

Abstract
Aberrant activation of the canonical Wnt/β-catenin pathway occurs in almost all colorectal cancers and contributes to their growth, invasion and survival. Although dysregulated β-catenin activity drives colon tumorigenesis, additional genetic perturbations are required to elaborate fully malignant disease. To identify genes that both modulate β-catenin activity and are essential for colon cancer cell proliferation, we conducted two loss-of-function screens in human colon cancer cells and compared genes identified in these screens with an analysis of copy-number alterations in colon cancer specimens. One of these genes, CDK8, which encodes a member of the mediator complex, is located at 13q12.13, a region of recurrent copy number gain in a substantial fraction of colon cancers. Suppression of CDK8 expression inhibited proliferation in colon cancer cells characterized by high levels of CDK8 and β-catenin hyperactivity. CDK8 kinase activity was necessary for β-catenin driven transformation and expression of several β-catenin transcriptional targets. Together these observations suggest that therapeutic interventions targeting CDK8 may confer clinical benefit in β-catenin-driven malignancies.
The Wnt/β-catenin pathway is implicated in over 90% of colon cancers and in a fraction of other human malignancies. Loss of the tumor suppressor APC or activating CTNNB1 (β-catenin) mutations results in constitutive activity of the β-catenin-T cell factor (TCF) transcriptional complex, which drives adenoma formation1,2. Although mutations in TP53 or K-RAS cooperate with dysregulated β-catenin signaling to program a fully malignant phenotype3, these mutations are found in less than half of β-catenin-driven colon cancers4.
To identify oncogenes that modulate β-catenin-dependent transcription and regulate colon cancer cell proliferation, we conducted two RNAi-based loss-of-function screens. We engineered DLD1 colon cancer cells, which harbor APC deletions and depend on β-catenin for proliferation5, to stably express “TOPFLASH” β-catenin-luciferase and “FOPFLASH” mutant-Renilla reporter constructs6,7 (DLD1Rep). Suppression of β-catenin expression in DLD1Rep cells by three β-catenin-specific short hairpin RNAs (shRNA) markedly reduced the TOPFLASH/FOPFLASH ratio (Fig. 1a), confirming that reporter activity requires β-catenin expression. We then screened DLD1Rep cells with a shRNA library containing 4849 shRNAs that target 1000 genes, including 95% of the human kinome6. We found 34 genes whose expression was necessary for β-catenin activity, including two known β-catenin regulators, CSNK1G38 and CSNK1E9 (Fig. 1b and Supplementary Table 1).
Figure 1. RNAi screens to identify genes essential for colon cancer cell proliferation and β-catenin activity.
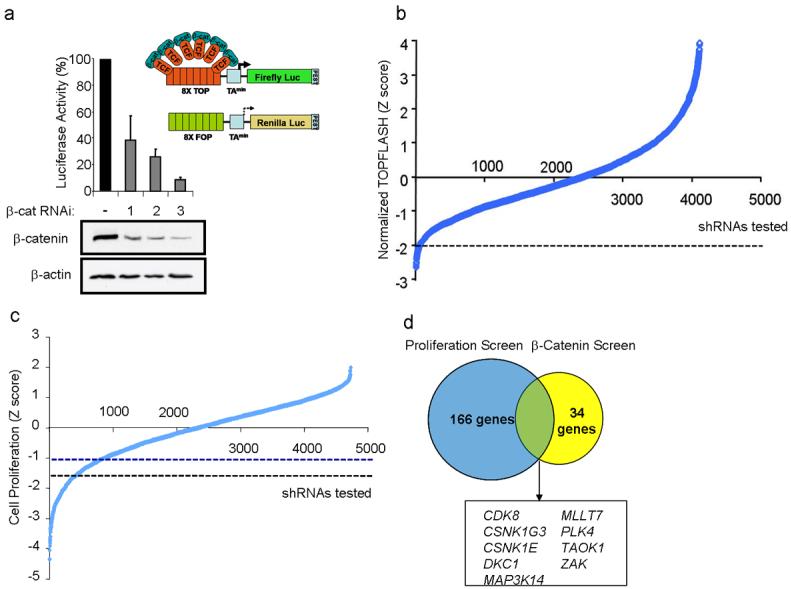
(a) Schematic of the DLD1Rep cell line showing the engineered 8X TOPFLASH and 8X FOPFLASH elements and relative TOP/FOP activity in the DLD1Rep cell line. (b) Distribution curve showing Z-scores representing β-catenin activity for all shRNAs tested in the DLD1Rep screen. shRNAs that reduced FOPFLASH levels to near background or activated FOPFLASH more than 2 standard deviations (SD) above mean were excluded (FOP ≤800 and FOP ≥2600 luciferase units). 2 of 5 CDK8-specific shRNAs were excluded on this basis. shRNA that induced Z-scores > 4 are not shown. Dashed line indicates Z score cutoff for shRNAs scored as hits. (c) Distribution curve showing Z-scores representing cell proliferation for shRNAs tested in HCT116 cells. This screen contained shRNAs targeting 1004 genes, and there was 92% overlap between the screens in (b) and (c). Blue and black dashed lines indicates Z score cutoff for shRNAs scored as hits. (d) Venn Diagram representation of 9 genes that reduced both β-catenin activity and colon cancer cell proliferation.
In parallel, we performed an arrayed, kinase-enriched shRNA screen in another β-catenin-dependent colon cancer cell line, HCT116, to identify genes essential for cancer cell proliferation. We identified 166 candidate genes necessary for proliferation using the criteria that at least two shRNAs targeting the same gene induced a decrease in proliferation. Among the genes identified in this screen were the oncogenes KRAS and MYC (Fig. 1c and Supplementary Table 2). Compilation of genes from the two screens revealed nine whose suppression affected both β-catenin transcriptional activity and colon cancer cell proliferation (Fig. 1d).
To determine whether any of these genes are amplified in colon cancers, we used single-nucleotide polymorphism (SNP) arrays and the GISTIC10,11 statistical method to conduct a genome-wide analysis of autosomal copy number (CN) alterations in primary resection specimens from 123 human colorectal adenocarcinomas (Fig. 2a). Among the nine genes identified by our RNAi screens, only CDK8 resides within a particularly significant amplicon at 13q12.13-13q12.2 (false discovery rate (FDR) =1×10−29) (Fig. 2a). These findings confirm recent reports that a large portion of chromosome 13 is amplified in colon cancers12,13. Fifty-eight of 123 (47%) samples harbored this region of CN gain (Fig. 2b and Supplementary Table 3).
Figure 2. Amplification and overexpression of CDK8 defines a subset of colon cancers.
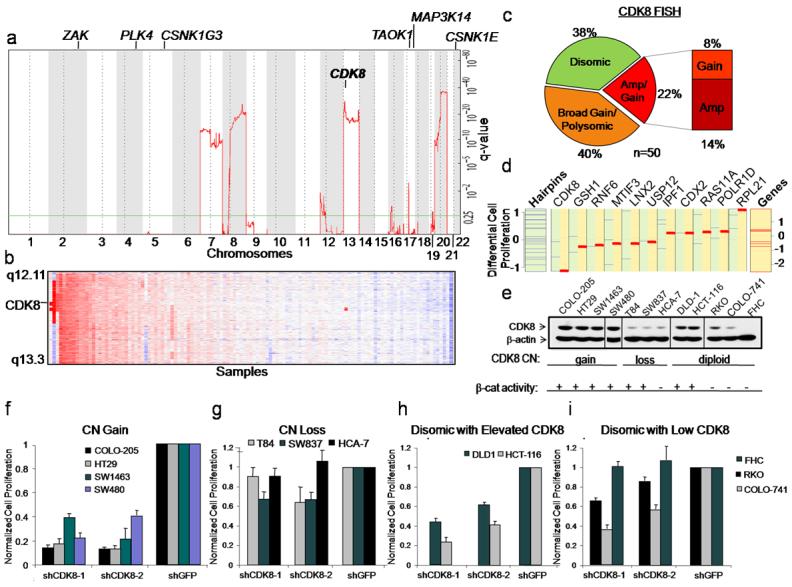
(a) Significance (y-axis) of recurrent amplifications at loci across the 22 autosomes (x-axis) identified by GISTIC analysis. Chromosomal location of RNAi hits is indicated above plot. (b) Heat map showing clustering of SNP array data, based on chromosome 13q12 copy number in 123 colon cancer specimens. Red indicates allelic gain, blue denotes loss. (c) Pie chart depicts the percentage of colon tumors exhibiting CDK8 CN gain, chromosome 13 polysomy or disomy (n=50) by FISH. Cutoff criteria for FISH are shown in Supplementary Table 4. (d) GSEA comparative analysis of suppressing resident genes in the minimal 13q12 region. Blue lines represent differential scores of cell proliferation effects for each validated shRNA that suppressed target gene expression by greater than 70%. The leftmost column demonstrates the total pool of validated hairpins for all genes on 13q12. Red lines represent a normalized enrichment score for each gene that takes into account the cell proliferation effects of all shRNAs and scores the specificity of the effects in cell lines that harbor or lack 13q gain. (e-i) Immunoblot analysis of CDK8 expression in 12 colon cancer cell lines. β-actin serves as a loading control (e). Effect of CDK8 suppression on proliferation of colon cancer cells that harbor chromosome 13 gain (f), harbor chromosome 13 loss (g), are disomic with higher CDK8 protein expression (h) and are disomic with lower CDK8 protein expression (i). Bar graph depicts cell proliferation normalized to the shGFP control for triplicate determinations. Error bars represent mean ± SD for a representative experiment performed in triplicate.
To confirm these findings, we performed fluorescence in situ hybridization (FISH), using probes specific for CDK8 and RB1 (chromosome 13 control probe), on a tissue microarray (TMA) carrying 50 evaluable colon cancer specimens. We detected CDK8 CN gain in 31 of 50 (62%) cases. 20 out of these 50 cases exhibited gains in both CDK8 and RB1 indicative of polysomy and consistent with recent observations linking RB/E2F1, β-catenin and CDK814. We also found CDK8 amplifications in 7 of these tumors (defined as CDK8:Control ratio ≥2) and low to moderate level CN gain (CDK8:Control ratio >1 and <2) in an additional 4 specimens (Fig. 2c and Supplementary Table 4). Immunohistochemical analysis of CDK8 expression in the same 50 specimens revealed elevated protein levels in 13 of 50 (26%) colon cancer samples, including those that showed CDK8 CN gain (Supplementary Fig. 1 and Supplementary Table 4). These observations indicate that CDK8 is amplified and overexpressed in a substantial fraction of colon cancers.
The minimal region shared by these tumors encompasses 16 annotated genes (Supplementary Fig. 2a). To determine if CDK8 is the primary target of this amplicon, we first assessed expression of these genes in colon cancer cells harboring chromosome 13q CN gain and found that 4 of the genes were not expressed (Supplementary Figure 2b). We suppressed the expression of the remaining 12 genes in four cell lines, two (HT29, COLO-205) that harbor 13q CN gain and two (SW837, T84) that show 13q loss (Supplementary Fig. 3a, b). To analyze the screen results on a per-gene basis in cell lines with either 13q12 CN gain or deletion, we used an adaptation of the GSEA15 method and found that CDK8 was the only gene in this region required for proliferation of cell lines harboring 13q gain (FDR=0.24) (Fig. 2d and Supplementary Table 5). These observations suggested that colon cancer cells harboring 13q12.2 amplification are particularly dependent on CDK8 expression for proliferation.
We then analyzed CDK8 CN gain and protein expression in a panel of 12 colon cancer lines. Four (COLO-205, HT29, SW1463, and SW480)16 of these 12 lines were found to harbor CDK8 gain (Supplementary Fig. 3a, b), and these cell lines exhibited the highest levels of CDK8 protein (Fig. 2e). Two additional colon cancer cell lines disomic for CDK8 (DLD1 and HCT116) also exhibited elevated CDK8 protein levels (Fig. 2d and Supplementary Fig.3a, b). Suppression of CDK8 expression induced substantially decreased proliferation in all six cell lines with elevated CDK8 protein levels (Fig. 2f,g) but inhibited proliferation rates in the cell lines with lower CDK8 protein levels to a lesser degree (Fig. 2h,i). Suppressing CDK8 in colon cancer cells reduced the fraction of cells in G1 and S phase, increased the number of aneuploid cells, and dramatically slowed cell proliferation without inducing apoptosis (Supplementary Fig. 4), similar to what was observed upon suppression of β-catenin. These observations demonstrate that colon cancer cells that express elevated CDK8 levels are highly dependent on its expression for proliferation.
To determine if CDK8 induces cell transformation, we overexpressed wild type CDK8 or a previously reported kinase inactive, substitution mutant (D173A; CDK8-KD)17 in immortal murine fibroblasts (NIH 3T3) (Fig. 3a). CDK8 expression induced focus formation, anchorage-independent colony growth, and tumor formation in immunodeficient animals (Fig. 3b-e), whereas the CDK8-KD mutant failed to transform the cells. These observations confirm that CDK8 is a bona fide oncogene, whose kinase activity is necessary for oncogenic activity.
Figure 3. CDK8 and transformation.
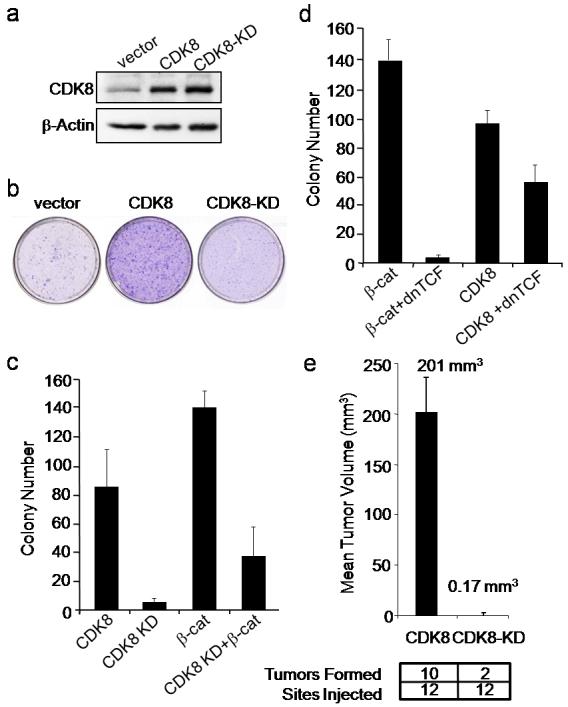
(a) Immunoblot analysis showing CDK8 and CDK8 kinase-dead (CDK8-KD) expression in NIH-3T3 cells. (b) Focus formation assay of NIH-3T3 cells expressing CDK8 or CDK8-KD. (c) CDK8 kinase activity drives anchorage independent (AI) growth and is necessary for β-catenin mediated AI growth. AI colony growth in NIH-3T3 cells infected with the indicated retroviral vectors. (d) β-catenin suppression only partial blocks CDK8 induced AI growth. Dominant negative TCF (dnTCF) was introduced in the presence of β-catenin or CDK8. AI colony growth was performed as indicated in (c). (e) CDK8 kinase activity drives tumor formation. Mean tumor volume from subcutaneous tumors formed NIH-3T3 cells expressing CDK8 or CDK8-KD constructs in immunodeficient mice. The difference in tumor formation between CDK8 and CDK8-KD was statistically significant, as assessed by unpaired t-test (p value = 0.0001). All experiments were performed in triplicate, and mean ± SD is shown.
To dissect the relationship between CDK8 and β-catenin activity, we measured endogenous β-catenin activity in the 12 cell lines used above. The RKO, COLO-741, HCA-7 and FHC cell lines do not harbor known APC or β-catenin mutations18,19,20 and, as predicted, exhibited low levels of β-catenin activity. Of these four cell lines, suppression of CDK8 induced a substantial proliferation effect only in COLO-741 (Fig. 2h and Supplementary Fig. 5a). Similarly, of the 12 cell lines tested, the six cell lines with highest CDK8 elevated levels showed a greater dependence on β-catenin for proliferation (Supplementary Fig. 5b).
CDK8 is a cyclin-dependent kinase member of the mediator complex, which couples transcriptional regulators to the basal transcriptional machinery21. To explore the role of CDK8 in modulating β-catenin transcriptional activity, we confirmed that suppressing CDK8 with two independent, CDK8-specific shRNAs (shCDK8-1, shCDK8-2) in an additional cell line, SW480, also reduced β-catenin-dependent transcriptional activity (Fig. 4a). CDK8 kinase activity depends on the co-factor Cyclin C22, and we found that Cyclin C knockdown preferentially affected colon cancer cell lines with chromosome 13q gain (Supplementary Fig. 6a, b). To test whether CDK8 kinase activity is required to regulate β-catenin activity, we expressed wild-type CDK8 or CDK8-KD in DLD1Rep cells carrying a shRNA targeting the 3'-untranslated region, shCDK8-1 and found that only wild-type CDK8 partially rescued the effects of suppressing endogenous CDK8(Supplementary Fig. 6c). These observations demonstrate that the kinase activity of CDK8 is necessary for both CDK8-induced transformation and β-catenin driven transcription.
Figure 4. CDK8 mediates transcription of β-catenin driven downstream target genes.
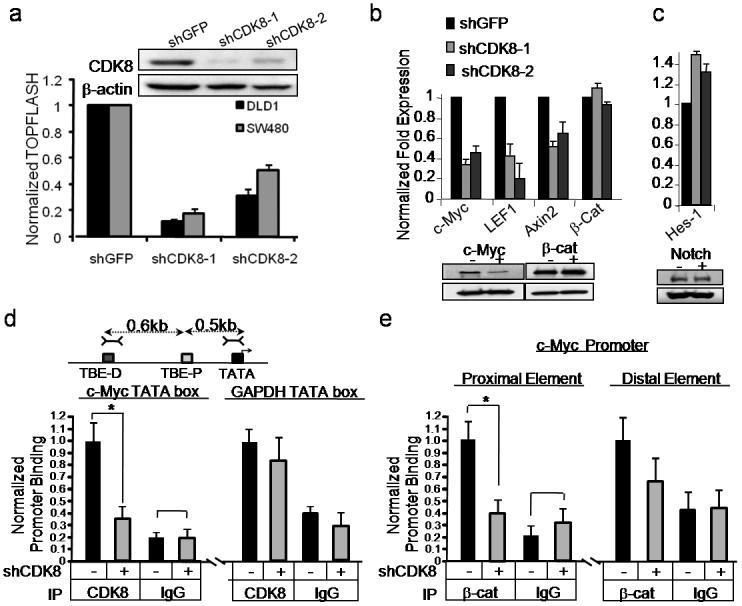
(a) Bar graph depicts β-catenin activity as normalized TOPFLASH (TOPFLASH/FOPFLASH) after introduction of CDK8-specific shRNAs. An immunoblot shows CDK8 protein levels at time of assay. β-actin serves as a loading control. (b, c) Bar graphs shows mRNA abundance of endogenous β-catenin and Notch transcriptional targets after CDK8 suppression in COLO-205 and DLD1 cells. Myc, β-catenin, and Notch expression changes were assessed by immunoblotting. β-actin expression serves as a loading control. (d) CDK8 binds the MYC promoter. Schematic representation depicts the location of proximal and distal TCF binding elements (TBE, gray boxes) and MYC promoter TATA box. Dashed Lines indicate the distance from TBEs to TATA box. Hatched lines depict the PCR primers used for chromatin immunoprecipitation (ChIP). Bar graph shows CDK8 binding to the MYC TATA box in COLO-205 colon cancer cells treated with the indicated shRNAs and assayed by ChIP. (d, e) Asterisk indicates statistically significance as assessed by unpaired student t-test (p value ≤ 0.013) and p ≤ 0.14 for the control IgG for (d) and (p value ≤ 0.005) and p ≤ 0.34 for the IgG control. For (e). (e) Bar graph depicts β-catenin binding to the indicated MYC promoter elements in the presence or absence of CDK8. ChIP assays were performed as above. All experiments were conducted at least two times in triplicate. Error bars represent the mean ± SD for a representative experiment performed in triplicate. Asterisk indicates a statistically significance as assessed by unpaired student t-test
The TCF-β-catenin complex regulates expression of several genes implicated in colon cancer, including MYC23, Axin226, and LEF127. Suppression of CDK8 in DLD1 and COLO-205 cells reduced expression of each of genes (Fig. 4b). In contrast, we failed to observe changes in the expression of Notch or HES-1 (Fig. 4c), previously reported targets of CDK824. Thus, CDK8 modulates a subset of β-catenin driven genes previously implicated in cancer23,25,26.
We then performed chromatin immunoprecipitation (ChIP) near two verified β-catenin/TCF binding elements (TBE)27 in the MYC promoter, as an example of a β-catenin regulated gene, to test whether CDK8 directly modulates MYC expression at the promoter level. We found CDK8 associated with the MYC promoter (Fig. 4d). We therefore asked if loss of CDK8 binding at the MYC promoter affects the ability of β-catenin to bind at the proximal and distal TBEs. Suppression of CDK8 expression reduced the amount of β-catenin bound to the proximal element within the MYC promoter but had little effect on the amount associated with the distal element (Fig. 4e). These observations implicate CDK8 and the mediator complex21 as a direct regulator of β-catenin-driven transcription.
To test whether CDK8 activity is required for β-catenin driven transformation, we expressed the dominantly interfering CDK8-KD mutant28 in transformed NIH-3T3 cells expressing a constitutively active β-catenin mutant (Fig. 3c). Disruption of CDK8 activity inhibited β-catenin driven transformation, whereas a dominantly interfering TCF construct, previously shown to inhibit β-catenin-induced cellular transformation29, only partially abrogated CDK8-mediated transformation (Fig. 3d). These observations suggest that while CDK8 is required for β-catenin mediated transformation, the full capacity of CDK8 to transform cells may extend beyond its ability to activate β-catenin.
In summary, we have used an integrated approach to identify CDK8 as an oncogene in a substantial fraction of colorectal cancers and demonstrate that its kinase activity is essential for its ability to regulate β-catenin dependent transcription and transformation. These observations indicate that CDK8 acts in part by co-activating β-catenin driven transcription in colon cancers characterized by both high CDK8 expression and β-catenin activity. Accordingly, therapeutic interventions that target the CDK8 kinase activity in such cancers may be of clinical value.
Methods Summary
Lentiviral infections were performed using pLKO.1 lentiviral shRNA constructs generated by the RNAi Consortium (TRC)6 and are listed in Supplementary Table 6. For high throughput screening, cells infected with lentiviruses were allowed to grow for 4 d (DLD1Rep) or 5 d (HCT116). Suppression of β-catenin activity was defined as the ability of at least one shRNA to decrease activity more than 2 standard deviations (SD) below the Z score. Suppression of proliferation was defined as the capacity for at least one shRNA to decrease proliferation more than 1.5 SD below the mean Z score and at least one additional shRNA targeting the same kinase to decrease proliferation more that 1 SD below the mean Z score. For tissue analyses, a Tissue Microarray (TMA) composed of human colon cancer tissue and matching patient normal colon was subjected to immunohistochemical staining. For FISH, BAC clones were hybridized in dual colors to four-micron TMA sections as follows: the RB1 probe labeled in SpectrumGreen was used as a chromosome 13 control probe and the RP11-726I20 BAC, spanning CDK8, was labeled with SpectrumOrange dUTP (both from Abbott Molecular/Vysis, Inc.).
Supplementary Material
Acknowledgements
We thank E. Shin for expert technical assistance in immunohistochemistry, M. Miri for assistance with sample collection, and G. Getz for assistance with SNP array analysis. This work was supported in part by a T32 NIH grant (R.F.) and a GI SPORE Career Development Grant #P50CA127003 (R.F.), a Harvard-MIT Clinical Investigator Training Program Fellowship (A.B), Department of Defense Prostate Cancer Postdoctoral Fellowships (I.G, S.Y), Warren-Whitman-Richardson, Hagerty Foundation Research Fellowships (I.D.) and K12 award (M.G.C.). J.B. is a Beatriu de Pinos Fellow of the Departament d'Educació i Universitats de la Generalitat de Catalunya. In accordance with Harvard Medical School guidelines, we disclose that W.C.H., M.M., M.L. and R.A.S. are consultants for Novartis.
The SNP data can be found at: http://research3.dfci.harvard.edu/cdk8colon/index.php
References
- 1.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–20. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 2.Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med. 2002;8:1323–7. doi: 10.1038/nm791. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 4.Smith G, et al. Mutations in APC, Kirsten-ras, and p53--alternative genetic pathways to colorectal cancer. Proc Natl Acad Sci U S A. 2002;99:9433–8. doi: 10.1073/pnas.122612899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van de Wetering M, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–50. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 6.Moffat J, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–98. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 7.Korinek V, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–7. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 8.Davidson G, et al. Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature. 2005;438:867–72. doi: 10.1038/nature04170. [DOI] [PubMed] [Google Scholar]
- 9.Hino S, Michiue T, Asashima M, Kikuchi A. Casein kinase I epsilon enhances the binding of Dvl-1 to Frat-1 and is essential for Wnt-3a-induced accumulation of beta-catenin. J Biol Chem. 2003;278:14066–73. doi: 10.1074/jbc.M213265200. [DOI] [PubMed] [Google Scholar]
- 10.Beroukhim R, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–12. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weir BA, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin ES, et al. Common and distinct genomic events in sporadic colorectal cancer and diverse cancer types. Cancer Res. 2007;67:10736–43. doi: 10.1158/0008-5472.CAN-07-2742. [DOI] [PubMed] [Google Scholar]
- 13.Tsafrir D, et al. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res. 2006;66:2129–37. doi: 10.1158/0008-5472.CAN-05-2569. [DOI] [PubMed] [Google Scholar]
- 14.Morris EJ,JJ, Y F, Di Stefano L, Herr A, Moon NS, Kwon EJ, Haigis KM, Naar AM, Dyson NJ. E2F1 represses β-catenin transcription and is antagonized by both pRB and CDK8. Submitted to Nature. 2008 doi: 10.1038/nature07310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garraway LA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–22. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 17.Gold MO, Rice AP. Targeting of CDK8 to a promoter-proximal RNA element demonstrates catalysis-dependent activation of gene expression. Nucleic Acids Res. 1998;26:3784–8. doi: 10.1093/nar/26.16.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci U S A. 1997;94:10330–4. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–4. [PubMed] [Google Scholar]
- 20.Rowan AJ, et al. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc Natl Acad Sci U S A. 2000;97:3352–7. doi: 10.1073/pnas.97.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conaway RC, Sato S, Tomomori-Sato C, Yao T, Conaway JW. The mammalian Mediator complex and its role in transcriptional regulation. Trends Biochem Sci. 2005;30:250–5. doi: 10.1016/j.tibs.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Tassan JP, Jaquenoud M, Leopold P, Schultz SJ, Nigg EA. Identification of human cyclin-dependent kinase 8, a putative protein kinase partner for cyclin C. Proc Natl Acad Sci U S A. 1995;92:8871–5. doi: 10.1073/pnas.92.19.8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sansom OJ, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–9. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 24.Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell. 2004;16:509–20. doi: 10.1016/j.molcel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 25.Yook JI, et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- 26.Murakami T, et al. Constitutive activation of Wnt/beta-catenin signaling pathway in migration-active melanoma cells: role of LEF-1 in melanoma with increased metastatic potential. Biochem Biophys Res Commun. 2001;288:8–15. doi: 10.1006/bbrc.2001.5719. [DOI] [PubMed] [Google Scholar]
- 27.He TC, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 28.Chiang MY, et al. Identification of a conserved negative regulatory sequence that influences the leukemogenic activity of NOTCH1. Mol Cell Biol. 2006;26:6261–71. doi: 10.1128/MCB.02478-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolligs FT, Hu G, Dang CV, Fearon ER. Neoplastic transformation of RK3E by mutant beta-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol Cell Biol. 1999;19:5696–706. doi: 10.1128/mcb.19.8.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.