

Abstract
Hepatocellular carcinoma (HCC) is a complex and heterogeneous tumor with several genomic alterations. There is evidence of aberrant activation of several signaling cascades such as EGFR, Ras/ERK, PI3K/MTOR, HGF/MET, Wnt, Hedgehog and apoptotic signaling, Recently a multikinase inhibitor, sorafenib, has shown survival benefits in patients with advanced hepatocellular carcinoma. This advancement represents a breakthrough in the treatment of this complex disease, and proves that molecular therapies can be effective in HCC. It is becoming apparent, however, that to overcome the complexity of genomic aberrations in HCC, combination therapies will be critical. Phase II studies have tested drugs blocking EGFR, VEGF/PDGFR and mTOR signaling. No relevant data has been produced so far in combination therapies. Future research is expected to identify new compounds to block important undruggable pathways, such as Wnt signaling, and to identify new oncogenes as targets for therapies through novel high-throughput technologies.
Recent guidelines have established a new frame for the design of clinical trials in HCC. Randomized phase II trials with a time to progression endpoint are proposed as pivotal for capturing benefits from novel drugs. Survival remains the main endpoint to measure effectiveness in phase III studies. Patients assigned to the control arm should receive standard-of-care therapy, that is, chemoembolization for patients with intermediate-stage disease and sorafenib for patients with advanced-stage disease. Biomarkers and molecular imaging should be part of the trials, in order to optimize the enrichment of study populations and identify drug responders. Ultimately, a molecular classification of HCC based on genome-wide investigations and identification of patient subclasses according to drug-responsiveness will lead to a more personalized medicine.
Keywords: Liver cancer; molecular pathogenesis; signaling pathway, targeted therapy; sorafenib; Tyrosine kinase inhibitors, phase II, Phase III, trial design, end-points
Introduction
Hepatocellular carcinoma (HCC) is a global health problem1-3. In the West, around 40% of patients are eligible for potential curative treatment (resection, transplantation or local ablation) and 20% for chemoembolization4-6. The absence of standard systemic therapy for advanced cases has changed with the recent positive randomized controlled trial (RCT) testing the multikinase inhibitor sorafenib, which represents a breakthrough in the management of this neoplasm7. The magnitude of the benefit obtained is similar of that observed with trastuzumab in breast cancer, bevacizumab in colon cancer or erlotinib in lung cancer with a decrease in the risk of death in the range of 25%-35%8-10. These results proving the efficacy of molecular targeted therapies for liver cancer have triggered the search of additional molecular agents to further expand patient's survival.
The current review summarizes the data about molecular pathogenesis and signal transduction pathways and potential therapeutic targets in HCC, analyzes the molecular therapies tested within clinical trials and present recommendations on end-points and trial design for testing new agents in this neoplasm.
Overview of Molecular Pathogenesis
The molecular pathogenesis of HCC is complex (see Review of Farazi et al, 11). This neoplasm arises in normal livers, abnormal but non-cirrhotic livers and in cirrhotic livers in 80% of cases as a result of different environmental risk factors4,5. Each of these scenarios involves different genetic and epigenetic alterations, chromosomal aberrations, gene mutations and altered molecular pathways.
Cirrhosis and pre-neoplastic stages
The sequence of molecular events leading to liver carcinogenesis is not well known11-14. The accumulation of genetic alterations driving cirrhotic liver to cancer is a multi-step process originated from stem cells or mature hepatocytes. HCV and HBV are critical insults for genetic damage. In patients with chronic HCV infection an increase in TGF-alpha and insulin-like growth factor-2 (IGF-2) contribute to accelerated hepatocyte proliferation11-14. Experimental studies suggest that HCV core protein acts as Wnt ligand, transactivates Ras signaling and inactivates p5315. In chronic HBV infection, non-random DNA integration of HBV leads to promoter activation of oncogenes, DNA rearrangement and chromosomal instability16. Simultaneously, oxidative stress resulting from chronic inflammation produces genomic and mitochondrial DNA damage 17. Genetic aberrations (allelic deletions) and epigenetic alterations (aberrant methylation) are present at pre-neoplastic stages. In this carcinogenic field, selected cell populations proliferate organized into dysplastic nodules. Telomerase activity progressively increases to allow uncontrolled cellular replication. Finally, activation of pathways related to survival and proliferation induce the malignant phenotype.
Hepatocellular carcinoma
According to Vogelstein18, solid tumors develop after dysruption of at least three critical intracellular signalling networks, whereas haemathological tumors develop after only one genomic hit, as in Chronic Myeloid Leukemia. Dysruption of specific pathways result from oncogene activation (generally one allele activation suffices) through point mutations, copy number alterations or epigenetic changes; or through inactivation of tumor suppressor genes (generally inactivation of two alleles required) resulting from loss of heterozigosity (LOH), point mutations, epigenetic silencing or aberrant transcription of dominant negative proteins. These concepts explain why single-target agents achieve complete remission in haematologic malignances (ie imatinib that blocks BCR/ABL fusion kinase), but not in solid tumors.
Hanahan and Weinberg19 proposed that signalling pathway dysruption in cancer can be grouped in six function capabilities, some of which need to be altered for cancer development: self-sufficiency in growth signals, insensitivity to anti-growth signals, evading apoptosis, limitless replicative potential, sustained angiogenesis and tumor invasion and metastases. Aiming to integrate these concepts into the molecular pathogenesis of HCC, we propose that there are common disturbances universal to all liver cancers on top of the more specific mechanisms (Figure 1): First, disturbances in the cell cycle regulation, reported in half of the cases, resulting from TP53 point mutations or LOH, silencing of p16 or Retinoblastoma genes or overexpression of cyclin D1 11-14. Second, aberrant angiogenesis, already present in early hepatocarcinogenesis, resulting from autocrine/paracrine secretion of VEGF, PDGF or angiopoietin-2 or high-level amplification of VEGFA gene. Third, evasion of apoptosis due to deregulation of intrinsic or extrinsic apoptotic pathways. Finally, reactivation of TERT which ensures limitless replicative potential.
Figure 1. Schematic representation of the molecular pathogenesis of HCC.

Two molecular types of alterations are delineated. Self-sufficiency in cell signals are the main responsible of initiation and proliferation of liver cancer. These alterations result from activation of pathways that might be activated only in specific subgroups of tumors: Wnt signaling, EGF-Ras-MAPK signaling, IGF signaling, mTOR signaling and others. Alterations present in almost all tumors involve limitless replicative potential resulting from activation of TERT, neoangiogenesis (per activation of VEGF,PDGF, angiopoetin and others), and insensitivity to anti-growth signals and checkpoint disruption (due to disturbances in TP53, p21,retinoblastoma,cyclin D1) 11-14,19.
The most important mechanism of liver cancer progression is cell proliferation. There is no dominant pathway responsible for it in HCC, but gene expression studies have resulted in major molecular classes of HCC, according to gene sets responsible for cell proliferation and survival20-23. One class features activation of Wnt signaling pathway with a high prevalence of mutations in CTNNB1. Mutations or deletions in CTNNB1 exon 3 prevent the ubiquitination of its protein product, beta-catenin, leading to its nuclear translocation and transactivation of target genes. Another class is associated with high proliferation, chromosomal instability and activation of RAS/MAPKK signaling, IGF signaling, c-met signaling or Akt/mTOR signaling. Features of stemness, suggesting progenitor cell origin, have been reported in this subclass. A third class is related to interferon signaling, associated with leukocyte infiltration23. Less clear is the role of NF-KB signaling, Jak-Stat, TGF-Beta and Hedgehog signaling activation. Other molecular mechanisms have been described as responsible of cell dissemination, invasiveness and metastasis, such as dys-regulation of nm23-H1, osteopontin (SPP1), ARHC (Rho C), MMP9 and MMP14 or TGF-beta mediated genes11-14. In parallel, an interleukin gene expression signature from the adjacent liver tissue has been linked to higher rates of venous invasion and metastasis, pointing to the importance of tumor microenvironment in the genesis and dissemination of HCC24.
Genetic Aberrations
High throughput technologies such as SNP-array, cDNA microarray and protein mass spectrometry have changed the molecular approach to liver cancer. Table 1 summarizes the most prevalent molecular aberrations in HCC such as alterations in gene expression, DNA amplifications/deletions, somatic mutations, and epigenetic changes which can be considered as potential therapeutic targets,
Table 1.
Key molecular aberrations described in hepatocellular carcinoma.
| FUNCTION | Gene | Gene Expression | Mutations/Copy alterations | Aberrant methylation |
|---|---|---|---|---|
| Growth factors and receptors | IGF-II | Increased | ||
| IGFR-II (M6PR) | Decreased | 25%/LOH 60% | ||
| EGF | Increased | |||
| EGFR | Increased | 0% | ||
| TGF-alpha | Increased | |||
| K-RAS | - | 11% (3-42%) | ||
| RASSF1 | Decreased | 85% | ||
| PIK3CA | - | 12% (0-35%) | ||
| PTEN | Decreased | 0-11% | ||
| HGF/c-MET | Increased | |||
| Proliferation and differentiation | β-catenin | Increased | 17% (0-44%); 58%,hepatoblastoma | |
| E-cadherin | Decreased | Hyper: 46% | ||
| c-myc | Increased | Hypo | ||
| APC | Decreased | 77% | ||
| Angiogenesis | VEGFA | Increased | Amplification (5%) | |
| VEGFR-2 | Increased | |||
| Angiopoietin-2 | Increased | |||
| Metastasis | MMP-14 | Increased | ||
| MMP-9 | Increased | |||
| Topoisomerase 2A | Increased | |||
| Osteopontin | Increased | |||
| Cell cycle | Rb | Increased | 15% | |
| cyclin D1 | Decreased | Amplification (7%) | ||
| p53 | Decreased | 27% (0-67%) | ||
| p16 | Decreased | 13% | Hyper: 56% | |
| p27kip | Decreased | |||
| Survivin | Increased |
Adapted from Villanueva et a14.
Structural alterations in HCC
Genome-wide alterations
Several candidate genes in hepatocarcinogenesis have emerged: c-myc (8q), Cyclin A2 (4q), Cyclin D1 (11q), Rb1 (13q), AXIN1 (16p), p53 (17p), IGFR-II/M6PR (6q), p16 (9p), E-Cadherin (16q), SOCS (16p) and PTEN (10q). Chromosomal amplification or deletions are identified in almost all tumors, being the most prevalent amplifications of 1q (58-78%), 6p, 8q, 17q, and 20q, and deletions in 4q, 8p, 13q, 16q and 17p12,20. High-level amplifications have been detected in 11q13 (5% of cases, regions encoding Cyclin D1) and 6p21 (4-6%; regions encoding VEGFA)20. Further research is needed to identify key oncogenes in these areas.
Mutations
Few somatic mutations have been described in HCC patients, an area that is expected to advance in the near future with the introduction of high-throughput sequencing technology14. TP53, a tumor suppressor gene involved in cell cycle control, DNA repair, apoptosis and differentiation, is mutated in 27% of cases (range 0-67%). Aflatoxin B exposure in Africa and Asia is associated with p53 G-to-T mutation at the third position of codon 249. Mutations in beta-catenin in exon 3 are present in 17% of cases of HCC, and in 60% of cases of hepatoblastoma. Less frequent somatic mutations have been described in AXIN1, PI3KA, and K-Ras. Conversely, prevalent mutations in other cancers, such as, EGFR, Her2/nu PTEN or H-Ras are marginal in HCC, while germline mutations have not been described.
Genomic instability
This has been described associated with telomere shortening, aberrant methylation and aberrations in mismatch repair genes. Telomerase activity is increased in nearly 90% of human HCCs and results from HBV integration in the TERT locus, amplification of the gene encoding the telomerase RNA component (TERC), or allelic loss of chromosome 10p, a region encoding a telomerase repressor25. High genomic instability has been associated with the proliferation subclass of HCC, and is more prevalent in HBV-related HCCs.
Epigenetic alterations
Epigenetic silencing of gene expression occurs by abnormal rmethylation of gene promoter regions. Liver cancer cells have certain areas of dense hypermethylation over a background of global hypomethylation. Hypermethylation affects CpG islands localized in promoter regions of tumor suppressor genes like p16INK4a, E-cadherin, NORE1A, RASSF1, IGFR-II/MP6 and BRCA111-14. Demethylation agents allow re-expression of these genes and restoration of their antineoplastic functions.
Dysrupted Signaling Pathways and Targeted Therapies
The molecular aberrations described harbor the main targets for liver cancer therapy: protein kinases14,26. Description of the whole kinome few years ago has facilitated the discovery of new oncology drugs. The key signal transduction pathways implicated in the pathogenesis of HCC are Wnt-βcatenin pathway, EGFR-RAS-MAPKK pathway, c-MET pathway, IGF signaling, Akt/mTOR signaling and VEGF and PDGFR signaling cascades (Figure 2). Targeted therapies developed for these pathways are summarized in Table 2. Other pathways involved in hepatocarcinogenesis such as Jak-STAT, TGF-β and Hedgehog need further attention to define their relevance and potential therapeutic interest.
Figure 2. Molecular targeted therapies in HCC.

Representation of monoclonal antibodies against ligands (VEGFR-bevacizumab, EGFR-cetuximab) tyrosine kinase inhibitors (VEGFR:sorafenib, sunitinib, PDFGR: sorafenib, sunitinib; EGFR: erlotinib, lapatinib, AEE788, Her2/nu: lapatinib, AEE788), ST kinase inhibitors (Raf: sorafenib; mTOR: rapamycin,everolimus, PI3K: XL-765) in preclinical studies or clinical trials in HCC. (Modified from Villanueva A et al. Clinical Opinion in Oncology, 2008)80
Table 2.
Molecularly targeted agents in clinical development in cancer
| Cancer cell function | Target | Agent (type) |
|---|---|---|
| Signal transduction | ||
| Growth factor receptors | ||
| EGFR | Gefitinib (TKI), Erlotinib (TKI), Cetuximab (MAb), Panitumumab (MAb) | |
| HER2 | Trastuzumab (MAb), Lapatinib (TKI) | |
| PDGFR | Imatinib (TKI), Sunitinb (TKI), Sorafenib (TKI) | |
| FLT3 | Lestaurtinib (TKI), PKC 412 (TKI), sunitinib | |
| IGFR1 | IMC-A12 (MAb), | |
| c-MET | SU11274, JNJ-38877605,ARQ197 | |
| c-KIT | Imatinib,dasatinib. | |
| Intracellular signalling | ||
| RAS | Farnesyl transferase inhibitor tipifarnib | |
| RAF | Sorafenib | |
| MEK | Vandetanib,AZD6244 | |
| mTOR | Temsirolimus, everolimus, rapamycin. | |
| Angiogenesis | ||
| Growth factors | ||
| VEGF | Bevacizumab (MAb), | |
| Growth factor receptors | ||
| VEGFR (1-3) | Sorafenib, sunitinib, Brivanib, cediranib, Valatanib, IMC1121B (MAb) | |
| PDGFR | Sorafenib, imatinib,sunitinib. | |
| Apoptosis | ||
| Intrinsic pathway | ||
| BCL2 | GX15-070, oblimersen | |
| Extrinsic pathway | ||
| Apo2L/TRAIL | Mapatumumab, Apomab,AMG-655, rhApo/TRAIL | |
| Protein turnover | Proteasome | Bortezomib* |
| Chromatin remodelling | ||
| HDAC | SAHA | |
| DNAmethyltransferase decitabine | ||
| Cell cycle | CDKs | Flavopiridol (CDKI) |
| Migration and invasion | SRC | Dasatinib, XL228 |
TKI: small molecules-tirosine kinase inhibitors.; MAb: monoclonal antibodies.
Other mechanism of action
Growth factor receptor signaling
EGFR-Ras-MAPKK pathway
The ligands EGF, HGF PDGF, VEGF among others activate the RAS/MAPK signaling pathway and induce transcription of genes of the AP1 family, such as c-fos and c-jun, which are key elements for cell proliferation27. The epidermal growth factor receptor (EGFR) is a member of a family of four related receptors (Her2/Neu, ErbB3 and ErbB4) that upon ligand binding trigger tyrosine kinase activity and consequently initiate signal transduction. Gain of function of EGFR classically occurs as a result of point mutations, amplification or increase in ligand-receptor interaction28.
Most of the agents under investigation block membranous tyrosine kinase receptors (TKRs) (Figure 2). Effective blockade of the EGFR signaling pathway can be achieved by the use of monoclonal antibodies against EGFR (cetuximab) or ErbB2/Her2/neu (trastuzumab). Cetuximab is FDA-approved for the treatment of head and neck29 and colo-rectal cancer30, and trastuzumab for overexpressing-Her2 metastatic breast cancer8. Alternatively, pathway activation can also be successfully inhibited by small molecules against the catalytic domain of EGFR like erlotinib, active in advanced stages of non-small cell lung cancer10, or against EGFR and Her2, such as lapatinib, which is effective in the treatment of breast cancer31.
In HCC, Ras/MAPK pathway activation might result from aberrant upstream signals (EGFR signaling, IGF signaling) or inactivation of tumor suppressor genes by aberrant methylation, as NORE1A32. Mutations of Raf and Ras are rare findings in HCC. Potent drugs blocking Ras/MAPK signaling are still at exploratory phase, except for sorafenib that has activity inhibiting B-Raf at nanomolar concentrations33.
c-MET signaling
Hepatocyte growth factor (HGF) is a critical molecule for hepatocyte regeneration after injury34. It is secreted by stellate cells and binds to the c-MET receptor. Aberrant activity of MET has been described in human cancers as a result of MET amplification, germline or somatic mutations, transcriptional up-regulation or HGF-dependent autocrine loops. Dysregulation of c-MET and HGF are common in HCC14,35, although the exact role of this pathway in the pathogenesis of HCC is not established. Several compounds have been developed targeting MET pathway, including antibodies against HGF or MET receptor, or selective small molecule MET inhibitors36, but none of them are yet in advanced stage of research in HCC.
IGF signaling
The insulin-like growth factor (IGF) family has a major role in fetal development, proliferation, differentiation, cell growth, apoptosis and has been implicated in multiple malignancies37. IGF I has been associated with high risk of neoplasm development, but not in HCC. In fact, experimental studies suggest the potential role of this molecule in improving cirrhosis. IGF-II is overexpressed in around 30% of human HCCs and the tumor suppressors IGFBP-1, -3, -4 are downregulated38. More important, the tumor suppressor IGFR-II, which main action consists in binding and degrading IGF-II, is downregulated in a subgroup of HCCs as a result of chromosomal detections in IGFR-II locus (6q) or point mutations. Several monoclonal antibodies and small molecules blocking IGF-1R are under early clinical investigations (A12, XL228) 39. The interaction between IGF and EGF signaling may act as a mechanism of resistance for some tumors. In fact the blockade of IGF-1R confers sensitivity to breast tumors resistant to trastuzumab.
PI3K/Akt/mTOR pathway
The PI3K/Akt/mTOR pathway plays a critical role in carcinogenesis40. Akt can be activated through a Tyrosine kinase receptor (EGF or IGF signaling) or through constitutive activation of PI3K or loss of function of the tumor suppressor gene PTEN by epigenetic silencing or somatic mutations. Mutations of PTEN might confer resistance to small molecules against EGFR. Although the role of pAkt in HCC needs further investigation, recent studies have suggested a worse prognosis for tumors with activated Akt41.
An important mediator of the PI3K-Akt pathway is mTOR, which acts as a central regulator of cell growth and proliferation, sensing nutritional status and allowing progression from G1 to S phase40. mTOR pathway is activated in a subset of HCCs, and its blockade with rapamycin or everolimus inhibits growth in HCC cell lines, and in experimental models42. Everolimus – a rapamycin analog- and termsirolimus are approved for the treatment of renal cancer. Novel compounds are currently being tested in early clinical trials. These molecules (rapamycin and analogues) are already approved as immunosuppressive treatments after liver transplantation.
Wnt-βcatenin pathway
The Wnt-βcatenin pathway is implicated in colon cancer and HCC. Unfortunately it constitutes the most undruggable pathway43. The signaling cascade is initiated extracellularly, when Wnt ligands stimulate the Frizzled receptors, which signals β-catenin to uncouple from E-cadherin. The activation of Wnt pathway induces translocation of β-catenin into the nucleus, where it regulates specific oncogenes, including c-myc, cyclin D, and survivin.
Activation of Wnt cascade has been shown in one third of HCCs, particularly in HCV-induced HCCs. Mutations in β-catenin occurs in around 17% of cases (ranging from 0% to 44%)14,44 (see Table 1); and association between Wnt pathway activation and the nuclear sequestration of β-Catenin has been described by immunohistochemistry20. Wnt canonical pathways can also be activated by aberrant methylation of the tumor supressors as APC and E-cadherin or by increase of autocrine/paracrine secretion of Wnt ligands. Non-canonical activation of Wnt in HCC deserves further research. New drugs targeting this pathway are under early clinical development.
Hedgehog signaling
The Hedgehog signaling pathway is fundamental to cell differentiation, regeneration, and stem cell biology. Overexpression of Sonic Hedgehog has been recently described in 60% of 115 human HCC and its effective blockage leads to downregulation of Gli-related target genes45. Moreover, tumorigenic activation of Smo can mediate c-myc overexpression, which plays an important role in hepatocarcinogenesis.
Apoptotic pathways
Apoptosis is one of the mechanisms leading to cell death when cells have sustained damage to their DNA or cytoskeleton46. Evasion of this mechanism is one of the hallmarks of cancer19. Several pro-apoptotic receptor agonist targeting extrinsic apoptosis pathway (including the ligand recombinant human Apo2L/TRAIL) and intrinsic apoptotic pathway (BCL-2 antisense) are in development. Synergistic effects have been described with the combination of small molecules and agonists of Apo2L/TRAIL pathway in experimental models (Avi). For instance, sorafenib has shown that sensitizes resistant human cancer cells to TRAIL-induced death, providing the rationale for testing these combinations46.
Pre-clinical Models for Testing Molecular Drugs
The available experimental HCC models for testing new drugs have been reviewed elsewhere47. Since there is no single dominant molecular pathogenesis underlying all HCCs, different models will be required to mimic different HCC subclasses. The xenograft model has demonstrated several advantages: tumors are rapidly and easily induced, and their subcutaneous location enables direct measurement of growth. Novel models are emerging: orthotopic implantation of intact fragments of human cancer into the corresponding organ of immunodeficient rodents; xenograft models generated from cultured cancer stem cells, the key target cells to assess efficacious drugs; or mouse cell lines in immunocompetent mice with underlying liver fibrosis19. However, the most ambitious approach would be to test novel drugs in genetically engineered mice recapitulating specific pathway abnormalities (such as double transgenic TGF/c-MYC48, transgenic of PDGFR49, or transgenic for beta-catenin) in animals with an underlying fibrotic milieu. None of the latest models are currently ready for the conventional experimental studies.
Correlation between pre-clinical findings and clinical trials
Among 1,000 drugs discovered, only 10 will be tested in clinical trials and only 1 will obtain regulatory approval. Data supporting early clinical testing should reflect good pre-clinical data on safety and PK profiles, pharmacodinamics showing prove of target abrogation, and adequate therapeutic index: reduction of viable tumor and expansion of survival. In HCC, the only drug approved so far, sorafenib, showed appealing pre-clinical and early clinical trial data. While drugs showing pre-clinical appealing results have been validated in early phase II studies (bevacizumab, erlotinib, rapamycin analogs, sunitinib), others have shown ineffective results (gefitinib, cetuximab and bortezomib). Divergent results between preclinical models and clinical outcomes can be due to the heterogeneity of tumors in humans versus in cell lines; the molecular aberrations of the cell line chosen, ectopic versus orthotopic location of tumor, dosage and scheduling of the compounds, and variability in selected endpoints. However, the greatest discrepancies are likely due to critical differences in the microenvironment. This is particularly relevant to HCC11, which arises in an environment of inflammation and fibrosis.
Standard Therapies for HCC
Despite recent improvements in surveillance programs and diagnostic tools allowing identification of small suspicious nodules, only 30-40% of HCC patients are eligible for curative treatments5. In well-selected patients, resection and liver transplantation provide 5-year survival rates of 70% whereas local ablation with radiofrequency reaches 50%5,6. It is assumed that these treatments change the natural history of the disease. Tumor relapse complicates half of the patients at 3 years, and none of the 14 RCTs published so far provide a strong rationale to establish a standard adjuvant therapy50.
In the last 25 years, many antitumoral agents have been evaluated in patients with unresectable HCCs5,6. Among these patients, though, at least three subgroups can be identified: patients at an intermediate stage (median survival 16 months, untreated; BCLC B), patients at advanced stage (median survival, 6-7 months, untreated; BCLC C) and patients at end-stage disease (median survival <3 months; BCLC D) 5. These outcomes have been validated within the largest RCT ever reported in HCC. It tested Seocalcitol, a vit D-like compound, vs placebo conducted in 746 patients51. The trial was negative and the drug albeit ineffective was safe. The median survival of the 370 patients at intermediate stage was around 16 months, whereas it was of 5.6 months in the 376 patients at advanced stage (Figure 3). These figures should be considered as comparators for outcome analysis of phase II investigations.
Figure 3. Natural history of patients with HCC according to the BCLC staging system.
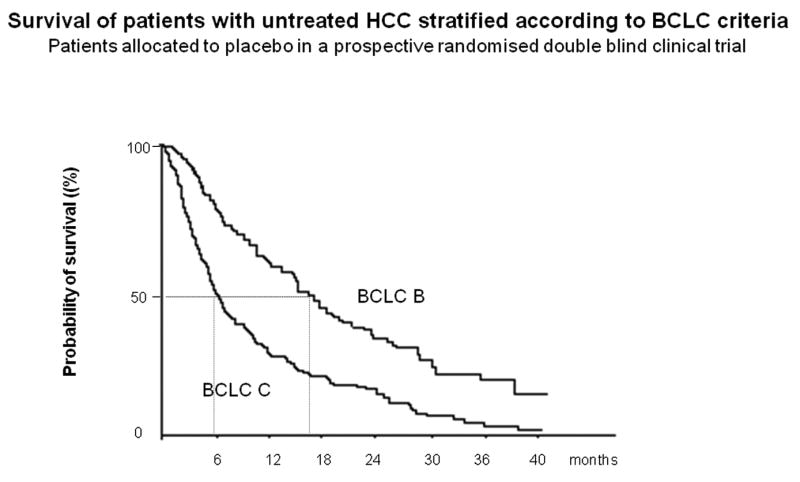
Natural history of 746 patients included in the seocalcitol vs placebo randomized controlled trial for hepatocellular carcinoma. The study was negative, and drug was considered not effective, but not toxic. (A). Natural history of the 370 patients belonging to the BCLC B staging group. Median survival ranged from 15. 8 months and 15.1 months, in the placebo group and seocalcitol group, respectively. Thus, it can be estimated that the expected survival for this subgroup is around 16 months. (B) Natural history of the 376 patients belonging to the BCLC C staging group. Median survival ranged from 5.7 months and 5.6 months, in the placebo group and seocalcitol group, respectively. Thus, the median survival for trial design of the target population of BCLC C patients is around 6 months51.
There are approximately 80 randomized controlled clinical trials assessing transarterial chemoembolization (TACE), intraarterial and systemic chemotherapy, hormonal treatments, immunomodulators, and internal and external radiotherapy6,52. Only chemoembolization (TACE) with gelfoam and doxorubicin or cisplatin when compared with control or subobtimal therapies has shown survival advantages in two RCTs and meta-analysis of pooled data of 6 RCTs conducted before 20036. As a consequence, TACE is considered the standard treatment for patients with intermediate stage according to the BCLC classification (asymptomatic multinodular tumors without vascular invasion; BCLC B) in American guidelines4 and in the recently reported AASLD-JNCI guidelines for trial design53. After this treatment, a median survival of 20 months is expected. Research in this area should focus on evaluating the usefulness of combined therapies or new therapeutic regimes to enhance the initial response.
In 2006, no drug was effective as first line treatment for patients with advanced HCC (stage C, BCLC classification: patients with symptomatic tumors (ECOG 1-2) and/or vascular invasion and/or extrahepatic disease), which represent 40% -70% of the whole HCC population. Several systemic therapies, including chemotherapy (PIAF regime, doxorubicin, epirubicin, cisplatinum, etc.), hormonal compounds (anti-estrogens, anti-androgens, octreotide), immunotherapy (interferon), and other compounds such as nolatrexed, an inhibitor of thymidylate synthase, seocalcitol -a vitamin-D like antiproliferative molecule- and a tubulin inhibitor (T-67) showed inconclusive or negative results. Two systematic reviews assessing all RCT assessing treatments for HCC confirmed this negative evidence6,52.
The Advent of Molecular Therapies in HCC
Molecular targeted therapies have created an encouraging trend in the management of cancer. More than 40 molecular targeted therapies have been tested during recent years (Table 2) and more than ten of them have improved the survival of patientswith breast, colorectal, non-small cell lung, renal and head and neck cancer (Table 3) 8-10,29-31,54-59. This is now also the case for HCC.
Table 3.
Comparison of clinical benefits obtained from sorafenib in HCC with other molecular targeted therapies in solid cancers. Magnitude of benefit is described as Hazard ratio (HR, 95% confidence interval).
| Cancer type | Line of systemic treatment | 1st end-point
HR (95%CI) |
2nd end-point
HR (95%CI) |
|---|---|---|---|
| Treatment assessed (n) | |||
| Hepatocellular carcinoma (advanced) | |||
| Sorafenib (n=229) | 1st | Survival | TTP |
| Placebo (n=303) | 0.69 (0.55-0.86) | 0.57(0.44-0.74) | |
| (Llovet JM et al, NEJM 2008) 7 | [3 months]* | ||
| Colo-rectal cancer (metastatic) | |||
| IFL+ bevacizumab (n=402) | 1st | Survival | PFS |
| IFL (iridotecan,fluorouracil,leucovorin) (n=411) | 0.66 (NA) | 0.54 (NA) | |
| (Hurwitz H et al, NEJM 2004) 9 | [4.7 months] | ||
| Cetuximab (n=287) | 2nd | Survival | PFS |
| Best supportive care (n=285) | 0.77 (0.64-0.92) | 0.68(0.57-0.80) | |
| (Jonker D, NEJM 2007) 30 | [1.5 months] | ||
| Lung cancer | |||
| Paclitaxel+Carboplatin+bevacizumab (434) | 1st | Survival | PFS |
| Paclitaxel+Carboplatin (n=444) | 0.79 (0.69-0.93) | 0.66 | |
| (Sandler A, NEJM 2006) 54 | [2 months] | ||
| Erlotinib (n=488) | 3rd | Survival | PFS |
| Placebo (n=243) | 0.70 (0.58-0.85) | 0.61 (0.51-0.74) | |
| (Shepherd F, NEJM 2005) 10 | [2 months] | ||
| Breast cancer | |||
| Early cancer | |||
| Paclitaxel+trastuzumab (n=1672) | 1st | DFS | Survival |
| Doxo+ciclofosfamida (n=1679) | 0.48 (0.39-0.59) | 0.67 (0.48-0.93) | |
| (Romond E et al, NEJM 2005) 8 | [NA] | ||
| Advanced cancer (HER2+) | |||
| Chemotherapy+ trastuzumab (n=235) | 1st | TTP | Survival |
| Chemotherapy (n=234) | 0.51 (0.41-0.63) | 0.8 (0.64-1) | |
| (Slamon C et al, NEJM 2001) 55 | [4.8 months] | ||
| Placlitaxel+ bevacizumab (n=347) | 1st | PFS | Survival |
| Placlitzaxel (n=326) | 0.60 (0.51-0.70) | 0.88 (NA) | |
| (Miller K et al, NEJM 2007) 56 | [no differences] | ||
| Capecitabine+ lapatinib (n=163) | 3rd | TTP | Survival |
| Capecitabine (n=161) | 0.49(0.34-0.71) | 0.92(0.58-1.46) | |
| (Geyer C et al, NEJM 2006) 31 | [no differences] | ||
| Renal Cancer | |||
| Sorafenib (n=451) | 1st | PFS | Survival |
| Placebo (n=452) | 0.44 (0.35-0.55) | 0.77 (0.63-0.95) | |
| (Escudier et al, NEJM 2006) 54 | [no differences] | ||
| Sunitinib (n=375) | 1st | PFS | Survival |
| Interferon-alpha (n=375) | 0.42 (0.32-0.54) | 0.65 (0.45-0.94) | |
| (Motzer et al, NEJM 2006) 58 | {no differences] | ||
| Termsirolimus (n=209) | 1st | Survival | PFS |
| Interferon-alpha (n=207) | 0.73 (0.58-0.92) | (NA) | |
| Termsirolimus+ interferon (n=210) | [2.6 months] | ||
| (Hudes, NEJM 2007) 59 | |||
| Head & neck (advanced) | |||
| Radiotherapy+cetuximab (n=211) | 1st | TTP | Survival |
| Radiotherapy (n=213) | 0.68 (0.52-0.89) | 0.74 (0.57-0.97) | |
| (Bonner J et al, NEJM 200629) | [19.7 months] |
Absolute gains in months of life expectancy are only displayed if the primary end-point is survival.
OS: Overall survival, DFS: disease-free survival; TTP: time to progression; HR: Hazard ratio (risk of death at any given point in the curve: 1=control; treatment 0.69 = 31% decrease of risk of death)
Results of phase III studies: the case of sorafenib
Sorafenib is a multikinase inhibitor with reported activity against Raf-1, B-Raf, VEGFR2, PDGFR, c-Kit receptors, among others receptor tyrosine kinases and serine threonine kinases33. Sorafenib improves progression free survival in renal cancer57. Preclinical studies show potent activity decreasing HCC cell viability and inducing apoptosis in vitro, and antitumoral activity in xenograft models. Subsequently, a phase II trial involving 137 patients with advanced HCC showed that sorafenib induced less than 5% of partial responses, but the observed median survival of 9.2 months with a median time to progression of 5.5 months60 was classified as a signal of potential clinical benefit, since the expected median survival of these patients is of 6 months. Interestingly, patients with activation of RAS/MAPK pathway assessed by positive staining for p-ERK had a longer time to progression (178 days versus 46 days).
Consequently, a large phase III clinical trial (SHARP) was conducted in 602 patients with advanced HCC. The study was stopped at the interim analysis because of survival advantages favoring sorafenib (n=299) vs placebo (n=303) 7. Based on 321 deaths, the hazard ratio sorafenib/placebo was 0.69 (95% CI: 0.55, 0.86; p=0.0005), representing a 31% decrease in the risk of death with a median survival for sorafenib arm of 10.6 months vs 7.9 months for placebo (Figure 4). In addition, sorafenib showed a significant benefit in terms of time to progression (TTP) assessed by independent radiological review with a median TTP of 5.5 months for sorafenib and 2.8 months for placebo. Overall, the most frequent adverse events were diarrhea, fatigue, weight loss and hand-food skin reaction. Grade 3/4 adverse events such as diarrhea (sorafenib vs placebo:11% vs 2%) and hand-foot skin reaction (8% vs 1%) were more frequent with sorafenib. Drug discontinuation due to sorafenib adverse events was of 15%, but drug-related adverse events were considered manageable and no death related with toxicity was described.
Figure 4. Survival curves of patients randomized to sorafenib or placebo (SHARP trial).

The median overall survival of patients randomized to sorafenib (299 patients) was 10.7 months compared with 7.9 months for placebo (303 patients). (Hazard ratio for death, 0.69; 95% CI, 0.55–0.87). (Reproduced with permission from Llovet JM et al, New England Journal of Medicine 2008) 7.
The strength of evidence provided by this trial is the highest level according to the NCI classification61 (1iA = randomized, double-blinded placebo-controlled trial targeting survival), and the magnitude of its efficacy in terms of survival compares well with other established molecular targeted therapies (Table 3). In the SHARP trial, the hazard ratio (HR) for death was 0.69 (a 31% decrease of the hazard of death). The absolute translation in months of life gained for such HR depends upon the median survival of the target population. In this trial, the net survival benefit was of about 3 months. If the magnitude of difference was maintained with an HR of 0.69 in patients at intermediate stage, for instance, the net survival improvement might reach 6 months. Table 3 shows that the effect of sorafenib compares well with other well-established treatments, such trastuzumab in breast cancer8, bevacizumab in colorectal cancer9 or erlotinib in non-small cell lung cancer10, where the decrease in the hazard of death is about 25-35% (HR 0-65-0.75).
The consequences of the trial are three-fold. First, sorafenib has become the standard of care for patients with advanced HCC and also for those progressing after loco-regional therapies. This statement has been endorsed by a panel of experts in trial design in HCC convened by the AASLD (as a result of a Single Topic Conference also endorsed by EASL and AGA) 53, and has modified the accepted treatment schedule described in Figure 5. Hence, sorafenib is established as the control arm for future trial design in this setting. Second, the study proofs that targeted therapies are active in this otherwise chemo-resistant tumor. Thus, this opens an avenue for combination therapies blocking different pathways. Finally, the positive result in advanced cases paves the way for testing this compound in the adjuvant setting, i.e. after potential curative treatments, and area that constitutes an unmet need in liver cancer management.
Figure 5. Barcelona-Clinic Liver Cancer (BCLC) staging classification and treatment schedule.

Patients with very early HCC (stage 0) are optimal candidates for resection. Patients with early HCC (stage A) are candidates for radical therapy (resection, liver transplantation [LT], or local ablation [via percutaneous ethanol injection {PEI} or radiofrequency ablation{RF}]). Patients with intermediate HCC (stage B) benefit from chemoembolization (TACE). Patients with advanced HCC, defined as presence of macroscopic vascular invasion, extrahepatic spread and/or cancer-related symptoms-ECOG 1-2 (stage C), benefit from sorafenib. Patients with end-stage disease (stage D) receive symptomatic treatment. Treatment strategy will transition from one stage to another upon treatment failure or contraindications for the procedures. (Reproduced with permission from Llovet JM et al, J Natl Cancer Inst 200853).
In parallel to this trial, a randomized phase II study in 97 patients was conducted combining doxorubicin and sorafenib vs doxorubicin alone62. Objective response rates were similar in both groups (less than 5%), and overall median survival was of 13.7 months in the combination arm compared with 6.5 months in the doxorubicin arm. Median progression free-survival was 6.9 months vs 2.8 months, respectively. Cardiotoxicity reflected by a relevant decrease of the ejection fraction was significantly higher in the combination arm62. All these data needs to be interpreted with caution, and more studies are warranted to clarify whether the combination therapy has true synergistic activity or if sorafenib is efficacious in HCC patients despite receiving doxorubicin. Cardiac toxicity needs to be evaluated in future studies.
Results from Phase II clinical trials
Several molecular therapies have been tested within phase II trials (Table 4). Most studies have been conducted during the last 5 years and targeted protein kinases, the major drug targets in oncology63. Agents are grouped according to the main targets: 1. Anti-EGFR: erlotinib, gefitinib, lapatinib and cetuximab; 2.Anti-angiogenic agents: bevacizumab, sorafenib, sunitinib, vatalanib, cediranib, and combinations; 3. MTOR inhibitors: everolimus, termsirolimus; 4. Other agents: c-met inhibitors, IGFR1 inhibitors and Wnt inhibitors.
Table 4.
Targeted therapies reported in phase II and III clinical trials in HCC.
| Drug (n) | Target population | Summary of results |
|---|---|---|
| PHASE III | ||
| - Sorafenib (n=299) vs placebo (n=302) [Llovet et al, NEJM 2008] 7 | ||
| BCLC C (82%), B (18%) | Survival : 10.7mo vs 7.9mo (p<0.001) | |
| HR for death:0.69 (0.55-0.87) | ||
| TTR: 5.5mo vs 2.8mo (p<0.001) | ||
| HR for progression :0.58 (0.45-0.74) | ||
| Other phase III studies ongoing: | ||
| Advanced HCC: | Acyclic retinoids vs placebo | |
| Sunitinib vs sorafenib. | ||
| Sorafenib vs sorafenib+ erlotinib | ||
| Adjuvant treatment: | Sorafenib vs placebo | |
| PI-88 vs placebo | ||
| PHASE II | ||
| Single agent | ||
| - Erlotinib [ n=38; Philip et al, JCO, 2005] 64 | ||
| Unresectable HCC | Survival: 13mo | |
| TTR: 3.2mo | ||
| [n=40; Thomas, Cancer, 2007] 65 | Survival: 6.2mo | |
| - Gefitinib [n=31; O'Dwyer JCO-A2006] 66 | ||
| Unresectable HCC | Survival: 6.5mo | |
| PFS:2.8mo | ||
| - Cetuximab [n=30, Zhu et al, Cancer 2007] 67 | ||
| Unresectable HCC | TTP:2mo | |
| - Sunitinib [n=26, Zhu et al,JCO-A2007] 75 | ||
| Unresectable HCC | Survival: 11.6mo | |
| TTR: 4.1mo | ||
| [n=37, Faivre et al, JCO-A2007] 74 | ||
| - Bevacizumab [ n=33; Schwartz et al, JCO-A2006] 69 | ||
| Advanced non-M1 HCC | TTR: 6.5mo | |
| [n=30; Malka et al, JCO-A2007] 70 | ||
| Combination therapies | ||
| - Erlotinib+ bevacizumab [n=29, Thomas et al, JCO-A2007] 73 | ||
| Unresectable HCC | Survival: 19mo | |
| - Sorafenib +doxorubicin (n=47) vs doxorubicin+placebo (n=49) [Abou-Alfa et al, ECCO 2007] 62 | ||
| Unresectable HCC | Survival: 13.7mo vs 6.5mo (p=0.004) | |
| TTR: 8.6mo vs 4.8mo (0.07) | ||
| -Bevacizumab+ capecitabine [n=45, Hsu et al. JCO-2007] 71 | ||
| Unresectable HCC | Survival: 10.7 months | |
| PFS: 4.1 months | ||
| -Bevacizumab-gemcitabine [N=X, Zhu et al JCO 2006] 72 | ||
| - Other phase II studies ongoing: | ||
| Bevacizumab, Lapatininb, Bivranib, Cediranib, Everolimus (RAD001), TSU-68, PI-88 | ||
TTP: Time to progression, PFS: progression free survival
*Natural history of patients at intermediate stage (BCLC B) is of median survival of 16 months, while natural history of patients at advanced stage (BCLC C) is of median survival of 6 months.
Anti-EGFR agents
The types of molecules targeting EGFR and Her2/nu are detailed in Table 2. Three tyrosine kinase inhibitors targeting EGFR have been tested in HCC: erlotinib and gefitinib targeting EGFR and lapatinib targeting both EGFR and Her2/nu. Cetuximab, a monoclonal antibody against EGFR has also been assessed.
Erlotinib has shown activity both in pre-clinical and clinical studies. The first report testing erlotinib at 150 mg daily in HCC included 38 patients with intermediate/advanced HCC (39% of which with extrahepatic metastases) described a low response rate (9%) and 6-month progression-free-survival in 32% of patients (12 out of 38) 64. The median survival was of 13 months, which can be explained by the action of the drug, but also because the target population was different than the conventional advanced HCC population (42% of patients without underlying liver disease). In a second study including 40 patients, the median overall survival was of 25 weeks (95%CI, 18-42 weeks) 65. Gefitinib prevented HCC development in experimental models, but a study in 31 patients reported a median survival of 6.5 months and the authors concluded that the drug had no activity66. Although Her2/neu overexpression and EGFR mutations are uncommon events in HCC a dual receptor blockade lapatinib (EGFR and Her2) is being tested in experimental models of HCC and early clinical trials.
Pre-clinical studies reported antiproliferative effects and some pro-apoptotic activity of cetuximab, a chimeric monoclonal antibody against EGFR, in cell lines. Two studies have tested cetuximab as a single agent. None of them showed any objective responses67,68 and while reported a median TTP of 2 months in 32 patients, the second described a median survival of 9.6 months. This conflicting data indicate that the true efficacy of this compound remains to be elucidated.
Inhibitors of angiogenesis
HCC is a notoriously hypervascular malignancy, even at very early stages of the disease. Overexpression of pro-angiogenic factors like VEGF, PDGF and angiopoietin 2 has been demonstrated in HCC11-14. Experimental studies demonstrate that targeting angiogenesis is of major relevance in HCC. The main compounds tested in clinical trials are monoclonal antibodies (bevacizumab) and of small molecules (sunitinib, sorafenib, bivranib, vatalanib, cediranib).
Bevacizumab is a humanized monoclonal antibody against VEGF approved for the treatment of breast cancer and liver metastasis of colorectal cancer. The mechanism of action of this compound is controversial, since it might act neutralizing VEGF but also normalizing the vasculature to recover the normal perfusion of tumors with aberrant vascularization. A phase I/II trial in 33 selected patients without portal vein invasion revealed modest antitumoral activity (10% objective response) with a median time to progression of 6.5 months69. Of note is that 2 patients developed major gastrointestinal bleeding, one of them leading to death. In a French study including 30 patients, 3 patients also presented bleeding complications70. Combinations of bevacizumab with chemotherapy (gemcitabine, oxaliplatin, capecitabine), obtain objective responses of 10-20% with median survivals of 9-10 months71,72. A study in 27 patients assessing combination of bevacizumab with erlotinib reported a surprisingly high median survival of 19 months73. Six patients showed objective responses (20%). These interesting data needs to be confirmed within extensive randomized phase II studies, because it might reflect the synergistic activity of the combination, but also a selection bias due to the recruitment of patients in the ill-defined category of unresectable HCC, or short follow-up.
Two phase II studies have examined the safety and efficacy of the multikinase inhibitor sunitinib in advanced HCC74,75, a drug already approved for renal cell carcinoma and gastrointestinal stromal tumors (GIST). Sunitinib inhibits VEGFR1 and 2, PDGFR, c-Kit and FLt3, among other kinases. The first European/Asian study treated 37 patients with a daily dose of 50mg/day and reported 1 partial response and 13 showed stable disease74. Of note, however, was that four patients suffered from treatment-related adverse events leading to death due to gastrointestinal bleeding, encephalopathy and hepatorenal syndrome. The US study treated 26 patients at 37.5 mg/day and showed a median progression-free survival of 4.5 months, overall survival of 11.6 months, and more manageable side effects, although 1 death due to bleeding was reported75. The worse safety profile identified with this drug in HCC compared with other tumors –renal, GIST- points to the need of being cautious in the management of very potent anti-angiogenic agents in cirrhotic patients. Three anti-angiogenic and multikinase agents, brivanib, cediranib and vatalanib are being tested currently and data are eagerly awaited.
PI3K/Akt/mTOR inhibitors
Approximately 50% of patients with HCC have activation of the mTOR pathway as assessed by immunohistochemical analysis of phosphorylated S642. This activation may be the result of increase signaling due to overexpression of ligands (i.e. EGF, IGF1, IGF2) or may be due to mutations in oncogenes (PI3KCA) or tumor suppressor genes (PTEN). Some of these patients have a simultaneous activation of Akt. Rapamycin is an inhibitor of mTOR activity approved as immunosuppressant in liver transplantation. A tempting strategy is to use it as first line anti-rejection therapy in the setting of liver transplantation for HCC, but results of randomized studies are needed to support this strategy. Preclinical studies with other analogues of rapamycin (i.e. everolimus, temsirolimus) have shown activity as single agents in xenograft models 42. and phase II studies are currently testing these compounds alone or in combination with sorafenib.
Other molecular agents
IGF signaling activation has been implicated in the pathogenesis of HCC38. Drugs blocking the receptor IGFR1, such as the monoclonal antibody A12, decrease tumor growth and improve survival of HCC xenografts39, providing the rationale to test them in clinical trials. Studies with small molecules and monoclonal antibodies against IGFR1 are ongoing. Similarly, there is rationale to test c-MET inhibitors.
Wnt pathway is activated in at least 30% of HCC20,22 but, unfortunately, there are not yet drugs available that effectively block its activation without significant side effects. Molecular targets of this pathway include Wnt ligands, Frizzled receptors and the oncogene beta-catenin. Preclinical studies have shown activity with different compounds that are currently tested. Similarly, there are active drugs inhibiting the proteasome activation, but the results of bortezomib, which is approved for multiple myeloma, are not encouraging. Telomerase, thought to be essential in cancer cell immortality, is a potential target in HCC25; there are some ongoing studies applying TERT immunization. Finally, trials with drugs blocking Hedgehog pathway and HDAC inhibitors are expected to be tested in the near future.
Rationale for combination therapies
Combination of molecular therapies is expected to improve the outcome benefits already obtained with sorafenib, but this is highly complex matter (see review Dancey76). There is rationale for blocking complementary pathways activated in HCC. This is the case with anti-antiangiogenic agents and blockers of cell proliferation, such as EGFR, MET and IGFR inhibitors. An alternative strategy is to combine therapies abrogating complementary intracellular signaling, such as RAS or MTOR inhibitors with cell proliferation inhibitors. Similarly, pro-apoptotic agents might synergize with cell proliferation inhibitors.
Biomarkers of response and resistance to molecular therapies are needed to provide further rationale for combination therapies. Several mechanism of resistance described in other solid tumors might also apply to HCC. For instance, resistance to EGFR inhibitors is mediated by mutations in downstream oncogenes (RAS) or tumor suppressors (PTEN) or focal amplifications of MET. Similarly, resistance to MTOR inhibitors might be mediated by a negative loop activating IGFR signaling. Understanding these mechanisms will guide drug combinations. Despite some combinations are scientifically sound, toxicity remains as the main practical limitation and safety data from phase I/II studies is mandatory prior launching phase III initiatives53.
Design of Clinical Trials for Molecular Targeted Therapies
The increasing amount of clinical trials ongoing in HCC has raised the need to have a common frame to test novel drugs accepted by all disciplines. As a consequence, new guidelines on the design of clinical trial and end points in HCC have been reported by a multidisciplinary panel of experts convened by the AASLD53. These statements will evolve as new evidence becomes available, including more precise information on natural history of HCC, new drugs or predictive biomarkers. The main recommendations are summarized below.
Survival and time to recurrence were proposed as primary end-points for phase III studies assessing primary and adjuvant therapies, respectively. Composite end-points such as disease free survival (DFS) or progression free survival (PFS) are vulnerable in HCC research, particularly when the target population is ill-defined, and should be included as secondary end-points. Quality of life assessment in HCC research suffers from the lack of a reliable, standardized and adequately validated questionnaire, and thus it is currently recommended as ancillary information.
Randomized phase II studies were considered pivotal prior to conduct phase III trials in HCC. These studies classically consider response rate as the gold-mark for efficacy. However, the advent of molecular compounds has changed the paradigm of trial design77. Clinically significant survival advantages have been reported with tumor responses less than 10%, as is the case of erlotinib in non-small cell lung cancer (NSCLC)10, temsirolimus in renal cancer59, bevacizumab in metastatic colorectal cancer9 and sorafenib in liver cancer7. Thus, response rate (RR) was formally discouraged as a reliable end-point to capture benefit in phase II studies. Time to progression (TTP) was endorsed as the most reliable time to event end-point in phase II studies. Although robust data correlating surrogate end-points (RR, TTP, PFS) and overall survival are lacking, the results from the SHARP trial testing sorafenib suggest a direct association between TTP and survival7. Assessment of tumor response and time to progression should follow the amendments of RECIST endorsed by the AASLD-JNCI panel53. Finally, Phase I studies should specifically include HCC patients with Child-Pugh'A cirrhosis in order to estimate the exact dose, toxicity and liver-related event risk not captured by phase I studies including patients with a variety of neoplasms. This will minimize the non-desirable events as a result of the natural history of cirrhosis that might lead to death due to bleeding, hepatorenal syndrome, infections and other complications78. This population selection will ensure that more than 90% of deaths in the first two years will be due to tumor progression78, and this is of relevance when using non-recommended composite end-points, such as PFS, as the contamination of the end-point with deaths unrelated with the effect of the drug should be tightly controlled.
The panel endorsed the BCLC staging classification for the selection of the target population and/or for the stratification of patients prior randomization (Figure 5). In consequence, the selection of unresectable HCC patients is discouraged for entering patients in phase II studies. By using a common classification, it will be possible to easily interpret the value of outcome data generated by different investigators. Table 4 reinforces the need of properly characterize patients recruited in phase II research studies.
Trial design in HCC is summarized in Table 5. The control arm for clinical trials should be the standard of care, meaning chemoembolization for intermediate HCCs and sorafenib for advanced cases53. Therefore, for the assessment of first-line systemic treatments for advanced HCC a design adding a new agent to sorafenib versus sorafenib alone is recommended. Comparison of single agents head to head with the standard of care therapy might jeopardize the recruitment of patients due to ethical reasons, unless the novel agent showed very promising efficacy in early phase II studies. For second-line treatments, the new agent should be randomized against placebo/best supportive care, and the selection criteria should include patients with contraindications or failures to sorafenib. Randomized studies testing molecular targeted therapies should optimally include biomarker analysis (tissue and/or serum samples) to enable the identification of molecular markers of response and for pharmacokinetic purposes, as reported in other cancers.
Table 5.
Proposal of phase IIII trial design in HCC following the AASLD-JNCI guidelines53.
| HCC subclass
(standard of care) |
Testing novel drugs
1st line treatment |
2nd line treatment* |
|---|---|---|
| BCLC 0 or A –Early stages
(Resection, Transplantation Local ablation) |
Adjuvant: drug vs placebo | - |
| BCLC B-Intermediate stage
(Chemoembolization-TACE) |
TACE vs TACE + drug | - |
| TACE vs drug or device** | ||
| BCLC C- Advance stage
(Sorafenib) |
Sorafenib vs Sorafenib +drug | Drug vs placebo |
| Sorafenib vs drug** |
In case of failure to standard of care
Head to head comparisons with standard of care are only justified if phase II data is very promising.
Future Prospects
The positive results obtained by the multikinase inhibitor sorafenib in HCC represent a breakthrough in the understanding of the disease, a landmark advancement in improving survival for patients for whom no effective therapy was available, and the prove of principle that targeted therapies have a role in this otherwise treatment-resistant neoplasm. Nonetheless, this represents a first step in the way towards disease curation. The molecular complexity of HCC warrants further efforts in the direction of combining therapies targeting enriched populations, a natural approach to a more personalized medicine76. Although some biomarkers and gene signatures have emerged to identify biologically homogeneous groups of tumors20-22, identification of drug-responders in HCC is far from an achieved goal, and should represent a priority for future clinical and translational research79.
Several lines of research should be now expedited. First, our understanding of the pathogenesis of liver cancer is still very elementary. The genomic alterations unravelled so far represent a minor part of the key hits required for HCC initiation and progression. Multidisciplinary translational research in the setting of international consortiums is required to analyze relevant information form tissue banks linked with clinical annotated data and for providing a molecular classification of the disease. Novel high-throughput technologies will more likely identify new oncogenes as targets for treatment. Experimental models recapitulating known hepatocarcinogenic aberrations in humans need to be developed to test new compounds targeting specific mutations or oncogenic addiction loops46-48. Cancer HCC stem cell lines should be developed to test novel drugs. Early and late clinical trials properly designed to capture benefits from these new compounds have to be launched in those areas where unmet clinical needs are already identified53. These include development of adjuvant therapies after resection and local ablation, and combination treatments with the standard of care in intermediate stages (chemoembolization) and advanced stages (sorafenib). These studies should include thorough biomarkers approaches to identify treatment responders to specific drugs, the first step towards personalized medicine80. Ultimately, outcome advantages should also take into account cost-benefit perspectives in the decision-making process81.
Acknowledgments
Grant Support: Josep M. Llovet is Professor of Research–Institut Català de Recerca Avançada (ICREA, IDIBAPS, Hospital Clínic Barcelona), and has grants from National Institute of Health -NIDDK 1R01DK076986-01 and National Institute of Health (Spain) grant I+D Program, grant: SAF-2007-61898 and Samuel Waxman Cancer Research Foundation. Dr J. Bruix: National Institute of Health, Spain (FIS Program, grant number: PI 05/150). CIBERehd is funded by the Instituto de Salud Carlos III.
Abbreviations
- HCC
hepatocellular carcinoma
- HCV
Hepatitis C virus
- TKR
Tyrosine Kinase Receptor
- CGH
Comparative genomic hybridization
- LOH
Loss of heterozygosity
- SNP
Single nucleotide polymorphism
- HBV
Hepatitis B virus
- EGF
Epidermal growth factor
- VEGF
Vascular endothelial growth factor
- IGF
Insulin-like growth factor
- TTP
Time to progression
- PFS
Progression free survival
- OS
overall survival
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.El Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340:745–50. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 3.Sherman M. Hepatocellular carcinoma: epidemiology, risk factors, and screening. Semin Liver Dis. 2005;25:143–54. doi: 10.1055/s-2005-871194. [DOI] [PubMed] [Google Scholar]
- 4.Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–36. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 5.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. The Lancet. 2003;362:1907–17. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 6.Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology. 2003;37:429–42. doi: 10.1053/jhep.2003.50047. [DOI] [PubMed] [Google Scholar]
- 7.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, Cosme A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008 doi: 10.1056/NEJMoa0708857. in press. [DOI] [PubMed] [Google Scholar]
- 8.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Jr, Davidson NE, Tan-Chiu E, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673–84. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 9.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 10.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005 Jul 14;353(2):123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 11.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 12.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–46. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 13.Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5:215–219. doi: 10.1016/s1535-6108(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 14.Villanueva A, Newell P, Chiang D, Friedman S, Llovet JM. Genes and signaling pathways involved in the pathogenesis of HCC. Sem Liv Dis. 2007;27:55–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 15.Branda M, Wands JR. Signal transduction cascades and hepatitis B and C related hepatocellular carcinoma. Hepatology. 2006;43:891–902. doi: 10.1002/hep.21196. [DOI] [PubMed] [Google Scholar]
- 16.Ferber MJ, Montoya DP, Yu C, Aderca I, McGee A, Thorland EC, Nagorney DM, et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene. 2003;22:3813–20. doi: 10.1038/sj.onc.1206528. [DOI] [PubMed] [Google Scholar]
- 17.Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26:2166–76. doi: 10.1038/sj.onc.1210279. [DOI] [PubMed] [Google Scholar]
- 18.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 19.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 20.Chiang D, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, Cozza A, et al. Focal gains of VEGFA and molecular classification of HCC. Cancer Res. 2008 doi: 10.1158/0008-5472.CAN-08-0742. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–6. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 22.Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, Herault A, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 23.Breuhahn K, Vreden S, Haddad R, et al. Molecular profiling of human hepatocellular carcinoma defines mutually exclusive interferon regulation and insulin-like growth factor II overexpression. Cancer Res. 2004;64:6058–64. doi: 10.1158/0008-5472.CAN-04-0292. [DOI] [PubMed] [Google Scholar]
- 24.Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, Kammula US, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99–111. doi: 10.1016/j.ccr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 25.Farazi PA, Glickman J, Jiang S, Yu A, Rudolph KL, DePinho RA. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res. 2003;63:5021–7. [PubMed] [Google Scholar]
- 26.Roberts LR, Gores GJ. Hepatocellular carcinoma: molecular pathways and new therapeutic targets. Semin Liver Dis. 2005;25:212–25. doi: 10.1055/s-2005-871200. [DOI] [PubMed] [Google Scholar]
- 27.Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–57. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 28.Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol. 2005;23:2445–59. doi: 10.1200/JCO.2005.11.890. [DOI] [PubMed] [Google Scholar]
- 29.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 30.Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 31.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–43. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 32.Calvisi DF, Ladu S, Gorden A, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130(4):1117–28. doi: 10.1053/j.gastro.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–44. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 34.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43(2 Suppl 1):S45–53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 35.Takami T, Kaposi-Novak P, Uchida K, Gomez-Quiroz LE, Conner EA, Factor VM, Thorgeirsson SS. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine induced hepatocarcinogenesis. Cancer Res. 2007;67:9844–51. doi: 10.1158/0008-5472.CAN-07-1905. [DOI] [PubMed] [Google Scholar]
- 36.Comoglio P, Giordano S, Trusolino L. Drug development of MET inhibitors:targeting oncogene addition and expedience. Nat Rev Drug Dis. 2008;7:504–16. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 37.Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–18. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- 38.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787–800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 39.Tovar V, Alsinet C, Solé M, Cabellos L, Lázaro A, Villanueva A, Schwartz M, et al. Role of Insulin-like growth factor signaling pathway in hepatocellular carcinoma. Molecular targeted therapies blocking IGF pathway in vitro and in vivo. J Hepatol. 2008 [Google Scholar]
- 40.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 41.Schmitz KJ, Wohlschlaeger J, Lang H, Sotiropoulos GC, Malago M, Steveling K, Reis H, et al. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J Hepatol. 2008;48(1):83–90. doi: 10.1016/j.jhep.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 42.Villanueva A, De Feo A, Chen Y, Yea S, Fiel I, Thung S, Schwartz M, Roayaie S, Schwartz M, Friedman SL, Llovet JM. Role of dual TKI and an mTOR inhibitor in blocking PI3K pathway in HCC. Gastroenterology. 2008 submitted. [Google Scholar]
- 43.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. Moon Nat Rew Genetics 2008 WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5(9):691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 44.Laurent-Puig P, Legoix P, Bluteau O, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120(7):1763–73. doi: 10.1053/gast.2001.24798. [DOI] [PubMed] [Google Scholar]
- 45.Osipo C, Miele L. Hedgehog signaling in hepatocellular carcinoma: novel therapeutic strategy targeting hedgehog signaling in HCC. Cancer Biol Ther. 2006;5(2):238–9. doi: 10.4161/cbt.5.2.2566. [DOI] [PubMed] [Google Scholar]
- 46.Ashkenazi A, Herbst R. To kill a tumor cell: the potential of proapoptotic receptor agonist. J Clin Invest. 2008;118:1979–1990. doi: 10.1172/JCI34359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newell P, Villanueva A, Friedman S, Koike K, Llovet JM. Experimental models in hepatocellular carcinoma. J Hepatol. 2008;48:858–879. doi: 10.1016/j.jhep.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JS, Chu IS, Mikaelyan A, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36:1306–11. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]
- 49.Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, Odell MM, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102:3389–94. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Llovet JM, Schwartz M, Mazzaferro V. Resection and liver transplantation for hepatocellular carcinoma. Semin Liver Dis. 2005;25:181–200. doi: 10.1055/s-2005-871198. [DOI] [PubMed] [Google Scholar]
- 51.Beaugrand M, Sala M, Degos F, et al. Treatment of advanced hepatocellular carcinoma by seocalcitol : an International randomized double-blind placebo-controlled study in 747 patients. J Hepatol. 2003;42:17A. [Google Scholar]
- 52.Lopez P, Villanueva A, Llovet JM. Up-dated systematic review of randomized controlled trials in hepatocellular carcinoma. 2002-2005. Alim Pharmacol Ther. 2006;23:1535–47. doi: 10.1111/j.1365-2036.2006.02932.x. [DOI] [PubMed] [Google Scholar]
- 53.Llovet JM, Di Bisceglie A, Bruix J, Kramer B, Lencioni R, Zhu A, Sherman M, Schwartz M, Lotze M, Talwalkar J, Gores GJ. Design and end-points of clinical trials in HCC. J Natl Cancer Inst. 2008;100(10):698–711. doi: 10.1093/jnci/djn134. on behalf of Panel of Experts in HCC. [DOI] [PubMed] [Google Scholar]
- 54.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 55.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 56.Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–76. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 57.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–34. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 58.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 59.Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–81. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 60.Abou-Alfa GK, Schwartz L, Ricci S, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24:4293–300. doi: 10.1200/JCO.2005.01.3441. [DOI] [PubMed] [Google Scholar]
- 61.National Cancer Institute website US National Institute of Health. http://www.cancer.gov.
- 62.Abou-Alfa GK, et al. Preliminary results from a phase II, randomized, double-blind study of sorafenib plus doxorubicin versus placebo plus doxorubicin in patients with advanced hepatocellular carcinoma. Proc ECCO. 2007 Abstr 3500. [Google Scholar]
- 63.Cohen P. Protein kinases-The major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1(4):309–15. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 64.Philip PA, Mahoney MR, Allmer C, et al. Phase II study of Erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J Clin Oncol. 2005;23:6657–63. doi: 10.1200/JCO.2005.14.696. [DOI] [PubMed] [Google Scholar]
- 65.Thomas MB, Chadha R, Iwasaki M, Glover K, Abbruzzese JL. The combination of bevacizumab (B) and erlotinib (E) shows significant biological activity in patients with advanced hepatocellular carcinoma (HCC) J Clin Oncol. 2007;25:214s. doi: 10.1200/JCO.2008.18.3301. Abstract 4567. [DOI] [PubMed] [Google Scholar]
- 66.O'Dwyer PJ, Levy DE, Kauh JS, Fitzgerald DB, Benson AB., III Gefitinib in advanced unresectable hepatocellular carcinoma: results from the Eastern Cooperative Oncology Group's Study E1203. J Clin Oncol. 2006;24 Abs 4143. [Google Scholar]
- 67.Zhu AX, Stuart K, Blaszkowsky LS, Muzikansky A, Reitberg DP, Clark JW, Enzinger PC, et al. Cetuximab Phase 2 study of cetuximab in patients with advanced hepatocellular carcinoma. Cancer. 2007;110(3):581–9. doi: 10.1002/cncr.22829. [DOI] [PubMed] [Google Scholar]
- 68.Gruenwald V, Gebel M, et al. A phase II open-label study of cetuximab in unresectable hepatocellular carcinoma: final results. J Clin Oncol. 2007;25 Abs 4598. [Google Scholar]
- 69.Schwartz JD, Lehrer D. Bevacizumab in unresectable hepatocellular carcinoma (HCC) for patients without metastasis and without invasion of the portal vein. J Clin Oncol. 2006;25 Abs 4144. [Google Scholar]
- 70.Malka DC, Farace F, et al. Bevacizumab in patients with advanced hepatocellular carcinoma (HCC): preliminary results of a phase II study with circulating endothelial cell (CEC) monitoring. J Clin Oncol. 2007;25 Abs 4570. [Google Scholar]
- 71.Sun W, Haller DG, Myculowycz K, Rosen M, Soulen M, Capparo M, Faust T, et al. Combination of capecitabine,oxaliplatin,with bevacizumab in treatment of advanced HCC. J Clin Oncol. 2007;25 Abs 4574. [Google Scholar]
- 72.Zhu AX, Blaszkowsky LS, Ryan DP, et al. Phase II study of gemcitabine and oxaliplatin in combination with bevacizumab in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24:1898–903. doi: 10.1200/JCO.2005.04.9130. [DOI] [PubMed] [Google Scholar]
- 73.Thomas MB, Iwasaki M, Glover K, Abbruzzese JL. The combination of bevacizumab (B) and erlotinib (E) shows significant biological activity in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2007;25 doi: 10.1200/JCO.2008.18.3301. Abs 4567. [DOI] [PubMed] [Google Scholar]
- 74.Faivre S, Raymond E, Douillard JY, et al. Assessment of safety and drug-induced tumor necrosis with sunitinib in patients with unresectable hepatocellular carcinoma (HCC) J Clin Oncol. 2007;25:149s. Abstract 3546. [Google Scholar]
- 75.Zhu AX, Sahani DV, di Tomaso E, et al. A phase II study of sunitinib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2007;25:213s. Abstract 4637. [Google Scholar]
- 76.Dancey JE, Chen HX. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat Rev Drug Discov. 2006;5:649–59. doi: 10.1038/nrd2089. [DOI] [PubMed] [Google Scholar]
- 77.Millar AW, Lynch KP. Rethinking clinical trials for cytostatic drugs. Nat Rev Cancer. 2003;3:540–5. doi: 10.1038/nrc1124. [DOI] [PubMed] [Google Scholar]
- 78.D'Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44:217–31. doi: 10.1016/j.jhep.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 79.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 80.Villanueva A, Toffanin S, Llovet JM. Linking Molecular Classification of Hepatocellular Carcinoma and Personalized Medicine: Preliminary Steps. Curr Opin Oncol. 2008;20:444–53. doi: 10.1097/CCO.0b013e328302c9e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Neumann PJ, Rosen AB, Weinstein MC. Medicare and cost-effectiveness analysis. N Engl J Med. 2005;353:1516–22. doi: 10.1056/NEJMsb050564. [DOI] [PubMed] [Google Scholar]