceeds the presumptive protective level of 0.5 IU/ml. By day 14, the response peaked at 19.5 IU/ml before trailing off at day 28 to 9.0 IU/ml. Only 103 ffu/mouse of SPBN-ΔP-RVG was needed to induce protective VNA titers by day 14 (0.5 IU/ml) and day 28 (1.2 IU/ml). Lower VNA titers were observed in mice immunized with SPBN-ΔP at all doses and time points tested compared with those from the SPBN-ΔP-RVG-immunized mice (Figure 6A). The higher VNA titers from SPBN-ΔP-RVG-immunized mice equated to increased survivorship after pathogenic challenge (Figure 6B). Between 60-100% of the mice immunized with 103 ffu of SPBN-ΔP-RVG survived pathogenic CVS-N2c challenge 4-6 weeks post-immunization, while none of the mice that received equivalent doses of SPBN-ΔP survived. Of note, at 5 days post-challenge, a rapid and robust VNA recall response was observed for mice immunized with SPBN-ΔP-RVG, generating neutralization titers between 11 and 33 times greater than pre-challenge levels (Figure 6C). In the case of mice immunized with only 103 ffu of SPBN-ΔP-RVG, VNA titers increased over 150-fold by day 35 post-challenge compared with pre-challenge data. Therefore, only low doses of SPBN-ΔP-RVG are needed to induce rapid and potent anti-RV antibody responses, which indicates its potential as a preventative or therapeutic vaccine against RV.
Figure 6.
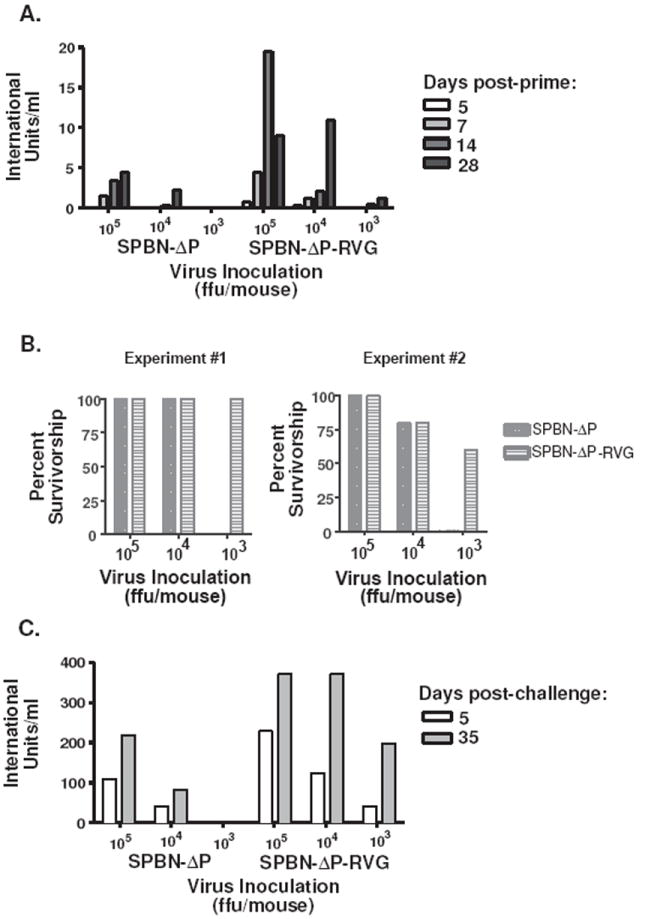
SPBN-ΔP-RVG induces strong virus neutralization antibody titers and provides protection against pathogenic RV challenge with as little as 103 ffu/mouse. The mice described in Figure 5 were challenged i.m. 4 (Experiment #1) or 6 (Experiment #2) weeks post-immunization with 100 LD50 pathogenic CVS-N2c. Pre-challenge virus neutralizing antibody titers, expressed in international units/ml, are shown (A), and survivorship was recorded (B). Virus neutralizing antibodies were also determined from all mice on day 5 post-challenge and from mice that survived at day 35 post-challenge.
SPBN-ΔP-RVG induces highly effective anti-viral IgG2a antibodies
The potential existed that SPBN-ΔP-RVG was modulating the quality of the induced antibody response when compared with SPBN-ΔP. Indeed, mice immunized with SPBN-ΔP-RVG showed higher IgG2a-specific antibodies with 105 (p=.1502), 104 (p<0.0001) or 103 (p<0.0001) ffu/mouse compared with those mice that received SPBN-ΔP (Figures 7A and 7C). Conversely, mice immunized with SPBN-ΔP showed slightly higher IgG1 antibody response compared with those mice immunized with SPBN-ΔP-RVG at doses of 105 (Figures 7A and 7C). These differences were not significant. At a dose of 104/mouse, the difference between the two groups was significant (p=0.0093). Only low levels of IgG1 were detected from mice immunized with SPBN-ΔP-RVG. The immune response induced by the prime inoculation largely affected the immune response post-challenge (Figure 7B and 7D). Of note, it was previously reported that an increase in the expression of RV G on the surface of cells infected with attenuated vaccine strains of RV induces apoptosis that results in increased immunity (27). However, the expression level of RV G on the surface of cells infected with SPBN-ΔP-RVG is lower than on cells infected with SPBN (Figure 8), indicating that factors other than expression level might be involved in the increased immunity induced by SPBN-ΔP-RVG.
Figure 7.
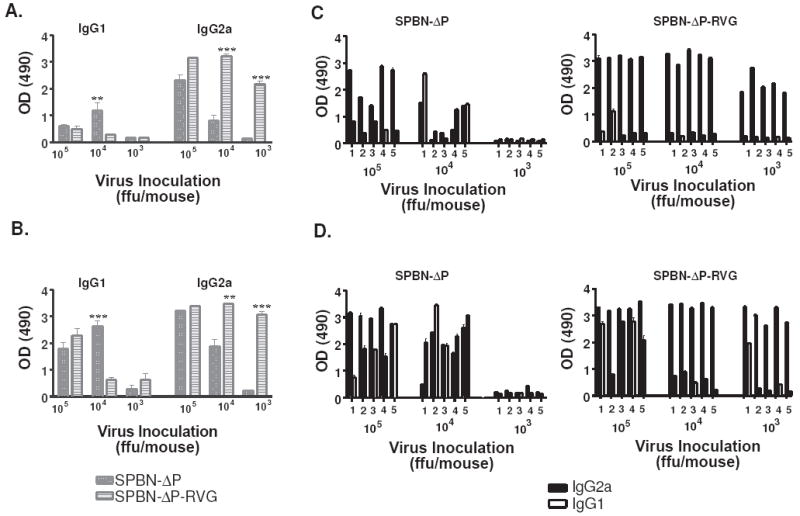
SPBN-ΔP-RVG induces an IgG2a-biased antibody response, which is amplified post-challenge. Sera from mice collected 14 days post-immunization (A) or 5 days post-challenge (B) were analyzed by ELISA for IgG1- or IgG2a-specific antibodies against RV G at a sera dilution of 1:50. The average and SEM antibody results from each group of mice (n=5) (A and B) or from individual mice (C and D) are shown. Data were analyzed using the unpaired t-test, 95% confidence (see the text for p values; (***p<0.001; **p=0.001-0.01; *p=0.01-0.05).
Figure 8.
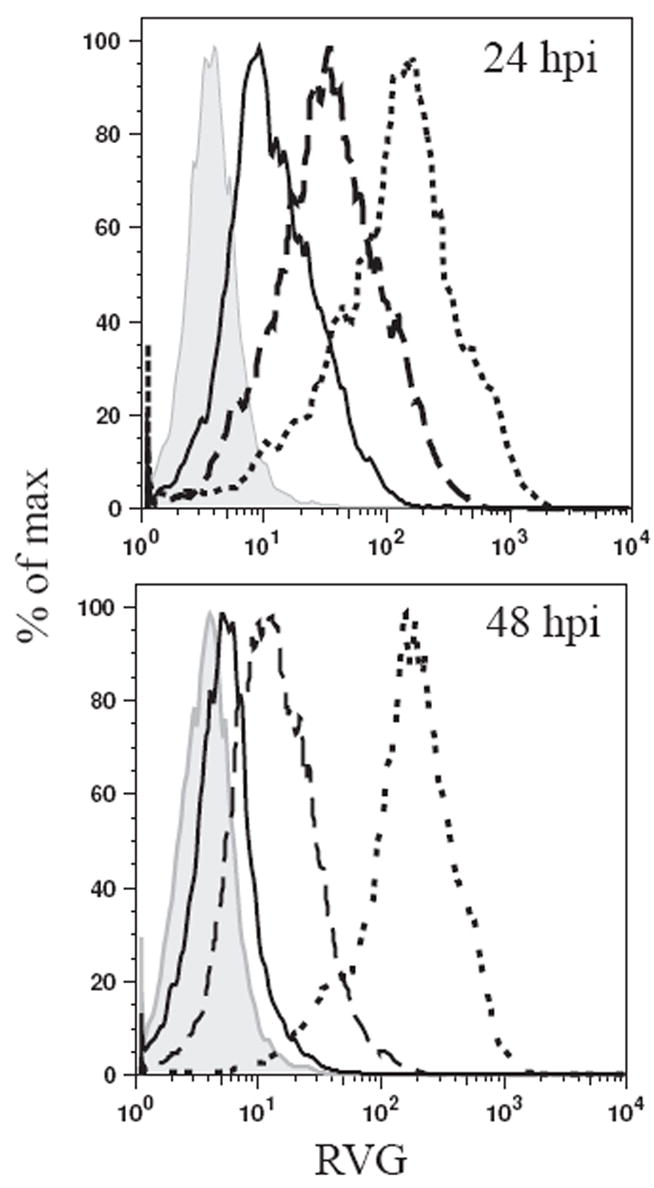
Input RV P is sufficient for cell surface expression of RV G. NA cells were infected with SPBN (dotted line), SPBN-ΔP (solid line) or SPBN-ΔP-RVG (dashed line) at an moi of 3, or left uninfected (solid histogram). At 24 (top) and 48 h p.i. (bottom), cells were incubated with a polyclonal rabbit anti-RV G antiserum, followed by a Cy-2-conjugated anti-rabbit antibody. Surface expression was determined by flow cytometry, and fluorescence intensity was plotted.
Safety and spread SPBN-ΔP virus in Rag2 immune-deficient mice
Next, we wanted to determine whether a P-deleted RV vector has the potential to reach the CNS regardless of clinical outcome. Rag2 mice inoculated with SPBN or SPBN-333, as controls, started to loose weight around day 8 and 17, respectively. SPBN- and SPBN-333-inoculated mice were euthanized on day 12 and 24 post-infection, respectively, at signs of rabies infection (Figures 9A and 9B). At day 24 post-infection, mice inoculated SPBN-ΔP were also euthanized. Mice inoculated with SPBN-ΔP did not show any signs of rabies infection, nor did any mouse from this group loose weight during this time (Figures 9C). The muscle, spinal cord and brain were collected from each animal, and total RNA was isolated from these organs. As noted in Figure 9A, viral genomic and messenger RNA were detected in the muscle, spinal cord and brain of all SPBN-infected mice 12 dpi. Genomic and messenger RNA were detected in the muscle, spinal cord and brain of the two mice that showed symptoms of rabies infection after SPBN-333 infection (Figure 9B). Two other mice inoculated with SPBN-333 showed messenger RNA in the spinal cord, but none was detected in the brain RNA. Importantly, no genomic or messenger RNA was detected in mice immunized with SPBN-ΔP at day 24 post-infection in any tissue tested. These results show that the vectors are safe for use in immune-deficient individuals, and there appears to be no CNS-involvement after peripheral infection with SPBN-ΔP.
Figure 9.
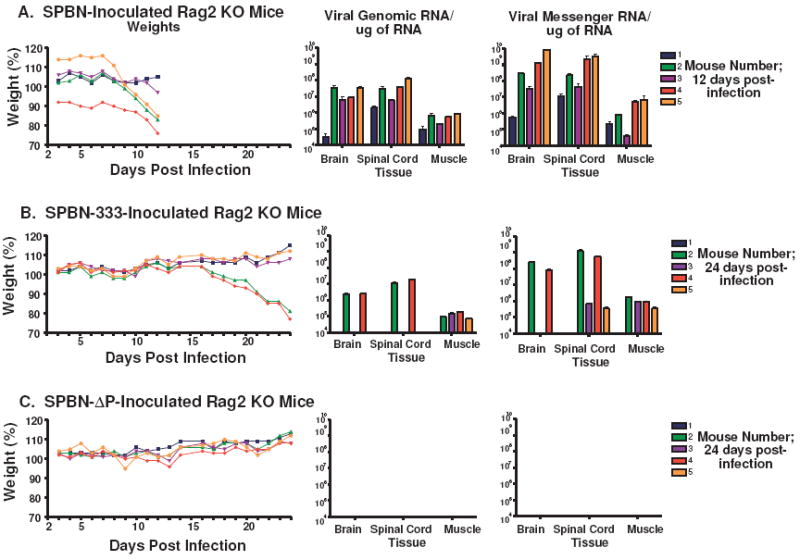
SPBN-ΔP does not spread to the spinal cord or brain of infected RAG2 KO mice. Groups of 5 RAG2 KO mice were inoculated i.m. with 105 ffu of either SPBN (A), SPBN-333 (B) or SPBN-ΔP (C) and their weights were recorded (left panel). Once one or more animal(s) in a group became moribund due to rabies infection, all mice in that group were euthanized. Mice inoculated with the highly attenuated SPBN-333 vector served as a guide for the time point to collect samples from mice inoculated with SPBN-ΔP since it was not known whether SPBN-ΔP would induce visible signs of rabies infection. Total RNA was isolated from muscle, brain and spinal cord and viral RT-PCR and quantitative real-time PCR was used to amplify and quantify viral genomic (middle panels) or messenger (right panels) RNA from individual samples.
Discussion
We previously described several highly attenuated RV-based vaccine vehicles where we genetically eliminated single and multiple pathogenic markers (2). Data from these studies indicate that very safe and effective RV-based vaccine vectors can be generated. Nonetheless, there are concerns for the use of even highly attenuated live replication-competent viral vaccines in humans. One alternative is the use of replication-deficient RV vectors that lack one of its five essential structural genes. Such replication-deficient viral vectors hold great promise as potential vaccines against a wide variety of infectious diseases and cancer due to safety concerns. Nonetheless, it is expected that even more potent immune responses will be required to simplify current vaccine regimens or for the development of new vaccines for infectious diseases where current vaccines are not available.
While the magnitude of a specific immune response is generally regarded as beneficial when developing a new vaccine, qualitative attributes also appear to be important. This was exemplified by SPBN-ΔP and SPBN-ΔP-RVG. UV-inactivated SPBN-ΔP induced primarily an IgG1 antibody response, which resulted in only minimal protection against pathogenic RV challenge. Live SPBN-ΔP, on the other hand, induced a mixture of both IgG1 and IgG2a, which is more effective in providing protection after challenge. Based on the finding that the IgG2a antibody response increased the immunogenicity and protective ability of our vectors, we tested a vector that we speculated would modulate the immune response towards an IgG2a-dominated antibody response. To this end, we showed that the expression of two RV G genes from SPBN-ΔP-RVG induced an antibody subclass profile that was largely devoid of IgG1 but contained primarily IgG2a. In addition, a detailed analysis of the kinetics of the IgG2a-induced immune response and VNA titers showed that this vector was capable of inducing a potent immune response within 5 days post-inoculation and was observed with only 105 ffu/mouse. In addition, only 103 ffu/mouse were necessary to induce a strong humoral response within 7 days post-inoculation that protected between 60 and 100% of mice after pathogenic challenge. The effectiveness of this vaccine vector at low doses is an important feature for effective large-scale vaccine production.
There are several questions raised by the data that are currently under investigation. First, what is the underlying mechanism that modulates the immune response to induce this protective IgG2a antibody subclass? Antigen dose is a potential explanation, however, we do not see a dose-dependency with respect to the type of antibody subclass induced in mice immunized with different concentrations SPBN-ΔP-RVG. The IgG2a antibody responses were remarkably similar in mice that received 105, 104, and 103 ffu/mouse of SPBN-ΔP-RVG. In addition, RV G expression levels by SPBN-ΔP-RVG was intermediate to expression by SPBN or SPBN-ΔP, both of which induce a mixed IgG1/IgG2a antibody response. Therefore, it might be expected that if dose was responsible for the isotype profile, SPBN-ΔP-RVG would induce a mixture as well, and not the IgG2a-dominated response observed. This leads us to believe that factors other than antigen dose are involved in the immune modulating effect.
At this point, the role that the deletion of RV P may play in SPBN-ΔP-RVG’s rapid and potent IgG2a-biased immune response is unclear. RV P has recently been shown to inhibit the IFNα/β response in infected cells (8). Since P-deleted viruses are devoid of this function, the potential exists that the induced IFNα/β contributes to the immune modulating effect of SPBN-ΔP-RVG. However, the immune modulating effect observed with SPBN-ΔP-RVG was more pronounced than with SPBN-ΔP, both of which are lacking the P gene. Therefore, while IFNα/β might be contributing to the modulating effect, still other factors appear to be involved. Nonetheless, we are currently investigating the contribution IFNα/β on P-deleted virus-induced immune responses.
Another potential explanation comes from recent studies that show the induction of apoptosis from DNA vectors correlated with a modulation of the immune response towards either a Th-1 or Th2-type response, depending on the intrinsic nature of the vaccine strategy (41, 42). The induction of what is termed “bland” apoptosis (42, 43) allows for antigen expression to occur before apoptotic bodies are formed. Extensive apoptosis was found to interfere with the induced immune response, presumably since apoptosis occurs before antigen presentation. It was previously shown that over expression of RV G on the cell surface induces apoptosis and significantly increases anti-RV-specific immune responses (27). This study, and others (27, 44-49), clearly demonstrate the link between the induction of apoptosis and anti-RV immunity. However, data from Figure 8 indicate that expression level from replication-deficient vectors itself might not be sufficient to induce apoptosis, and that other factors would be involved for SPBN-ΔP-RVG.
The second question raised by our data is why an IgG2a-dominated antibody response is more effective against RV than an IgG1-biased or an IgG1/IgG2a-mixed antibody response. While many vaccines are effective by inducing an immune response that closely mimics a natural infection (50), there are cases where this is not sufficient, and alternative antibody responses might be needed (51). Vaccination of mice with live or inactivated attenuated RV induced the production of IgG1 or IgG1/IgG2a antibody responses, respectively. However, our data suggests that SPBN-ΔP-RVG, which induces an IgG2a-dominated response, is very effective. It was recently shown that an important parameter for the effectiveness of different IgG subclasses relies on binding efficiencies to either activating or inhibiting Fc receptors (FcRs) (52, 53). Antibodies provide a “bridge” that links the antigen to FcRs on phagocytes and other immune cells [for review, see (54)] and regulate antibody-mediated responses [for review, see (55)]. Except for NK cells that express only a low affinity activating FcR, other innate immune effector cells co-express both activating and inhibitory FcRs and the differential binding affinities of the antibody isotype, and subsequent signaling cascade, determines the magnitude of the induced response. With respect to mice, IgG1 antibodies bind to the inhibitory FcR, FcγRIIB, with a much higher affinity than to activating FcR, which, for IgG1, is FcγRIII (52, 53). Due to a higher binding efficiency to FcγRIIB than to FcγRIII, IgG1 provides a dampening effect that tightly controls the resulting effector functions (53). Conversely, IgG2a binds with much higher affinity to activating FcγRIV than inhibitory FcγRIIB. In addition, it appears that Th1 induction increases the expression of both IgG2a and its activating FcγRs (FcγRI and FcγRIV), which amplifies the IgG2a-mediated effector function (52). This is consistent with the findings that IgG2a antibodies are potent inducers of anti-viral effector functions. Therefore, a vaccine that can elicit potent IgG2a antibodies, while minimizing or eliminating IgG1 responses, may prove beneficial. Indeed, the data presented here indicates that this may be a strategy for an improved vaccine for RV and possibly for other infectious diseases.
The third question raised by our data is whether these vectors would make effective vaccine vectors against other infectious diseases. It was unclear at the onset of these studies whether the P-deleted viruses would be able to express sufficient antigen to induce a protective immune response. SPBN-ΔP-RVG appears to be providing a mechanism to overcome any hindrance that reduced antigen expression may place on the induced immune response. These vectors induce rapid and potent immune responses not only against RV G, which might be expected due to increased antigen expression, but also against RNP. It would appear that SPBN-ΔP-RVG is modulating the immune response that could increase immunity against a variety of antigens. This is encouraging for the use of SPBN-ΔP-RVG as a vaccine vector for delivering genes to target other infectious diseases where a potent IgG2a-biased antibody response would be beneficial.
We are currently addressing these questions experimentally. However, the rapid and potent IgG2a-dominated immune response induced by even low doses of SPBN-ΔP-RVG makes this vector a very promising pre- or post-exposure vaccine vector against RV infection for use in both developed and developing countries.
Acknowledgments
We would like to thank Dawn S. Traynor and Andrew Bonner for excellent technical assistance.
This study was supported by NIH grants AI049153 to MJS and AI070252 to JPM. JPM is also sponsored by the Infectious Disease Society of America (IDSA) and the National Foundation for Infectious Diseases (NFID) Wyeth Young Investigator Award In Vaccine Development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Knobel DL, Cleaveland S, Coleman PG, et al. Re-evaluating the burden of rabies in Africa and Asia. Bull World Health Organ. 2005;83(5):360–8. [PMC free article] [PubMed] [Google Scholar]
- 2.McGettigan JP, Pomerantz RJ, Siler CA, et al. Second-generation rabies virus-based vaccine vectors expressing human immunodeficiency virus type 1 gag have greatly reduced pathogenicity but are highly immunogenic. Journal of Virology. 2003;77(1):237–44. doi: 10.1128/JVI.77.1.237-244.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKenna PM, Koser ML, Carlson KR, et al. Highly attenuated rabies virus-based vaccine vectors expressing simian-human immunodeficiency virus89.6P Env and simian immunodeficiency virusmac239 Gag are safe in rhesus macaques and protect from an AIDS-like disease. Journal of Infectious Diseases. 2007;195(7):980–8. doi: 10.1086/512243. [DOI] [PubMed] [Google Scholar]
- 4.Pirofski LA, Casadevall A. Use of licensed vaccines for active immunization of the immunocompromised host. Clinical Microbiology Reviews. 1998;11(1):1–26. doi: 10.1128/cmr.11.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conzelmann KK, Cox JH, Schneider LG, Thiel HJ. Molecular cloning and complete nucleotide sequence of the attenuated rabies virus SAD B19. Virology. 1990;175(2):485–99. doi: 10.1016/0042-6822(90)90433-r. [DOI] [PubMed] [Google Scholar]
- 6.Dietzschold B, Rupprecht CE, Fu ZF, Koprowski H. Rhabdoviruses. In: Fields BN, editor. Fields Virology. Philadelphia, PA: Lippincott-Raven Publishers; 1996. pp. 1137–52. [Google Scholar]
- 7.Rose JK, Schubert M. Rhabdovirus genomes and their products. New York: Plenum Publishing Corp; 1987. [Google Scholar]
- 8.Brzozka K, Finke S, Conzelmann KK. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. Journal of Virology. 2005;79(12):7673–81. doi: 10.1128/JVI.79.12.7673-7681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shoji Y, Inoue S, Nakamichi K, Kurane I, Sakai T, Morimoto K. Generation and characterization of P gene-deficient rabies virus. 2004 Jan 5;:295–305. doi: 10.1016/j.virol.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Morimoto K, Shoji Y, Inoue K. Characterizatio of P gene-deficient rabies virus: propagation, pathogenicity and antigenicity. Virus Research. 2005;111:61–7. doi: 10.1016/j.virusres.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Lafon M, Lafage M, Martinez-Arends A, et al. Evidence for a viral superantigen in humans. Nature. 1992;358(6386):507–10. doi: 10.1038/358507a0. see comments. [DOI] [PubMed] [Google Scholar]
- 12.Lodmell DL, Smith JS, Esposito JJ, Ewalt LC. Cross-protection of mice against a global spectrum of rabies virus variants. Journal of Virology. 1995;69(8):4957–62. doi: 10.1128/jvi.69.8.4957-4962.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lodmell DL, Ewalt LC. Rabies vaccination: comparison of neutralizing antibody responses after priming and boosting with different combinations of DNA, inactivated virus, or recombinant vaccinia virus vaccines. Vaccine. 2000;18(22):2394–8. doi: 10.1016/s0264-410x(00)00005-0. [DOI] [PubMed] [Google Scholar]
- 14.Li WH, Zhang Y, Wang SH, Liu L, Yang F. Recombinant replication-defective adenovirus based rabies vaccine. Chung Kuo i Hsueh Ko Hsueh Yuan Hsueh Pao Acta Academiae Medicinae Sinicae. 2003;25(6):650–4. [PubMed] [Google Scholar]
- 15.Lees CY, Briggs DJ, Wu X, et al. Induction of protective immunity by topic application of a recombinant adenovirus expressing rabies virus glycoprotein. Veterinary Microbiology. 2002;85(4):295–303. doi: 10.1016/s0378-1135(01)00523-5. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Zhang Y, Wang S. Immune response of mice to replication-defective recombinant adenovirus containing glycoprotein gene of rabies virus 3aG strain. Chinese Journal of Experimental & Clinical Virology. 2001;15(1):61–5. [PubMed] [Google Scholar]
- 17.Vos A, Neubert A, Pommerening E, et al. Immunogenicity of an E1-deleted recombinant human adenovirus against rabies by different routes of administration. Journal of General Virology. 2001;82(Pt 9):2191–7. doi: 10.1099/0022-1317-82-9-2191. [DOI] [PubMed] [Google Scholar]
- 18.Lodmell DL, Ray NB, Parnell MJ, et al. DNA immunization protects nonhuman primates against rabies virus. Nature Medicine. 1998;4(8):949–52. doi: 10.1038/nm0898-949. [DOI] [PubMed] [Google Scholar]
- 19.Lodmell DL, Ray NB, Ewalt LC. Gene gun particle-mediated vaccination with plasmid DNA confers protective immunity against rabies virus infection. Vaccine. 1998;16(23):115–8. doi: 10.1016/s0264-410x(97)88325-9. [DOI] [PubMed] [Google Scholar]
- 20.Lodmell DL, Ray NB, Ulrich JT, Ewalt LC. DNA vaccination of mice against rabies virus: effects of the route of vaccination and the adjuvant monophosphoryl lipid A (MPL) Vaccine. 2000;18(1112):1059–66. doi: 10.1016/s0264-410x(99)00352-7. [DOI] [PubMed] [Google Scholar]
- 21.Lodmell DL, Parnell MJ, Bailey JR, Ewalt LC, Hanlon CA. One-time gene gun or intramuscular rabies DNA vaccination of non-human primates: comparison of neutralizing antibody responses and protection against rabies virus 1 year after vaccination. Vaccine. 2001;20(56):838–44. doi: 10.1016/s0264-410x(01)00392-9. [DOI] [PubMed] [Google Scholar]
- 22.Ray NB, Ewalt LC, Lodmell DL. Nanogram quantities of plasmid DNA encoding the rabies virus glycoprotein protect mice against lethal rabies virus infection. Vaccine. 1997;15(8):892–5. doi: 10.1016/s0264-410x(96)00281-2. [DOI] [PubMed] [Google Scholar]
- 23.Fu ZF, Wunner WH, Dietzschold B. Immunoprotection by rabies virus nucleoprotein. Current Topics in Microbiology & Immunology. 1994;187:161–72. doi: 10.1007/978-3-642-78490-3_9. [DOI] [PubMed] [Google Scholar]
- 24.Fu ZF, Dietzschold B, Schumacher CL, Wunner WH, Ertl HC, Koprowski H. Rabies virus nucleoprotein expressed in and purified from insect cells is efficacious as a vaccine. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(5):2001–5. doi: 10.1073/pnas.88.5.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aspden K, Passmore JA, Tiedt F, Williamson AL. Evaluation of lumpy skin disease virus, a capripoxvirus, as a replication-deficient vaccine vector. Journal of General Virology. 1985;84(Pt 8):1985–96. doi: 10.1099/vir.0.19116-0. [DOI] [PubMed] [Google Scholar]
- 26.Xiang ZQ, Gao GP, Reyes-Sandoval A, Li Y, Wilson JM, Ertl HC. Oral vaccination of mice with adenoviral vectors is not impaired by preexisting immunity to the vaccine carrier. Journal of Virology. 2003;77(20):10780–9. doi: 10.1128/JVI.77.20.10780-10789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faber M, Pulmanausahakul R, Hodawadekar SS, et al. Overexpression of the rabies virus glycoprotein results in enhancement of apoptosis and antiviral immune response. Journal of Virology. 2002;76(7):3374–81. doi: 10.1128/JVI.76.7.3374-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGettigan JP, Sarma S, Orenstein JM, Pomerantz RJ, Schnell MJ. Expression and immunogenicity of human immunodeficiency virus type 1 Gag expressed by a replication-competent rhabdovirus-based vaccine vector. Journal of Virology. 2001;75(18):8724–32. doi: 10.1128/JVI.75.18.8724-8732.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dietzschold B, Wunner WH, Wiktor TJ, et al. Characterization of an antigenic determinant of the glycoprotein that correlates with pathogenicity of rabies virus. Proceedings of the National Academy of Sciences of the United States of America. 1983;80(1):70–4. doi: 10.1073/pnas.80.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schnell MJ, Foley HD, Siler CA, McGettigan JP, Dietzschold B, Pomerantz RJ. Recombinant rabies virus as potential live-viral vaccines for HIV-1. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(7):3544–9. doi: 10.1073/pnas.050589197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mebatsion T, Weiland F, Conzelmann KK. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. Journal of Virology. 1999;73(1):242–50. doi: 10.1128/jvi.73.1.242-250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schnell MJ. Viral vectors as potential HIV-1 vaccines. FEMS Microbiology Letters. 2001;200(2):123–9. doi: 10.1111/j.1574-6968.2001.tb10703.x. [DOI] [PubMed] [Google Scholar]
- 33.Morimoto K, Hooper DC, Carbaugh H, Fu ZF, Koprowski H, Dietzschold B. Rabies virus quasispecies: implications for pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(6):3152–6. doi: 10.1073/pnas.95.6.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGettigan JP, Koser ML, McKenna PM, et al. Enhanced humoral HIV-1-specific immune responses generated from recombinant rhabdoviral-based vaccine vectors co-expressing HIV-1 proteins and IL-2. Virology. 2006;344(2):363–77. doi: 10.1016/j.virol.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Tan GS, Preuss MA, Williams JC, Schnell MJ. The dynein light chain 8 binding motif of rabies virus phosphoprotein promotes efficient viral transcription. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(17):7229–34. doi: 10.1073/pnas.0701397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dulina AV, Morogova VM, Shafeeva RS, Pogrebniak EM. Antigenic activity and the reactogenicity of a concentrated, purified, UV-inactivated cultured rabies vaccine. Zh Mikrobiol Epidemiol Immunobiol. 1983;(9):47–50. [PubMed] [Google Scholar]
- 37.Thoulouze MI, Lafage M, Montano-Hirose JA, Lafon M. Rabies virus infects mouse and human lymphocytes and induces apoptosis. Journal of Virology. 1997;71(10):7372–80. doi: 10.1128/jvi.71.10.7372-7380.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.WHO. Recommendations for inactivated rabies vaccine for human use produced in cell substrates and embryonic eggs. WHO Technical Report Series No 941. 2007 [Google Scholar]
- 39.Markine-Goriaynoff D, van der Logt JT, Truyens C, et al. IFN-gamma-independent IgG2a production in mice infected with viruses and parasites. Int Immunol. 2000;12(2):223–30. doi: 10.1093/intimm/12.2.223. [DOI] [PubMed] [Google Scholar]
- 40.Rupprecht CE, Hanlon CA, Blanton J, et al. Oral vaccination of dogs with recombinant rabies virus vaccines. Virus Research. 2005;111(1):101–5. doi: 10.1016/j.virusres.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 41.Sasaki S, Amara RR, Oran AE, Smith JM, Robinson HL. Apoptosis-mediated enhancement of DNA-raised immune responses by mutant caspases. Nature Biotechnology. 2001;19(6):543–7. doi: 10.1038/89289. see comment. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki S, Xin KQ, Okudela K, Okuda K, Ishii N. Immunomodulation by apoptosis-inducing caspases for an influenza DNA vaccine delivered by gene gun. Gene Therapy. 2002;9(12):828–31. doi: 10.1038/sj.gt.3301696. [DOI] [PubMed] [Google Scholar]
- 43.Restifo NP. Vaccines to die for. Nature Biotechnology. 2001;19(6):527–8. doi: 10.1038/89255. comment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morimoto K, Hooper DC, Spitsin S, Koprowski H, Dietzschold B. Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. Journal of Virology. 1999;73(1):510–8. doi: 10.1128/jvi.73.1.510-518.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pulmanausahakul R, Faber M, Morimoto K, et al. Overexpression of cytochrome C by a recombinant rabies virus attenuates pathogenicity and enhances antiviral immunity. Journal of Virology. 2001;75(22):10800–7. doi: 10.1128/JVI.75.22.10800-10807.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lay S, Prehaud C, Dietzschold B, Lafon M. Glycoprotein of nonpathogenic rabies viruses is a major inducer of apoptosis in human jurkat T cells. Annals of the New York Academy of Sciences. 1010 doi: 10.1196/annals.1299.108. [DOI] [PubMed] [Google Scholar]
- 47.Baloul L, Lafon M. Apoptosis and rabies virus neuroinvasion. Biochimie. 2003;85(8):777–88. doi: 10.1016/s0300-9084(03)00137-8. [DOI] [PubMed] [Google Scholar]
- 48.Megret F, Prehaud C, Lafage M, et al. Immunopotentiation of the antibody response against influenza HA with apoptotic bodies generated by rabies virus G-ERA protein-driven apoptosis. Vaccine. 2005;16(23):5342–50. doi: 10.1016/j.vaccine.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 49.Lay S, Prehaud C, Dietzschold B, Lafon M. Glycoprotein of nonpathogenic rabies viruses is a major inducer of apoptosis in human jurkat T cells. Annals of the New York Academy of Sciences. 2003;1010:577–81. doi: 10.1196/annals.1299.108. [DOI] [PubMed] [Google Scholar]
- 50.Hilleman MR. Six decades of vaccine development--a personal history. Nature Medicine. 1998;4(5 Suppl):507–14. doi: 10.1038/nm0598supp-507. [DOI] [PubMed] [Google Scholar]
- 51.Burton DR, Parren PW. Vaccines and the induction of functional antibodies: time to look beyond the molecules of natural infection? Nature Medicine. 2000;6(2):123–5. doi: 10.1038/72200. [DOI] [PubMed] [Google Scholar]
- 52.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity. 2005;23(1):41–51. doi: 10.1016/j.immuni.2005.05.010. see comment. [DOI] [PubMed] [Google Scholar]
- 53.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310(5753):1510–2. doi: 10.1126/science.1118948. see comment. [DOI] [PubMed] [Google Scholar]
- 54.Woof JM, Burton DR. Human antibody-Fc receptor interactions illuminated by crystal structures. Nature Rev Immunol. 2004;4(2):89–99. doi: 10.1038/nri1266. [DOI] [PubMed] [Google Scholar]
- 55.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8(1):34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]