

Abstract
Human immunodeficiency virus type 2 (HIV-2)/simian immunodeficiency virus SIVSM Vpx is incorporated into virion particles and is thus present during the early steps of infection, when it has been reported to influence the nuclear import of viral DNA. We recently reported that Vpx promoted the accumulation of full-length viral DNA following the infection of human monocyte-derived dendritic cells (DCs). This positive effect was exerted following the infection of DCs with cognate viruses and with retroviruses as divergent as HIV-1, feline immunodeficiency virus, and even murine leukemia virus, leading us to suggest that Vpx counteracted an antiviral restriction present in DCs. Here, we show that Vpx is required, albeit to a different extent, for the infection of all myeloid but not of lymphoid cells, including monocytes, macrophages, and monocytoid THP-1 cells that had been induced to differentiate with phorbol esters. The intracellular localization of Vpx was highly heterogeneous and cell type dependent, since Vpx localized differently in HeLa cells and DCs. Despite these differences, no clear correlation between the functionality of Vpx and its intracellular localization could be drawn. As a first insight into its function, we determined that SIVSM/HIV-2 and SIVRCM Vpx proteins interact with the DCAF1 adaptor of the Cul4-based E3 ubiquitin ligase complex recently described to associate with HIV-1 Vpr and HIV-2 Vpx. However, the functionality of Vpx proteins in the infection of DCs did not strictly correlate with DCAF1 binding, and knockdown experiments failed to reveal a functional role for this association in differentiated THP-1 cells. Lastly, when transferred in the context of a replication-competent viral clone, Vpx was required for replication in DCs.
Members of the simian immunodeficiency virus (SIV) SIVSM/human immunodeficiency virus type 2 (HIV-2) lineage carry vpx, a unique open reading frame that most likely evolved via the duplication of the primate lentivirus vpr (42). Vpx is incorporated into virion particles via the p6 domain of Gag and is thus present during the early steps of infection, when it probably exerts most of its functions (1, 37). The main described function of Vpx has been the nuclear import of viral DNA, similarly to HIV-1 Vpr. In support of this function, Vpx has been shown to be important for the infection of nondividing macrophages but not of cycling cells (7, 12, 38), and several studies have revealed a correlation between the ability of particular Vpx mutants to promote nuclear import and their nuclear localization (5, 11, 26, 32).
However, despite these reports, the exact function of Vpx remains debated, as a few studies failed to reveal a nuclear localization of Vpx (22, 46), while others also reported a requirement for Vpx in the infection of cycling cells, suggesting a role distinct from nuclear import (2, 14, 18, 21, 23, 33, 43, 47).
We have recently reported that although dispensable for the infection of primary lymphocytes, Vpx was required for the infection of human monocyte-derived immature dendritic cells (DCs) by members of the SIVSM/HIV-2 lineage (16, 28). In these cells, Vpx acted prior to nuclear import and promoted the accumulation of full-length viral DNA (17). These early findings have been recently confirmed in two independent studies also indicating a clear effect of Vpx at the reverse transcription step in macrophages (13, 40).
Surprisingly, when provided to DCs via noninfectious virion-like particles (VLPs), Vpx exerted a similar positive effect on viral DNA accumulation following infection with retroviruses as distinct as primate and nonprimate lentiviruses and gammaretroviruses (16). The fact that Vpx had similar effects during the infection of DCs with a number of divergent retroviruses led us to propose that Vpx could act on cellular factors rather than directly on viral components by countering a restrictive activity that specifically limits lentiviral infection in these cells. The recently described involvement of components of the Cul4-based E3 ubiquitin ligase complex in the function of Vpx, namely, the damaged DNA-binding protein 1 (DDB1) subunit and the DDB1- and CUL4-associated factor 1 (DCAF1) adaptor, is in agreement with this hypothesis (38, 40), as it raises the possibility that Vpx may use this complex to target a cellular factor during viral infection.
Here, we present evidence indicating that the requirement for Vpx is a characteristic, although variable, feature in the infection of all primary myeloid cells by SIVMAC and that this phenotype can be reproduced in differentiated monocytoid THP-1 cells. An extensive mutagenesis analysis of Vpx revealed important residues required for the protein's functionality that were conserved between SIVMAC and HIV-2 Vpx proteins. The intracellular localization of wild-type (WT) and mutant Vpx proteins was more heterogeneous than previously reported, and important differences between the intracellular distribution of Vpx in HeLa cells and that in DCs were observed. As a first step toward the molecular dissection of the function of Vpx, we determined that the association with the DCAF1 adaptor is a common feature of Vpx proteins belonging to the SIVSM/HIV-2 and SIVRCM lineages. However, the correlation between DCAF1 binding and the functionality of Vpx was not strict, as the Vpx protein of HIV-2ROD retained complete functionality despite an important decrease in its ability to associate with DCAF1. Conversely, the Vpx protein of SIVRCM did associate with DCAF1 but could not rescue the infectivity defect of Vpx-deficient SIVs. In addition, knockdown experiments with DCAF1 as well as with the DDB1 component of the Cul4-based E3 ubiquitin ligase complex in differentiated THP-1 cells failed to reveal a functional role for this association, at least during the early steps of viral infection of this cell type. Finally, we determined that Vpx was also required in a cell-type-specific manner when transferred in the context of a replication-competent viral clone.
MATERIALS AND METHODS
Cells.
Human primary monocytes were obtained from peripheral blood mononuclear cells of healthy donors at the Etablissement Français du Sang de Lyon (17). Monocytes were purified after Ficoll and Percoll purification followed by negative selection to more than 95% purity (MACS microbeads; Miltenyi Biotec). The differentiation of monocytes in immature DCs or macrophages was achieved upon culture for 4 days in granulocyte-macrophage colony-stimulating factor and interleukin 4 (IL-4) at 100 ng/ml (both from AbCys, Paris, France) or granulocyte-macrophage colony-stimulating factor, respectively. Primary blood lymphocytes (PBLs) were obtained after Ficoll purification. PBLs were stimulated for 24 h with phytohemagglutinin (PHA) (Sigma) and IL-2 (AIDS Reagent and Reference Program of the NIH) prior to infection. All cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (5% for DCs) and with the cytokine used for their differentiation. The human monocytic THP-1 cell line was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% modified Eagle's medium nonessential amino acids. Differentiation was achieved after cell stimulation for 24 h with phorbol-12-myristate-13-acetate (PMA) at 100 nM (Sigma). Human 293T cells were maintained in complete Dulbecco's modified Eagle's medium in 10% fetal calf serum.
LVs and DNA constructs.
The HIV-1 and SIVMAC251 (SIVMAC in the text) lentiviral vectors (LVs) were obtained following calcium phosphate DNA transfection of 293T cells with a packaging construct carrying gag-pro-pol, tat, and rev (8.91 for HIV-1 and SIV15− for SIVMAC), a miniviral genome bearing a green fluorescent protein (GFP) reporter expression cassette, and a plasmid coding the vesicular stomatitis virus G envelope protein (VSVg) (Fig. 1) (28, 31). Noninfectious VLPs used as carriers of Vpx and added in trans during the early steps of infection of DCs were similarly produced by the transfection of 293T cells in the absence of the viral genome (Vpx-VLPs) (17).
FIG. 1.

Schematic representation of the LVs used here. The LVs used in this study were pseudotyped with the VSVg envelope. HIV-1 and SIVMAC vectors were produced upon the transfection of packaging (8.91 and SIV15−, respectively)-, transfer miniviral genome-, and VSVg-encoding DNAs into 293T cells. mi-shRNAs specific for DDB1, DCAF1, or luciferase (Luc) were expressed in the context of an HIV-1 vector from the pAPMmi-shRNA construct, which allows puromycin selection of transduced cells. The structure of the replication-competent viral clone HIV-2GL-AN is also shown. SD, splice donor; SA, splice acceptor; cPPT, central polypurine tract; RRE, Rev-responsive element; U3*, self-inactivating U3.
N-terminally tagged FLAG-Vpr and FLAG-Vpx mutant proteins were obtained by standard molecular biology techniques and cloned in place of gfp in the pRRL.sin vector. The HIV-2ROD sequence (GenBank accession no. P06939) used here was obtained from Andrew Lever (University of Cambridge, United Kingdom). Myc-DCAF1 was a kind gift of Florence Margottin-Goguet (Cochin Institute, Paris, France) (24).
The cloning of microRNA-based short hairpin RNAs (shRNAs) specific for DCAF1 and DDB1 was performed in the context of an HIV-1 lentiviral construct that allows their stable expression upon viral transduction (pAPMshRNA). Vector pAPMshRNA contains an expression cassette for puromycin and another for the microRNA-based shRNAs, and its construction will be detailed elsewhere.
Primer sequences were designed through the Open Biosystems website facility. Three microRNA-based shRNAs per gene target were simultaneously used to silence DCAF1 and DDB1. The primers used are as follows: shDCAF1-1 (TGCTGTTGACAGTGAGCGCGGACTGGAGGTGATCATTAATTAGTGAAGCCACAGATGTAATTAATGATCACCTCCAGTCCATGCCTACTGCCTCGGA), shDCAF1-2 (TGCTGTTGACAGTGAGCGCGCACTTCAGATTATCATCAATTAGTGAAGCCACAGATGTAATTGATGATAATCTGAAGTGC TTGCCTACTGCCTCGGA), shDCAF1-3 (TGCTGTTGACAGTGAGCGACGAATATCCAGTATTGGTAAATAGTGAAGCCACAGATGTATTTACCA ATACTGGATATTCGGTGCCTACTGCCTCGGA), shDDB1-1 (TGCTGTTGACAGTGAGCGCGCCTGCATCCTGGAGTATAAATAGTGAAGCCAC AGATGTATTTATACTCCAGGATGCAGGCATGCCTACTGCCTCGGA), shDDB1-2 (TGCTGTTGACAGTGAGCGCCCTATCACAATGGTGACAAATTAGTGAAGCCACAGATGTAATTTGTCACCATTGTGATAGGTTGCC TACTGCCTCGGA), and shDDB1-3 (TGCTGTTGACAGTGAGCGCCCAGTTTCTGCAGAATGAATATAGTGAAGCCACAGATGTATATTCATTCT GCAGAAACTGGTTGCCTACTGCCTCGGA).
The above-mentioned primers were used as a matrix in a PCR with the following forward and reverse primers: miR30-XhoI (5′-GATGGCTGCTCG AGAAGGTATATTGCTGTTGACAGTGAGCG) and miR30-EcoRI (3′-GTCTAGAGGAATTCCGAGGCAGTAGGCA). The PCR products were then cloned as XhoI/EcoRI fragments directly into the APM vector.
The replication-competent HIV-2GL-AN proviral clone was obtained from A. Adachi (Tokushima, Japan) and was described elsewhere previously (43). Mutations were introduced by standard mutagenesis within the XbaI/NsiI fragment spanning vpx (nucleotides 5072 to 6264 according to sequence reported under GenBank accession no. M30851).
Viral production, single-round infections, and viral replication assays.
Virions were produced by calcium phosphate DNA transfection of 293T cells. For lentiviral vector production, packaging, transfer, and envelope-encoding plasmids were transfected at a ratio of 8:8:4. Vectors were purified from the supernatant of transfected cells by ultracentrifugation through a double-step sucrose cushion (45% and 25%, wt/vol) (15). Viral particles were then resuspended and normalized by their infectious titers on HeLa cells or by an exogenous reverse transcriptase (exo-RT) assay against standards of known infectivity. Transductions were carried out in the presence of 6 μg/ml of polybrene for 2 h prior to extensive cell washing. Macrophages and DCs were infected 4 to 5 days after differentiation, monocytes were infected right after thaw in the absence of exogenous cytokines, and PBLs were infected after 24 h of stimulation with PHA/IL-2. The percentage of infected cells was assessed 72 h postinfection by flow cytometry, with the exception of monocytes that were analyzed 5 to 7 days after infection, as described previously (3).
To evaluate the ability of Vpx to increase the infectivity of HIV-1 when provided in trans to DCs, preincubation assays were carried out as described previously (16). Briefly, Vpx-VLPs were produced as described above for LVs but in the absence of a viral genome. In a preincubation assay, these noninfectious VLPs were provided to DCs at the same time of infection with a GFP-encoding HIV-1 vector at a multiplicity of infection (MOI) of between 0.5 and 1 (17).
Replication-competent viruses were similarly produced by DNA transfection of 293T cells and normalized by exo-RT activity. Infections were carried out overnight prior to extensive cell washing. Every 3 to 4 days, aliquots of the supernatants of infected cultures were harvested and stored until the final analysis by exo-RT.
Antibodies.
Monoclonal antibodies used in Western blot (WB) analyses were obtained from the AIDS Reagent and Reference Program of the NIH (anti-SIV capsid, catalog no. 3537), Sigma (anti-FLAG epitope, clone M2, catalog no. F3165) (also used for immunoprecipitation), and Covance (anti-Myc, clone 9E10, catalog. no. MMS-150R). The anti-VprBP (DCAF1) and anti-DDB1 antibodies were purchased from Shangai Genomics (catalog no. SG4220-28) and Calbiochem (catalog no. PC718), respectively. The anti-actin antibody was obtained from Santa Cruz. For immunostaining, rabbit anti-FLAG (catalog no. F7425; Sigma) and mouse anti-Nup153 (MMS-102P; Covance) antibodies as well as Alexa 546- and 488-conjugated antibodies (Molecular Probes) were used.
Immunofluorescence.
HeLa cells were grown on coverslips and transfected by calcium phosphate with DNAs coding for FLAG-Vpx. DCs were infected with HIV-1 LVs coding for FLAG-Vpx and cytospun 4 to 7 days later. One day posttransfection or just after cytospin in the case of DCs, cells were fixed in 3% paraformaldehyde-phosphate-buffered saline (PBS), and free aldehydes were quenched with 50 mM NH4Cl-PBS. Cells were then permeabilized with 0.2% Triton X-100 for 5 min and blocked for 15 min in 3% (wt/vol) bovine serum albumin-PBS. After immunostaining, images were acquired using an Axiovert 100 M Zeiss LSM 510 confocal microscope.
Immunoprecipitation.
293T cells were transfected in six-well plates with 0.7 μg of plasmids coding for FLAG-Vpx and Myc-DCAF1. Thirty-six hours posttransfection, cells were lysed in 350 μl of SD buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 0.5% Triton X-100, protease inhibitor cocktail; Sigma), sonicated, and clarified by centrifugation. Cell lysates were then precleared by incubation with protein A-Sepharose beads for 1 h at +4°C. Upon bead removal, lysates were immunoprecipitated for 2 h with an anti-FLAG M2 antibody prior to the addition of protein A-Sepharose beads for 1 h. Beads were then washed four times in SD buffer-400 mM NaCl (50 mM Tris [pH 7.5], 400 mM NaCl, 0.5% Triton X-100) prior to WB analysis. Quantification of the amount of proteins was carried out using the Odyssey infrared imaging system and software (Li-Cor Biosciences), which allows a linear detection of protein signals over a broad range of protein concentration (4 logs). When indicated, cycling THP-1 cells were transduced with FLAG-vpx-carrying HIV-1 vectors at an MOI of 10 to 20. Coimmunoprecipitations were carried out 48 h after cell differentiation with PMA.
RESULTS
Vpx is required for the early steps of infection in myeloid blood cells and in differentiated THP-1 cells.
Vpx is required for infection by SIVSM/HIV-2 viruses, and it increases the infectivity of several lentiviruses when provided in trans via noninfectious VLPs on DCs (16, 17, 28). To determine if these properties were conserved among myeloid cells, GFP-encoding LVs and Vpx-carrying VLPs (Fig. 1) were used on DCs, primary monocytes, differentiated macrophages, PBLs, and monocytoid THP-1 cells (Fig. 2).
FIG. 2.
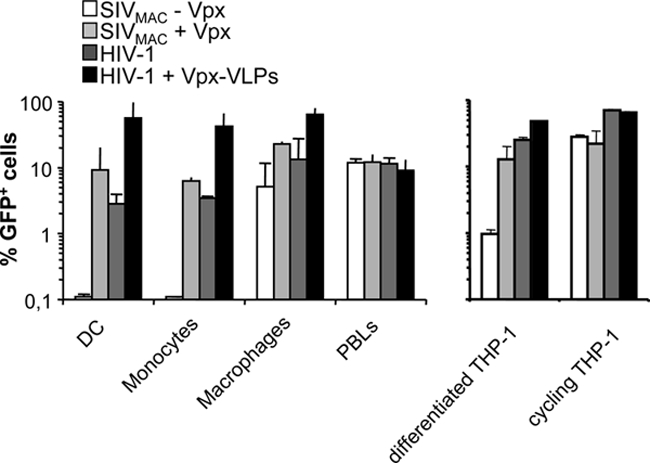
Vpx is required during the early steps of infection of myeloid blood cells and in differentiated THP-1 cells. Primary blood monocytes, macrophages, DCs, PHA/IL-2-stimulated PBLs, as well as cycling and differentiated THP-1 cells were compared for their susceptibilities to infection with SIVMAC vectors carrying or not carrying Vpx and for the positive effect of Vpx-VLP preincubation on infection with HIV-1. The different vectors were produced by the transfection of 293T cells and purified through a double-sucrose cushion. They were then normalized for their infectious titer on HeLa cells and used on the indicated cell type at an MOI of 1 for 2 h prior to extensive cell washing. Noninfectious Vpx-VLPs were produced in the absence of the viral genome and were quantified by exo-RT in comparison with standards of known infectivity. When indicated, Vpx-VLPs were added to DCs together with GFP-encoding HIV-1 vectors. The percentage of infected cells was determined 3 days later by flow cytometry or between days 5 and 7 in the case of monocytes. THP-1 cells were differentiated for 24 h with PMA, which induces their macrophage-like differentiation prior to infection. The graph presents data obtained from three independent experiments with cells obtained from different blood donors.
Vpx was not required for the infection of stimulated PBLs by SIVMAC, but it was absolutely required for the infection of monocytes and DCs. A less stringent requirement was observed in differentiated macrophages in which infection occurred in the absence of Vpx, although Vpx increased it by three- to fivefold (16). Similarly to what observed during infection with SIVMAC, Vpx-VLPs increased the infectivity of HIV-1 vectors by at least 10- to 30-fold in DCs and monocytes but only three- to fivefold in macrophages. As we reported previously, Vpx did not modify HIV-1 infection in stimulated PBLs (16).
Several myeloid/monocytic cell lines were screened for their ability to reproduce the requirement for Vpx during the early steps of viral infection (i.e., HL-60, U937, and THP-1 cells). Only PMA-differentiated THP-1 cells partially recapitulated the phenotype observed in primary myeloid cells. PMA treatment induces the adherence of THP-1 cells that acquire a macrophage-like phenotype. Under these conditions, Vpx optimized the infection of the SIVMAC vector (by 10-fold) but was not absolutely required for it, similarly to primary macrophages. When preincubation experiments were performed, Vpx-VLPs increased the infectivity of incoming HIV-1 vectors marginally but detectably (2- to 2.5-fold). In contrast, Vpx had no effect on viral infection in the absence of PMA. Thus, differentiated THP-1 cells recapitulated the behavior of primary myeloid cells with respect to Vpx, making it an appropriate model cell line with which to dissect its function (16).
Overall, this comparative analysis indicates that Vpx exerts a positive function on lentiviral infectivity during the infection of all myeloid cells and in at least one model cell line albeit with variations with respect to the magnitude of this effect. Given that the effect of Vpx was greater during the infection of DCs, we focused mostly on this cell type.
Identification of point mutations that impair the function of Vpx during early phases of infection of DCs.
To identify residues that are involved in the function of Vpx, alanine-scanning mutagenesis was carried out in the context of a FLAG-tagged SIVMAC Vpx fusion protein. Several mutations that were previously described to affect nuclear localization or to alter potential phosphorylation sites of Vpx were examined in this context, together with a deletion removing the C-terminal proline-rich tail of Vpx (ΔPro) (amino acids 1 to 101) (4, 26, 32, 35).
First, mutant proteins were analyzed for their ability to complement the infectivity defect of SIVMAC viruses devoid of Vpx (17). SIVMAC vectors were produced by the transfection of 293T cells together with plasmids encoding the different Vpx mutants. Virions were normalized by protein content, examined for the level of Vpx incorporation, and used in a single-cycle infection assay of DCs. All mutants were packaged into virion particles at similar levels, with the exception of the H39A, W49/53/56A, and ΔPro mutants, which were incorporated in smaller amounts. These viral particles were equally infectious on HeLa cells (data not shown), but they differed in their abilities to support the infection of DCs (Fig. 3A, top). Specifically, three Vpx mutants were indistinguishable from the WT (N26A, S52A, and S63/65A), three displayed two- to threefold-reduced infectivity (S13A, KK84/85A, and ΔPro), and three mutants displayed 30- to 50-fold-reduced infectivity (T17A, T28A, and GC86/87A). The remaining Vpx mutants lost all their ability to support the early steps of DC infection (100-fold lower than the WT and the STT13/17/28A, H39A, Y66/69/71A, W49/53/56A, and K68/77A mutants).
FIG. 3.

Identification of point mutations that modulate the functionality of Vpx in DCs. (A) Infectious GFP-encoding SIVMAC vectors incorporating WT or mutant FLAG-Vpx proteins were produced upon the cotransfection of 293T cells and purified through a double-sucrose cushion. The different viral preparations were normalized for their protein contents by exo-RT activity and then used to infect DCs in a single-round infection assay (SIVMAC infection) (top). Similarly, the ability of Vpx-VLPs to increase the infectivity of GFP-encoding HIV-1 vectors (HIV-1 infection) (bottom) was tested in a preincubation assay, as previously described (16). Briefly, Vpx-VLPs incorporating the indicated Vpx mutants were normalized by exo-RT and provided to DCs together with HIV-1. The percentage of infected GFP-positive cells was determined 3 days later by flow cytometry. Each graph presents data obtained from three independent experiments after normalization with WT SIVMAC Vpx. (B) Normalized amounts of SIVMAC virion particles were analyzed by WB using anti-FLAG and anti-capsid antibodies.
We ignore at present whether the lower level of incorporation of certain Vpx mutants is due to an impaired interaction with Gagp6. However, the fact that the ΔPro Vpx mutant retains WT functionality despite low levels of incorporation indicates that such low levels are sufficient for the protein's function during the early steps of infection.
Similar results were obtained for all mutants in preincubation assays that assessed the ability of Vpx proteins to increase the infectivity of HIV-1 vectors (Fig. 3A, bottom). The only exceptions were three mutants that displayed less drastic defects in preincubation assays than in the rescue of Vpx-deficient SIVMAC viruses (T17A, T28A, and GC86/87A). We believe that this may relate to the more severe phenotype of the latter.
Vpx displays a heterogeneous subcellular localization in HeLa cells.
Vpx was previously reported to localize to the nucleus (26, 32). To determine the intracellular localizations of the different Vpx mutants, HeLa cells were transfected with DNAs encoding FLAG-Vpx fusions and analyzed by confocal microscopy. The addition of N-terminal FLAG did not affect Vpx function or its localization compared to untagged Vpx (17) (data not shown).
HeLa cells were labeled 24 h after transfection with an anti-FLAG antibody together with an anti-nucleoporin 153 (Nup153) antibody to visualize the nuclear envelope. Under these conditions, the intracellular distribution of Vpx was highly heterogeneous (representative images of each pattern are shown in Fig. 4, with a graphical representation of their relative distributions). Contrary to data from previous reports, WT Vpx was present both within the nucleus and in an as-yet-unidentified perinuclear region of the cytoplasm (70% of cells). In 25% of the cells, Vpx was excluded from the nucleus, while in 1 to 2% of cells, it displayed a diffuse distribution (both cytoplasmic and nuclear).
FIG. 4.

Vpx displays a heterogeneous subcellular localization pattern in HeLa cells. HeLa cells were transfected with WT and mutant FLAG-Vpx expression constructs and analyzed by confocal microscopy 24 h later with anti-FLAG and anti-Nup153 (a nucleoporin localized at the nuclear membrane) antibodies. Pictures representative of the different localization patterns observed here are shown together with a graph presenting their relative distributions. At least 100 cells were scored for each mutant in at least two independent experiments.
These results were observed with both FLAG-tagged and untagged Vpx in Vero and 293T cells and also when confocal microscopy analysis was performed at 16 or 48 h after DNA transfection, suggesting that this distribution was not due to the amount of protein expressed intracellularly (data not shown).
Several Vpx mutants displayed a WT distribution albeit with small variations (S13A, T17A, T28A, STT13/17/28A, N26A, W49/53/56A, S52A, and S63/65A). In contrast, the remaining mutations skewed the relative intracellular localization of Vpx toward a more nuclear (H39A and Y66/69/71A from 38 to 58% of counted cells), diffuse (ΔPro from 1 to 2% to 50% of cells), or cytoplasmic (GC86/87A from 25% to 67% of cells) distribution. In addition to the above-described patterns, three Vpx mutant proteins (S52A, S63/65A, and Y66/69/71A) also displayed a concomitant punctuate cytoplasmic staining (in about 60% of cells) (not shown). Interestingly, the KK84/85A Vpx mutant that is functional in DC infection was present only in a punctuate pattern (85% of the cells).
Vpx displays a distinct subcellular localization in DCs.
Given that Vpx exerts cell-type-specific functions, we hypothesized that this protein could localize differently in DCs. Despite intensive effort, we were unable to reliably localize Vpx with incoming virion particles. Thus, we decided to examine the localization of Vpx in DCs after ectopic expression. To this end, DCs were infected with HIV-1-derived LVs expressing FLAG-tagged WT Vpx together with a restricted number of Vpx mutant proteins in place of GFP. To ease the detection of positive cells, transductions were carried out at MOIs that ensured the transduction of up to 95% of DCs and examined by confocal microscopy 4 to 7 days after infection (MOIs of 10 to 15, as assessed with parallel infection with GFP-encoding viruses). Under these conditions, WT Vpx was detected in only 5 to 10% of DCs and was present in a single circumscribed area of the cytoplasm, near the nucleus (Fig. 5). This pattern was consistently observed in all Vpx-positive cells for the WT as well as for the STT13/17/28A and ΔPro Vpx mutants (nonfunctional and largely functional in single-cycle infectivity assays, respectively). In contrast, the KK68/77A and KK84/85A Vpx mutant proteins presented a punctuated cytoplasmic distribution in DCs, similar to the one observed in HeLa cells. Several Vpx mutant proteins could not be detected by confocal microscopy even following the transduction of DCs with higher viral inputs (H39A, W49/53/56A, and GC86/87A).
FIG. 5.

The intracellular localization of Vpx is cell type dependent. DCs were infected with HIV-1-derived LVs encoding WT or mutant FLAG-Vpx proteins. Four to seven days later, DCs were centrifuged on slides, fixed, and stained with anti-FLAG and anti-Nup153 antibodies. The pictures depict typical localization patterns observed here (n = 100 scored cells in at least two independent experiments). The table summarizes the localization pattern observed upon the expression of a larger panel of SIVMAC Vpx mutants. ND, not detected.
Of note, the Nup153 staining in DCs was weaker than that in HeLa cells; it appeared punctuated on the nuclear envelope, and in some cases, strong staining was observed outside the nucleus. Extranuclear staining of nuclear pore components has been described previously and seems to represent specialized structures within the endoplasmic reticulum that act as storage for nuclear pore proteins (named annullate lamellae) (9). The different Nup153 stainings observed between HeLa cells and DCs may underline cell-type-specific differences both in the amount of such endoplasmic reticulum-associated storages as well as in nuclear pore density, as was described previously (8, 25). However, such staining was on average independent from the expression of Vpx.
Overall, our experiments revealed marked differences between the intracellular localization of WT Vpx in HeLa cells and that in DCs, but no clear correlation could be drawn between the protein's localization and functionality.
The DCAF1 adaptor associates with Vpx proteins derived from the SIVSM/HIV-2 and SIVRCM lineages.
Recently, the Cul4-based E3 ubiquitin ligase complex has been identified as being a cofactor in the G2/M cell cycle arrest induced by Vpr (6, 10, 20, 24, 36, 41, 44), and it has been shown to interact via its adaptor DCAF1 with Vpx by yeast two-hybrid (24) or coimmunoprecipitation (40) assays. Here, we asked whether Vpx proteins issued from members of the SIVSM/HIV-2 and SIVRCM lineages associated broadly with DCAF1 and compared this association to their functionality in DCs. To this end, we first tested the abilities of VLPs containing different Vpx proteins to increase the infectivity of HIV-1 vectors compared to that of VLPs devoid of Vpx. As we reported previously, the function of Vpx during infection is shared among proteins of the SIVSM/HIV-2 but not of the SIVRCM lineage (Fig. 6A) (17). The different Vpx proteins were then tested for their abilities to associate with tagged DCAF1 in HeLa cells. As a control, WT HIV-1 Vpr was analyzed in parallel with a Vpr mutant carrying a single point mutation (Q65A) that abolishes this association (24).
FIG. 6.

DCAF1 associates with Vpx proteins derived from the SIVSM/HIV-2 and SIVRCM lineages. (A) Normalized amounts of noninfectious VLPs incorporating Vpx proteins derived from SIVMAC, SIVRCM, or two viral clones of HIV-2, namely, HIV-2ROD and HIV-2GH-1, were used in a preincubation assay of DCs prior to infection with a constant amount of HIV-1 vectors encoding GFP. The graph presents the ratio of the relative increase in HIV-1 infection observed with or without the preincubation of DCs with Vpx-VLPs (17). All FLAG-Vpx proteins were efficiently incorporated into VLPs (not shown) (17). (B) 293T cells were transfected with the indicated expression constructs, lysed 36 h posttransfection, and immunoprecipitated with an anti-FLAG antibody. The immunoprecipitates were analyzed by WB using antibodies specific for the Myc and FLAG epitopes. Actin was used to normalize the different cellular lysates. (C) Quantification of the amount of viral proteins was obtained using the Odyssey infrared quantification system. The graph corresponds to data from an average of four independent experiments. (D) THP-1 cells were transduced with FLAG-vpx-carrying HIV-1 vectors and differentiated with PMA prior to immunoprecipitation with an anti-FLAG antibody and analysis by WB.
As expected, WT HIV-1 Vpr associated with DCAF1, and this interaction was lost upon a mutation of the Q65 residue (Fig. 6B). SIVMAC, SIVRCM, and HIV-2GH-1 Vpx proteins associated with DCAF1, while HIV-2ROD Vpx was severely diminished in its ability to interact with DCAF1. The amounts of proteins present prior to and after immunoprecipitation were quantified using the Odyssey infrared imaging system (Li-Cor Biosciences) (Fig. 6C). This analysis revealed that HIV-2ROD Vpx bound to DCAF1 with a reduction of more than 20-fold (Fig. 6B and C, lanes 4), in line with previous results (6, 10, 20, 24, 36, 41, 44). Given that HIV-2ROD Vpx is functional during the early steps of infection of DCs despite its much weaker association with DCAF1, our results raise the possibility that the association with DCAF1 may not be required for the function of Vpx. To confirm these results in cells relevant to Vpx's functions, cycling THP-1 cells were transduced with FLAG-vpx-carrying HIV-1 vectors at an MOI of between 10 and 20. Forty-eight hours post-PMA differentiation, cells were lysed and immunoprecipitated with an anti-FLAG antibody. Immunoprecipitates were then analyzed by WB for the presence of endogenous DCAF1 (Fig. 6D). The results obtained here indicate that the SIVMAC Vpx protein associates with DCAF1, while that of HIV-2ROD is severely diminished in its ability to interact with DCAF1, thus confirming the results obtained using 293T cells.
DCAF1 and DDB1 are dispensable for the function of Vpx during infection of differentiated THP-1 cells.
To test the functional relevance of the association between Vpx and the DCAF1/DDB1/Cul4A E3 ubiquitin ligase complex, we silenced DCAF1 as well as DDB1 in differentiated THP-1 cells, which are more amenable to gene silencing than primary myeloid cells.
DDB1 is an obligatory subunit of the Cul4-based E3 ubiquitin complex, and it was recently reported to be involved in the function of Vpx in macrophages (38). Silencing was obtained after the transduction of cycling THP-1 cells with HIV-1-derived vectors bearing puromycin and a microRNA-based shRNA hairpin expression cassette. After cell transduction and puromycin selection, THP-1 cells were differentiated and challenged with GFP-encoding vectors. Under these conditions, effective silencing of DCAF1 and DDB1 was obtained, but no significant changes in the susceptibility of silenced THP-1 cells to viral infection in comparison to that of control cells were observed (Fig. 7).
FIG. 7.

DCAF1 and DDB1 are dispensable for the effect of Vpx in differentiated THP-1 cells. DCAF1 and DDB1 (or luciferase [Luc] as a control) were silenced upon the transduction of cycling THP-1 cells with HIV-1 vectors carrying three different microRNA (mi)-based shRNAs per target gene followed by 1 week of selection of transduced cells with puromycin. THP-1 cells were then differentiated with PMA, analyzed by WB, and infected with the indicated viruses. The percentage of GFP-positive cells was measured 3 days later by flow cytometry. The graph presents data obtained with three independent experiments.
Effect of mutations in Vpx on HIV-2 viral replication.
To determine whether Vpx was also required in a cell-dependent manner in the context of viral replication, an HIV-2GL-AN proviral clone was used in place of SIVMAC for its higher replicative ability in primary human cells. HIV-2GL-AN is a chimera between two viral clones, HIV-2GH-1 and HIV-2ROD, and its Vpx coding sequence is derived from the former (43). Despite the elevated sequence identity between SIVMAC and HIV-2GH-1 Vpx (83% at the amino acid level), we reintroduced several of the mutations previously examined for SIVMAC Vpx in the context of a FLAG-tagged HIV-2GH-1 Vpx protein together with a novel mutant in which all lysine residues were mutated simultaneously (KKKR68/77/84/85A). The functionality of these mutants was assessed in a preincubation assay by evaluating their ability to increase the infectivity of HIV-1 vectors in DCs.
All the HIV-2GH-1 Vpx mutants behaved similarly to the corresponding SIVMAC mutants when their abilities to increase HIV-1 infection were analyzed in a preincubation assay (Fig. 8A). The only exception was the ΔPro mutant, which displayed a larger defect than the one observed for SIVMAC despite equivalent levels of incorporation into virion particles (Fig. 8B).
FIG. 8.

Mutagenesis of the HIV-2GH-1 Vpx protein and cell-type-specific phenotype of Vpx mutants during viral replication. (A) Point mutations previously examined in the context of SIVMAC Vpx were introduced into HIV-2GH-1 FLAG-Vpx and tested in a preincubation experiment with DCs. DCs were infected with Vpx-VLPs incorporating the different HIV-2 Vpx mutants together with gfp-carrying HIV-1 vectors. The percentage of GFP-positive cells was determined 3 days later by flow cytometry. The graph presents data obtained from two independent experiments. (B) The amount of Vpx protein incorporated into virion particles was assessed by WB. A few of these mutations were then transferred in the context of a replication-competent viral clone. (C and D) Jurkat cells (C) and DCs (D) were infected with normalized amounts of WT and Vpx mutant HIV-2GL-AN proviral clones together with a proviral clone bearing deletions in either vpr or vpx (43). Viral replication was measured by exo-RT in the supernatant of infected cultures at different time points after infection.
A few of these mutations were then introduced in the context of an HIV-2GL-AN molecular clone and compared to the WT and deletion mutants of Vpr and Vpx that were described previously (43). Viral particles produced upon the transfection of 293T cells and normalized for their protein content by exo-RT were used to infect lymphoid Jurkat T cells and DCs. Viral spread through the culture was evaluated by analyzing the accumulation of virion particles in the supernatant of infected cells over time by exo-RT (Fig. 8C and D). Jurkat cells supported a robust viral replication, and no differences were observed in the replication kinetics of WT, deletion mutants of Vpx or Vpr, or the different Vpx point mutants independently from the initial viral input (MOIs of 0.01 to 0.5) (not shown). Instead, the deletion of Vpx, but not of Vpr, abolished HIV-2 viral replication in DCs. Consistent with a lack of major defects in single-round infectivity assays, the KR84/85A and S52A Vpx mutant viruses replicated in DCs, contrarily to the ΔPro and KKKR68/77/84/85A mutant viruses (Fig. 8D).
Overall, these results extend the notion of the cell-type-specific function of Vpx not only during single-cycle infection assays but also during viral replication.
DISCUSSION
The data presented here underscore the preponderant role of Vpx during the infection of myeloid cells as opposed to lymphoid cells (in primary cells and in at least one cell line, THP-1), thus completing the emerging picture drawn from the previous work of several laboratories on the importance of this protein both ex vivo and in vivo (12, 13, 17, 19, 26, 28, 32, 38, 40, 45).
Within the myeloid cells tested, the requirement for Vpx varied from absolute to moderate in DCs and monocytes or in macrophages and THP-1 cells, respectively. Given that lentiviral infection of myeloid cells is modulated by the cell's differentiation status, the variations observed here and in other studies with respect to Vpx are likely the result of such differences (38, 40), reflecting a subtle and dynamic interplay between the cytoplasmic environment and Vpx.
Two major lines of evidence suggest that Vpx modulates the cytoplasmic environment to make it more permissive to viral infection. The positive effect of Vpx on viral DNA accumulation is cell type specific, and it is observed not only in cognate viruses but also in primate and nonprimate lentiviruses as well as gammaretroviruses (17). Given the low level of sequence conservation among these viruses, it is unlikely that Vpx acts on a common viral component. The recent observation that fusion between cells that are resistant and those that are permissive to infection with Vpx-deficient virus (i.e., macrophages and COS cells) (38) resulted in heterokaryons that were resistant to infection in the absence of Vpx further argues for the possibility that Vpx may interact with cell-type-specific factors.
Recently, members of the APOBEC family were described as being responsible for the poor susceptibility of DCs to HIV-1 infection through deamination (34). We have not observed a deamination of viral DNA following the infection of DCs, suggesting that APOBEC may not play a major role in the function of Vpx. However, at present, we cannot rule out the possibility that APOBEC may exert a restrictive activity independent from deamination.
In our previous work, we put forward the hypothesis that Vpx may impair the degradation of a cellular factor due to the comparable effects of proteasome inhibitors and Vpx on the viral infectivity of DCs (17). In view of the pleiotropic effects of proteasome inhibitors, the possibility exists that proteasome inhibition and Vpx act on two parallel pathways to increase viral infectivity. However, it is of interest that Vpx was recently reported to associate with the Cul4-based E3 ubiquitin ligase complex, which is involved in the degradation of a number of cellular proteins recruited via several adaptor proteins, termed DCAFs (38, 40). In the data presented here, we show that although the association with DCAF1 seems to be generally conserved in Vpx proteins of the SIVSM/HIV-2 and SIVRCM lineages, this binding does not strictly correlate with the function of Vpx. This is exemplified by the HIV-2ROD Vpx and SIVRMC Vpx proteins, which display an opposite relationship between DCAF1 binding and functionality, with the former being functional despite a 20-fold reduction in its ability to associate with DCAF1. These results together with knockdown experiments using THP-1 cells indicate that Vpx can function normally in spite of a severed association with DCAF1. At least two possibilities can explain these results. First, Vpx may exert a positive effect on viral infectivity through both a DCAF1-dependent and -independent mechanism and thus make use of DCAF1 during the infection of macrophages (39) but not during the infection of THP-1 cells (this study). These differences may relate to cell-type-specific functions of E3-ubiquitin ligase adaptors in the cell metabolism and physiology and may reflect an exquisite ability of Vpx to adapt to specific changes in the intracellular milieu.
Alternatively, the action of Vpx may require only very low levels of DCAF1. This hypothesis cannot be excluded at present, since, albeit feebly, HIV-2ROD Vpx binds DCAF1, and the complete ablation of dcaf1 seems lethal in cycling mouse embryonic fibroblasts (29), so it is possible that the cells expressing the lowest DCAF1 levels are eliminated during our knockdown studies. Under this hypothesis, our results would argue that since most of DCAF1 can be removed, Vpx acts on an enzymatic activity rather than directly competes with a cellular factor. Lastly, DCAF1 and DDB1 may provide redundant functions, a hypothesis that we could not test due to the lethality of the double knockdown (not shown).
We previously reported that Vpx thus acted on viral DNA formation prior to nuclear import (17). Although still controversial (7), a similar effect was recently confirmed by two independent studies (13, 40). The nuclear import and nuclear localization of Vpx have often been linked in the past (5, 24, 27). In our hands, the intracellular distribution of Vpx is more heterogeneous than previously appreciated (5, 10, 24, 27), and in DCs, where it exerts a specific function, Vpx is excluded from the nucleus, suggesting that nuclear localization is not important for the action of Vpx. Vpx has been reported to possess a nuclear export signal that can be disrupted upon the simultaneous mutations of three tryptophan residues at positions 49, 53, and 56 (39). However, a previous study failed to reveal a nuclear export signal in Vpx (5), and in our hands, the W49/53/56A Vpx mutant displays a WT intracellular distribution and is not retained exclusively in the nucleus, arguing against a nuclear export signal in Vpx.
Does the cell-type-specific intracellular distribution of Vpx relate to its function? Although we believe it likely, we could not gather a clear correlation between the functionality of several Vpx mutants and their intracellular distribution. However, it remains possible that a more specific colocalization of Vpx with known intracellular markers as well as a more extended analysis of Vpx mutants in primary DCs may reveal important information regarding the function of Vpx.
The structure of Vpx is presently unknown, but it has been modeled over the nuclear magnetic resonance structure of the related HIV-1 Vpr protein (27, 30). With all its limitations, this model suggests the presence of a three-helix bundle with a loop between helices 2 and 3 that is specific to Vpx. Some of the mutations examined here affect the interface between helices 2 and 3 and may thus impair Vpx functions by destabilizing the overall protein structure (amino acids 42 to 59 and 72 to 86, respectively) (for example, H39A and W49/53/56A). Others may affect the protein functionality by more specifically interfering with known properties of Vpx. Vpx has been shown to be phosphorylated on serine and threonine residues (35) and ubiquitinated (37). The fact that mutations within the N-terminal helix (amino acids 24 to 38) (STT13/17/28A) impair the action of Vpx is intriguing, as this suggests that Vpx is regulated by posttranslational modification such as phosphorylation and/or ubiquitination. Whether this is the case is unclear, as the phosphorylation of Vpx remains a controversial issue, but we are currently testing this hypothesis.
In conclusion, it is becoming clear that a restrictive activity is responsible for the poor susceptibility of DCs, and of myeloid cells more generally, to lentiviral infection. Given that Vpx is able to relieve it and to exert a positive effect on the infection of a number of lentiviruses, Vpx seems to be the appropriate tool to dissect a restriction that broadly targets lentiviral infection in myeloid cells.
Acknowledgments
We are indebted to Jeanine Bernaud for help with blood sample collection and to the service of microscopy of the IFR128 (Platim). We thank the AIDS Research and Reference Reagent Program of the NIH and A. Adachi and F. Margottin-Goguet for sharing of material.
This work received the support of Sidaction and the ANRS. J.L. was supported by a grant from the Swiss National Science Foundation (3100A0-113558) and grant RO1AI36199 from the NIAID, NIH.
Footnotes
Published ahead of print on 1 October 2008.
REFERENCES
- 1.Accola, M. A., A. A. Bukovsky, M. S. Jones, and H. G. Göttlinger. 1999. A conserved dileucine-containing motif in p6gag governs the particle association of Vpx and Vpr of simian immunodeficiency viruses SIVmac and SIVagm. J. Virol. 739992-9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akari, H., J. Sakuragi, Y. Takebe, K. Tomonaga, M. Kawamura, M. Fukasawa, T. Miura, T. Shinjo, and M. Hayami. 1992. Biological characterization of human immunodeficiency virus type 1 and type 2 mutants in human peripheral blood mononuclear cells. Arch. Virol. 123157-167. [DOI] [PubMed] [Google Scholar]
- 3.Arfi, V., L. Rivière, L. Jarrosson-Wuillème, C. Goujon, D. Rigal, J.-L. Darlix, and A. Cimarelli. 2008. Characterization of the early steps of infection of primary blood monocytes by human immunodeficiency virus type 1. J. Virol. 826557-6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belshan, M., L. A. Mahnke, and L. Ratner. 2006. Conserved amino acids of the human immunodeficiency virus type 2 Vpx nuclear localization signal are critical for nuclear targeting of the viral preintegration complex in non-dividing cells. Virology 346118-126. [DOI] [PubMed] [Google Scholar]
- 5.Belshan, M., and L. Ratner. 2003. Identification of the nuclear localization signal of human immunodeficiency virus type 2 Vpx. Virology 3117-15. [DOI] [PubMed] [Google Scholar]
- 6.Belzile, J. P., G. Duisit, N. Rougeau, J. Mercier, A. Finzi, and E. A. Cohen. 2007. HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4A(VPRBP) E3 ubiquitin ligase. PLoS Pathog. 3e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng, X., M. Belshan, and L. Ratner. 2008. Hsp40 facilitates nuclear import of the human immunodeficiency virus type 2 Vpx-mediated preintegration complex. J. Virol. 821229-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cordes, V. C., S. Reidenbach, and W. W. Franke. 1996. Cytoplasmic annulate lamellae in cultured cells: composition, distribution, and mitotic behavior. Cell Tissue Res. 284177-191. [DOI] [PubMed] [Google Scholar]
- 9.Daigle, N., J. Beaudouin, L. Hartnell, G. Imreh, E. Hallberg, J. Lippincott-Schwartz, and J. Ellenberg. 2001. Nuclear pore complexes form immobile networks and have a very low turnover in live mammalian cells. J. Cell Biol. 15471-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeHart, J. L., E. S. Zimmerman, O. Ardon, C. M. Monteiro-Filho, E. R. Arganaraz, and V. Planelles. 2007. HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol. J. 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Depienne, C., P. Roques, C. Creminon, L. Fritsch, R. Casseron, D. Dormont, C. Dargemont, and S. Benichou. 2000. Cellular distribution and karyophilic properties of matrix, integrase, and Vpr proteins from the human and simian immunodeficiency viruses. Exp. Cell Res. 260387-395. [DOI] [PubMed] [Google Scholar]
- 12.Fletcher, T. M., III, B. Brichacek, N. Sharova, M. A. Newman, G. Stivahtis, P. M. Sharp, M. Emerman, B. H. Hahn, and M. Stevenson. 1996. Nuclear import and cell cycle arrest functions of the HIV-1 Vpr protein are encoded by two separate genes in HIV-2/SIV(SM). EMBO J. 156155-6165. [PMC free article] [PubMed] [Google Scholar]
- 13.Fujita, M., M. Otsuka, M. Miyoshi, B. Khamsri, M. Nomaguchi, and A. Adachi. 2008. Vpx is critical for the reverse transcription of human immunodeficiency virus type 2 genome in macrophages. J. Virol. 827752-7756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibbs, J. S., D. A. Regier, and R. C. Desrosiers. 1994. Construction and in vitro properties of SIVmac mutants with deletions in “nonessential” genes. AIDS Res. Hum. Retrovir. 10607-616. [DOI] [PubMed] [Google Scholar]
- 15.Goujon, C., L. Jarrosson-Wuilleme, J. Bernaud, D. Rigal, J. L. Darlix, and A. Cimarelli. 2003. Heterologous human immunodeficiency virus type 1 lentiviral vectors packaging a simian immunodeficiency virus-derived genome display a specific postentry transduction defect in dendritic cells. J. Virol. 779295-9304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goujon, C., L. Jarrosson-Wuilleme, J. Bernaud, D. Rigal, J. L. Darlix, and A. Cimarelli. 2006. With a little help from a friend: increasing HIV transduction of monocyte-derived dendritic cells with virion-like particles of SIV(MAC). Gene Ther. 13991-994. [DOI] [PubMed] [Google Scholar]
- 17.Goujon, C., L. Riviere, L. Jarrosson-Wuilleme, J. Bernaud, D. Rigal, J. L. Darlix, and A. Cimarelli. 2007. SIVSM/HIV-2 Vpx proteins promote retroviral escape from a proteasome-dependent restriction pathway present in human dendritic cells. Retrovirology 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guyader, M., M. Emerman, L. Montagnier, and K. Peden. 1989. VPX mutants of HIV-2 are infectious in established cell lines but display a severe defect in peripheral blood lymphocytes. EMBO J. 81169-1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirsch, V. M., M. E. Sharkey, C. R. Brown, B. Brichacek, S. Goldstein, J. Wakefield, R. Byrum, W. R. Elkins, B. H. Hahn, J. D. Lifson, and M. Stevenson. 1998. Vpx is required for dissemination and pathogenesis of SIV(SM) PBj: evidence of macrophage-dependent viral amplification. Nat. Med. 41401-1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hrecka, K., M. Gierszewska, S. Srivastava, L. Kozaczkiewicz, S. K. Swanson, L. Florens, M. P. Washburn, and J. Skowronski. 2007. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. USA 10411778-11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kappes, J. C., J. A. Conway, S. W. Lee, G. M. Shaw, and B. H. Hahn. 1991. Human immunodeficiency virus type 2 vpx protein augments viral infectivity. Virology 184197-209. [DOI] [PubMed] [Google Scholar]
- 22.Kappes, J. C., J. S. Parkin, J. A. Conway, J. Kim, C. G. Brouillette, G. M. Shaw, and B. H. Hahn. 1993. Intracellular transport and virion incorporation of vpx requires interaction with other virus type-specific components. Virology 193222-233. [DOI] [PubMed] [Google Scholar]
- 23.Kawamura, M., H. Sakai, and A. Adachi. 1994. Human immunodeficiency virus Vpx is required for the early phase of replication in peripheral blood mononuclear cells. Microbiol. Immunol. 38871-878. [DOI] [PubMed] [Google Scholar]
- 24.Le Rouzic, E., N. Belaidouni, E. Estrabaud, M. Morel, J. C. Rain, C. Transy, and F. Margottin-Goguet. 2007. HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle 6182-188. [DOI] [PubMed] [Google Scholar]
- 25.Maeshima, K., K. Yahata, Y. Sasaki, R. Nakatomi, T. Tachibana, T. Hashikawa, F. Imamoto, and N. Imamoto. 2006. Cell-cycle-dependent dynamics of nuclear pores: pore-free islands and lamins. J. Cell Sci. 1194442-4451. [DOI] [PubMed] [Google Scholar]
- 26.Mahalingam, S., B. Van Tine, M. L. Santiago, F. Gao, G. M. Shaw, and B. H. Hahn. 2001. Functional analysis of the simian immunodeficiency virus Vpx protein: identification of packaging determinants and a novel nuclear targeting domain. J. Virol. 75362-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahnke, L. A., M. Belshan, and L. Ratner. 2006. Analysis of HIV-2 Vpx by modeling and insertional mutagenesis. Virology 348165-174. [DOI] [PubMed] [Google Scholar]
- 28.Mangeot, P. E., K. Duperrier, D. Negre, B. Boson, D. Rigal, F. L. Cosset, and J. L. Darlix. 2002. High levels of transduction of human dendritic cells with optimized SIV vectors. Mol. Ther. 5283-290. [DOI] [PubMed] [Google Scholar]
- 29.McCall, C. M., P. L. Miliani de Marval, P. D. Chastain II, S. C. Jackson, Y. J. He, Y. Kotake, J. G. Cook, and Y. Xiong. 2008. Human immunodeficiency virus type 1 Vpr-binding protein VprBP, a WD40 protein associated with the DDB1-CUL4 E3 ubiquitin ligase, is essential for DNA replication and embryonic development. Mol. Cell. Biol. 285621-5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morellet, N., S. Bouaziz, P. Petitjean, and B. P. Roques. 2003. NMR structure of the HIV-1 regulatory protein VPR. J. Mol. Biol. 327215-227. [DOI] [PubMed] [Google Scholar]
- 31.Naldini, L., U. Blomer, P. Gallay, D. Ory, R. Mulligan, F. H. Gage, I. M. Verma, and D. Trono. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272263-267. [DOI] [PubMed] [Google Scholar]
- 32.Pancio, H. A., N. Vander Heyden, and L. Ratner. 2000. The C-terminal proline-rich tail of human immunodeficiency virus type 2 Vpx is necessary for nuclear localization of the viral preintegration complex in nondividing cells. J. Virol. 746162-6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park, I. W., and J. Sodroski. 1995. Functional analysis of the vpx, vpr, and nef genes of simian immunodeficiency virus. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 8335-344. [PubMed] [Google Scholar]
- 34.Pion, M., A. Granelli-Piperno, B. Mangeat, R. Stalder, R. Correa, R. M. Steinman, and V. Piguet. 2006. APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J. Exp. Med. 2032887-2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajendra Kumar, P., P. K. Singhal, M. R. Subba Rao, and S. Mahalingam. 2005. Phosphorylation by MAPK regulates simian immunodeficiency virus Vpx protein nuclear import and virus infectivity. J. Biol. Chem. 2808553-8563. [DOI] [PubMed] [Google Scholar]
- 36.Schrofelbauer, B., Y. Hakata, and N. R. Landau. 2007. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc. Natl. Acad. Sci. USA 1044130-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Selig, L., J.-C. Pages, V. Tanchou, S. Prévéral, C. Berlioz-Torrent, L. X. Liu, L. Erdtmann, J.-L. Darlix, R. Benarous, and S. Benichou. 1999. Interaction with the p6 domain of the Gag precursor mediates incorporation into virions of Vpr and Vpx proteins from primate lentiviruses. J. Virol. 73592-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharova, N., Y. Wu, X. Zhu, R. Stranska, R. Kaushik, M. Sharkey, and M. Stevenson. 2008. Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 4e1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singhal, P. K., P. Rajendra Kumar, M. R. Subba Rao, and S. Mahalingam. 2006. Nuclear export of simian immunodeficiency virus Vpx protein. J. Virol. 8012271-12282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srivastava, S., S. K. Swanson, N. Manel, L. Florens, M. P. Washburn, and J. Skowronski. 2008. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 4e1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan, L., E. Ehrlich, and X. F. Yu. 2007. DDB1 and Cul4A are required for human immunodeficiency virus type 1 Vpr-induced G2 arrest. J. Virol. 8110822-10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tristem, M., C. Marshall, A. Karpas, and F. Hill. 1992. Evolution of the primate lentiviruses: evidence from vpx and vpr. EMBO J. 113405-3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ueno, F., H. Shiota, M. Miyaura, A. Yoshida, A. Sakurai, J. Tatsuki, A. H. Koyama, H. Akari, A. Adachi, and M. Fujita. 2003. Vpx and Vpr proteins of HIV-2 up-regulate the viral infectivity by a distinct mechanism in lymphocytic cells. Microbes Infect. 5387-395. [DOI] [PubMed] [Google Scholar]
- 44.Wen, X., K. M. Duus, T. D. Friedrich, and C. M. de Noronha. 2007. The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J. Biol. Chem. 28227046-27057. [DOI] [PubMed] [Google Scholar]
- 45.Wolfrum, N., M. D. Muhlebach, S. Schule, J. K. Kaiser, B. P. Kloke, K. Cichutek, and M. Schweizer. 2007. Impact of viral accessory proteins of SIVsmmPBj on early steps of infection of quiescent cells. Virology 364330-341. [DOI] [PubMed] [Google Scholar]
- 46.Wu, X., J. A. Conway, J. Kim, and J. C. Kappes. 1994. Localization of the Vpx packaging signal within the C terminus of the human immunodeficiency virus type 2 Gag precursor protein. J. Virol. 686161-6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu, X. F., Q. C. Yu, M. Essex, and T. H. Lee. 1991. The vpx gene of simian immunodeficiency virus facilitates efficient viral replication in fresh lymphocytes and macrophage. J. Virol. 655088-5091. [DOI] [PMC free article] [PubMed] [Google Scholar]