

Abstract
Excitatory amino acid transporters (EAATs) are the primary regulators of extracellular glutamate concentrations in the central nervous system. Their dysfunction may contribute to several neurological diseases. To date, five distinct mammalian glutamate transporters have been cloned. In brain, EAAC1 (excitatory amino acid carrier 1) is the primary neuronal glutamate transporter, localized on the perisynaptic membranes that are near release sites. Despite its potential importance in synaptic actions, little is known concerning the regulation of EAAC1 trafficking from the endoplasmic reticulum (ER) to the cell surface. Previously, we identified an EAAC1-associated protein, GTRAP3-18, an ER protein that prevents ER exit of EAAC1 when induced. Here we show that RTN2B, a member of the reticulon protein family that mainly localizes in the ER and ER exit sites interacts with EAAC1 and GTRAP3-18. EAAC1 and GTRAP3-18 bind to different regions of RTN2B. Each protein can separately and independently form complexes with EAAC1. RTN2B enhances ER exit and the cell surface composition of EAAC1 in heterologous cells. Expression of short interfering RNA-mediated knockdown of RTN2B decreases the EAAC1 protein level in neurons. Overall, our results suggest that RTN2B functions as a positive regulator in the delivery of EAAC1 from the ER to the cell surface. These studies indicate that transporter exit from the ER controlled by the interaction with its ER binding partner represents a critical regulatory step in glutamate transporter trafficking to the cell surface.
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system that contributes not only to the fast synaptic neurotransmission, but also to complex physiological process such as learning and memory (1, 2). However, excessive levels of extracellular glutamate are excitotoxic and lead to neuronal death in acute or chronic neural injury (3). The rapid clearance of glutamate from the extracellular space is accomplished by binding and subsequent uptake of glutamate by a family of Na+-dependent, high affinity glutamate transporters. In mammalian tissue, five subtypes of transporters have been identified and cloned: EAAT13 or GLAST1, EAAT2 or GLT-1, EAAT3 or EAAC1, EAAT4, and EAAT5, which exhibit an identity in amino acid sequence of about 50% among each other (4). Their dysfunction may contribute to neurological diseases: such as amyotrophic lateral sclerosis (ALS), stroke, epilepsy, and Alzheimer disease (5).
The EAAC1 subtype of transporter is enriched on the post-synaptic processes of pyramidal cells in cortex and hippocampus as well as in inhibitory interneurons (6, 7). There is evidence that EAAC1 limits spillover between excitatory synapses in hippocampus (8), and provides precursor for the synthesis of the inhibitory neurotransmitter, γ-aminobutyric acid (9, 10). In addition to these physiologic roles, some studies have shown that EAAC1 expression is altered under various pathologic conditions and can contribute to neurodegeneration (11).
Appropriate trafficking and targeting of EAAC1 to the surface of the perisynaptic region is thought to be crucial for synaptic function. A redistribution of EAAC1 from an intracellular pool to the cell surface has been reported in both long term potentiation and contextual fear conditioning (12), suggesting an association between regulated trafficking of EAAC1 and models of learning and memory. However, the mechanisms governing progression of transporters trafficking to the cell surface via the secretory pathway have not been identified.
We initiated the study to elucidate the mechanisms on EAAC1 trafficking by defining the EAAC1 interacting proteins. GTRAP3-18 was the first protein described as a binding partner of EAAC1, and is capable of reducing the activity of EAAC1 both in vivo and in vitro when induced (13). GTRAP3-18 resides in the ER and prevents complex oligosaccharide formation on EAAC1 in a dose-dependent manner by restricting EAAC1 ER exit.4 To further elucidate how EAAC1 trafficking is regulated in the early compartments of the secretory pathway, we sought to identify proteins that interact with GTRAP3-18 through a yeast two-hybrid approach. Here we report that a member of reticulon family protein RTN2B interacts with GTRAP3-18 and EAAC1. RTN2B facilitates the trafficking of EAAC1 out of the ER, whereas GTRAP3-18 retains EAAC1 in the ER and reduces its cell surface expression when overexpressed. Our results implicate that the surface composition of transporters may be adjusted by controlling their export from the ER.
EXPERIMENTAL PROCEDURES
Yeast Two-hybrid Screen
The yeast two-hybrid screen was performed using the HF7c′ yeast strain harboring the reporter genes HIS3 and β-galactosidase under the control of GAL4 activation. The 188 amino acids of full-length Gtrap3-18 were sub-cloned in-frame into pGBT9 (GAL4 binding domain vector, Clontech) and used to screen a rat brain cDNA library constructed in pGAD10 (GAL4 activation domain vector, Clontech). The plasmids were transformed into HF7c′ yeast cells and positive clones selected on triple-minus plates (Leu−, Trp−, His−) and assayed for β-galactosidase activity Positive clones were co-transformed with either the bait plasmid or the original pGAD10 vector into yeast cells to confirm the interaction. A screen of 1.5 × 106 double transformants on histidine-deficient medium approximately yielded 600 His-positive clones; 61 of which displayed β-galactosidase activity. Plasmids were rescued from each of these His-positive and β-galactosidase-positive clones and analyzed by restriction pattern and sequencing analysis.
CDNA Constructs
Rat Rtn2B 5′ gene-specific primer was designed based on cDNA sequence of mouse Rtn2B, which shares 97% identity to rat Rtn2B according to the 3′ sequence alignment. The 3′ gene-specific primer was designed based on sequence of one of the positive clones obtained from the yeast two-hybrid screen. PCR was performed using rat brain cDNA as a template. The 1410-bp product was subcloned into TA vector (pCR2.1, Invitrogen). For expression, full-length Rtn2B cDNA was subcloned into a pcDNA3.1D/V5-His-TOPO vector (Invitrogen). Truncation mutants of Rtn2B were constructed by PCR and deletions were introduced using QuikChange II XL Site-directed mutagenesis kit (Stratagene, La Jolla, CA). GFP-EAAC1 was generated by inserting the full-length EAAC1 in pEGFP-C1 vector (Clontech). CFP or YFP was fused to the NH2 terminus of GTRAP3-18 or RTN2B (Clontech). HA-tagged GTRAP3-18, Spinophilin protein fragment (1–221 aa), and Myc-tagged EAAC1 were described previously (13).4
Antibodies and Reagents
Antibodies were raised by bovine serum albumin-conjugated peptides. Rabbit anti-RTN2 antibody was generated by immunizing rabbits with the peptide corresponding to amino acids 451–469. The antibody was then affinity purified on a column of covalently coupled peptide. Chicken anti-RTN2B and anti-GTRAP3-18 were produced by Aves Labs (Tigard, OR) using the peptides against amino acids 30–47 of RTN2B and amino acids 14–28 of GTRAP3-18, respectively. The following antibodies were also used: rabbit anti-EAAC1 (6), anti-Calnexin (Stressgen, Victoria, BC Canada), anti-Bip (Stressgen), anti-GM130 (BD Biosciences), anti-RTN1 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-NogoA (Santa Cruz Biotechnology), anti-neuron-specific class III β-tubulin (R&D Systems, Minneapolis, MN), anti-actin (Sigma), anti-Myc (Roche Applied Science), rabbit anti-V5 (Novus, Littleton, CO), mouse anti-V5 (Invitrogen), and anti-HA (Covance, Richmond, CA). For the secondary antibodies, we used the Cy series (Jackson ImmunoResearch, West Grove, PA) and AlexaFluor series (Molecular Probes, Carlsbad, CA).
Cell Cultures
HEK 293 and COS7 cells were maintained according to standard protocols (ATCC), split 1 day before transfection and used at 50–70% confluence. Mammalian cells were transfected using FuGENE 6 reagent (Roche) according to the manufacturer’s directions. For fluorescence resonance energy transfer (FRET) experiments, the CaPO4 precipitation method was used to transiently transfect COS7 cells. Primary neuronal and mixed cultures were prepared as described (15). Briefly, cortex was dissected from E15–18 SD rats, dissociated in 0.25% trypsin, and cells were plated on poly-D-lysin/lamimin-coated coverslips (BD Biosciences) at a density of 300,000/well in 24-well plates.
Immunoprecipitation
Transiently transfected HEK 293T cells were solubilized with ice-cold IP buffer (50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 1% Triton X-100 with protease inhibitors) for 30 min with rotation and centrifuged to remove the cellular debris. Cell lysates were pre-cleared with Protein A/G Plus-agarose slurry (Santa Cruz Biotechnology) for 2 h at 4 °C. Aliquots (40 μl) of pre-clarified lysates (10% of total) were saved as input. Pre-clarified lysates were diluted with IP buffer and then incubated with 40 μl of a 50% HA beads (Covance) or V5 beads slurry (Novus) overnight at 4 °C. Beads were washed three times in IP buffer containing 100 mM NaCl and eluted with SDS sample buffer. Input and immunoprecipitated samples were analyzed by SDS-PAGE followed by Western blot. For whole brain immunoprecipitation, adult rat brain was washed with cold HB (homogenize buffer: 50 mM Tris-HCl, pH 7.4, 320 mM sucrose, 5 mM EDTA with protease inhibitors) three times and then homogenized. The homogenate was diluted with IP buffer containing Triton X-100 to a final concentration of 1% and solubilized at 4 °C for 3 h. The insoluble material was removed by centrifugation at 20,000 × g for 30 min at 4 °C. The supernatant fraction was subjected to pre-clarification and then incubated overnight at 4 °C with protein A-Sepharose beads and rabbit anti-EAAC1 or pre-immuno IgG, or with IgY precipitating agarose (Chemicon, Temecula, CA) and chicken anti-RTN2B or control non-immuno IgY. Beads were washed and bound proteins were eluted with SDS sample buffer.
Immunocytochemistry
Transfected COS7 cells were fixed in 4% paraformaldehyde in phosphate-buffered saline containing 4% sucrose, rinsed, permeabilized in 0.25% saponin in phosphate-buffered saline for 5 min, and then blocked with 5% normal goat serum in phosphate-buffered saline containing 0.05% saponin for 1 h. Cells were then incubated with appropriate primary antibodies for 1 h in 2% normal goat serum and 0.05% saponin in phosphate-buffered saline, washed, and probed with secondary antibodies for 1 h. Primary neurons or mixed cultures were grown on the coated coverslips. After 5–8 days, cells were fixed with 4% paraformaldehyde, washed, and blocked with 5% normal goat serum and 0.1% Triton X-100 before incubating them with primary antibodies at room temperature for 1 h. Cells were then washed and incubated with secondary antibodies for 30 min. Fluorescence images were acquired by confocal microscopy using a Zeiss LSM 510 laser scanning microscope.
FRET Microscopy
The three-filter method was used to calculate net FRET (16). The described system setup was additionally equipped with two Ludl filter wheels DC (to change between excitation/emission filters in less than 100 ms) to perform the three filter method (17). Images were taken using a double dichroic mirror for all measurements. Various combinations of excitation and emission filters were used to produce the CFP, YFP, and FRET images (ICFP, IYFP, and IFRET, respectively). Background fluorescence was subtracted from all images; net FRET (NFRET) was calculated using the equation: NFRET = IFRET − (α × IYFP) − (β × ICFP), where α and β represent the bleed-through values for YFP (0.235 ± 0.013; n = 6 experimental days) and CFP (0.638 ± 0.003; n = 6 experimental days). To gauge the system both with positive and negative controls for FRET imaging, we used a fusion protein of CFP and YFP, termed CYFP (18) and CFP and YFP vectors co-expressed, respectively. All experiments were performed with n = 6 experimental days.
Cell Surface Biotinylation
Biotinylation of cell surface proteins in 6-well plates was performed as described (19), using EZ-link Sulfo-NHS-Biotin and UltraLink immobilized Streptavidin beads (Pierce). Samples were boiled in SDS-PAGE sample buffer and subjected to Western blot analysis. Densitometry analysis was performed using Quantity One (Bio-Rad) software.
Glutamate Uptake Assay
Na+-dependent glutamate transport activity was measured (20) in transfected HEK 293 cells grown on 6-well plates. The wells were incubated with 1 ml of either sodium (Na+) or choline (Na−) Krebs buffer containing 10 μM glutamate and 0.5 μM L-[3H]glutamate for 4 min at 37 °C. Glutamate uptake was stopped on ice and by three rinses in ice-cold wash buffer (0.05 M Tris, 0.16 M NaCl, pH 7.4). Then cells were solubilized in 1 ml of 0.1 M NaOH, and 500 μl of lysate was analyzed for radioactivity in a scintillation counter. Na+-dependent uptake was defined as the difference in radioactivity accumulated in Na+-containing buffer and in choline (Na−)-containing buffer. Protein concentration was measured to ensure that the uptake activity was compared in the same amount of protein content.
siRNA Transfection
RNA interference was carried out using SMARTPool Rtn2B siRNA and negative control siRNA (Dharmacon, Lafayette, CO) as described (21). Transfections were performed with Lipofectamine 2000 (Invitrogen). The effect of siRNA on the expression of heterologous RTN2B in HEK 293 cells was assessed by immunoblotting with lysates from cells harvested 24 h after transfection. Neurons grown on coverslips were co-transfected with pEGFP-C2 plasmid (Clontech) and siRNA at 5 days in vitro. Following transfection, neurons were washed and cultured in neuronal growth medium for 48 h before being fixed and subjected to immunocytochemistry analysis. Quantification of the staining was performed using Metamorph software (Molecular Devices, Downingtown, PA).
Statistics
Statistical analyses of FRET microscopy and immunostaining intensity were performed using one-way analysis of variance followed by Tukey-Kramer multiple comparisons test using JMP statistics software (SAS Institute, Cary, NC). Quantifications of immunoblotting in the biotinylation experiment and glutamate uptake were also analyzed by analysis of variance followed by Tukey-Kramer multiple comparisons test. In some cases, Student’s t test was employed. All data are graphed or presented as mean ± S.E.
RESULTS
Identification of RTN2B as an Interacting Protein of EAAC1 and Its Binding Partner GTRAP3-18
Previous studies showed that the ER protein GTRAP3-18 associated with EAAC1 and negatively regulated EAAC1 glutamate uptake activity on the cell surface via a retention of EAAC1 in the ER (13).4 To further reveal the regulatory mechanism of EAAC1 trafficking between the ER and Golgi complex, we initiated a program to identify proteins interacting with GTRAP3-18. The 188 amino acids of full-length GTRAP3-18 were used as bait to probe a yeast two-hybrid rat brain cDNA library. Two of the clones isolated in this screen contained the sequences highly homologous to the COOH-terminal coding region of mouse brain reticulon Rtn2. Full-length rat Rtn2 cDNA (GI: 33415442) was isolated from rat brain cDNA. It encodes a protein of 469 aa, with a calculated relative molecular mass of 52 kDa.
Like other reticulon genes, the Rtn2 gene is transcribed into different mRNA variants, which share a common carboxyl-terminal segment. Three RTN2 proteins have been identified in human: RTN2A (545 aa, translated from mRNA contains exon 1 to 11), RTN2B (472 aa, translated from mRNA without exon 5), and RTN2C (205 aa, translated from mRNA initiated from exon 5 to 11) (22). In mouse, two major transcripts encode a short (RTN2C, 204 aa) and a long (RTN2B, 471 aa, mRNA contains no exon 5) protein, which has been found to be enriched in muscle and brain, respectively (23). The rat RTN2 cDNA we cloned shared 95.3% identity to the long mouse RTN2 brain transcript, so it was named RTN2B.
To confirm the yeast two-hybrid results, we first examined the interaction of RTN2B with GTRAP3-18 by FRET analysis in living mammalian cells. Three-filter FRET analysis revealed a statistically significant higher FRET signal of CFP-RTN2B with YFP-GTRAP3-18 compared with the negative control (Fig. 1, A and B), indicating that these two proteins interact in living cells.
FIGURE 1. Interactions of RTN2B with GTRAP3-18 and EAAC1.

A and B, interaction of RTN2B and GTRAP3-18 in living cells as shown by FRET. Three-filter FRET microscopy was performed on COS7 cells transfected with equal amounts of CFP-RTN2B and YFP-GTRAP3-18. A representative single experiment taken in the CFP, YFP, or FRET channel is shown (A). B, a summary of 6 experimental days with 25 to 35 evaluated regions. NFRET was calculated as described under “Experimental Procedures” (n = 6, *, p <0.0005). C and D, co-immunoprecipitation of GTRAP3-18, RTN2B, and EAAC1 from transfected HEK293 cells. Lysates from HEK 293 cells transfected with HA-GTRAP3-18, RTN2B-V5, and/or Myc-EAAC1, as indicated were immunoprecipitated with an HA (C) or a V5 (D) antibody. Cell lysates (Input: 10% of the total lysates) and precipitates (IP) were immunoblotted for HA, V5, and Myc. The complex oligosaccharide form and high mannose oligosaccharide form of EAAC1 were marked with open and filled arrowheads, respectively. E, immunoprecipitation of RTN2B, GTRAP3-18, and EAAC1 with anti-RTN2B or anti-EAAC1 antibodies from adult rat brain extracts. Blots were probed with RTN2B, GTRAP3-18 (arrow), EAAC1, and RTN1A antibodies.
The interactions between RTN2B and GTRAP3-18, as well as EAAC1, were further tested by in vitro and in vivo co-immunoprecipitation. Full-length Rtn2B was subcloned in-frame with a V5 tag at its COOH terminus. GTRAP3-18 and EAAC1 were NH2 terminally tagged with an HA and a Myc epitope, respectively. These constructs were cloned into mammalian expression vectors and expressed the expected protein products after transfection into HEK 293 cells (Fig. 1, C and D). On gradient gels, as other transporter isoforms (e.g. GLT1), EAAC1 migrated as double bands with close molecular mass, representing two oligosaccharide states (24). The upper band (marked with open arrowhead) was the complex oligosaccharide form (the mature form). When co-expressed with GTRAP3-18, EAAC1 was retained in the ER thus enriched in the high mannose oligosaccharide form (the immature form, marked with filled arrowhead, Fig. 1, C and D). In all Western blots shown in this report, more than 80% of EAAC1 migrates as dimers, which were presented in the figures. This is a well known observation in the electrophoretic studies of glutamate transporters (25). Monomer and multioligomers have the same results as the dimer. Transfected GTRAP3-18-HA displayed a double band pattern on 15% gel. We prepared lysates from the transfected cells, and precipitated GTRAP3-18 with an HA antibody and RTN2B with a V5 antibody. As shown in Fig. 1C, RTN2B-V5 and Myc-EAAC1 were co-immunoprecipitated with HA-GTRAP3-18 (Fig. 1, lanes 4 and 5). HA-GTRAP3-18 and Myc-EAAC1 were also found in RTN2B-V5 precipitates (Fig. 1D, lanes 7–9). At a similar expression level, the binding of RTN2B to an HA-tagged actin-binding protein, Spinophilin fragment (1–221 aa), our negative control, was not detected (Fig. 1, C, lane 6, and D, lane 10). Thus, RTN2B specifically interacted with GTRAP3-18 and transporter EAAC1 in transfected cells. Notably, the interactions of GTRAP3-18 and EAAC1 with RTN2B were independent of each other (Fig. 1, C, lane 4, and D, lanes 7 and 8).
To determine whether RTN2B associates with transporter EAAC1 in vivo, co-immunoprecipitation experiments were performed using detergent extracts from adult rat brain. EAAC1 was immunoprecipitated with an anti-EAAC1 antibody, along with RTN2B and GTRAP3-18. Meanwhile, using a chicken antibody raised against residues 30 – 48 on the NH2 terminus of RTN2B, we were able to co-immunoprecipitate EAAC1 and GTRAP3-18 with RTN2B. Specifically, RTN1A was not present in the EAAC1 or RTN2B immunoprecipitates (Fig. 1E). These data indicate that RTN2B associated with both EAAC1 and GTRAP3-18 in brain tissue. The consistent results of in vitro and in vivo immunoprecipitation also demonstrated that the epitope tags added to RTN2B, GTRAP3-18, and EAAC1 did not have an effect on their interaction.
The First Transmembrane Domain of RTN2B Binds to GTRAP3-18
Having established that GTRAP3-18 and EAAC1 interacted with RTN2B independently, we next investigated whether GTRAP3-18 and EAAC1 competed for the binding to RTN2B or they associated with different domains of RTN2B. RTN2B was predicted to have two transmembrane domains. The transmembrane domains and the 66-aa loop region in between, called reticulon-homology domain, shared homologies with other reticulon family members (26, 27). The NH2-terminal domain had three hydrophilic regions. To determine the region(s) on RTN2B responsible for EAAC1 and GTRAP3-18 binding, we generated NH2-terminal, COOH-terminal, or transmembrane domain-truncated constructs of RTN2B tagged with V5 epitopes (Fig. 2A). All of the RTN2B mutants were expressed at their expected sizes although some at apparently reduced levels compared with the full-length protein. In the immunoprecipitation blot using V5 antibodies, besides the bands migrating at the corresponding molecular weights, extra bands were observed in the full-length and some truncated RTN2B, except ΔC. As estimated by molecular weights, these bands likely represented the dimers of the RTN2B proteins (marked with * in red in Fig. 2, B and C). This observation is consistent with the previous reports that RTNs can form homo-oligomers (28, 29).
FIGURE 2. EAAC1 and GTRAP3-18 bind to different regions of RTN2B.

A, diagram of truncation constructs used for the co-immunoprecipitation experiments to identify the binding domains to EAAC1 and GTRAP3-18. All the constructs, including the full-length RTN2B were COOH terminally tagged with V5 epitopes. The Y-2-H fragment represented the construct obtained from the yeast two-hybrid screen for interaction with GTRAP3-18. Co-immunoprecipitation of HA-GTRAP3-18 (B) or Myc-EAAC1 (C) with truncation mutants of RTN2B-V5 from transfected HEK 293 cells. Cell lysates were precipitated with a V5 antibody. Input: 10% of the total lysates. Input and immunoprecipitated samples were analyzed by SDS-PAGE followed by Western blot. The bands migrating at the molecular weights corresponding to the possible dimers of RTN2B truncation mutants were marked with an * in red in the immunoprecipitation blots. Open and filled arrowheads indicated the complex oligosaccharide (mature) form and high mannose oligosaccharide (immature) form of EAAC1.
GTRAP3-18 displayed a significantly reduced expression level when co-expressed with RTN2B-ΔN2 (Fig. 2B, lane 3). It co-immunoprecipitated with all the RTN2B mutant proteins, except ΔTM1, demonstrating that the first transmembrane domain was required for GTRAP3-18 association. Also, RTN2B-ΔN (residues 239 – 469) co-precipitated with GTRAP3-18 (Fig. 2B, lane 11), consistent with the yeast two-hybrid screen result. Considering that both RTN2B-ΔC (residues 1–320) and RTN2B-ΔN (residues 239 – 469) bound to GTRAP3-18 (Fig. 2B, lanes 10 and 11), we concluded that the first transmembrane domain included in both constructs was sufficient for GTRAP3-18 interaction. Therefore, the first transmembrane domain was responsible for GTRAP3-18 binding.
NH2 Terminus of RTN2B Is Required for EAAC1 Binding
EAAC1 was co-precipitated with full-length RTN2B, as well as ΔTM1 (Fig. 2C, lanes 7 and 12). Whereas RTN2B-ΔN1, −ΔN2, and ΔN proteins were all readily recovered from the immunoprecipitation, we failed to detect EAAC1 protein in these precipitates. This suggested that the NH2-terminal domain of RTN2B was required for its association with EAAC1 (Fig. 2C, lanes 8, 9, and 11). There was a much smaller amount of RTN2B-ΔC expressed and recovered from the anti-V5 immunoprecipitation compared with the full-length and NH2-terminal-truncated RTN2B, making it difficult to draw a clear conclusion on whether the NH2 terminus (RTN2B-ΔC) was sufficient for EAAC1 interaction (Fig. 2C, lane 10). Nevertheless, RTN2B-ΔTM, which failed to bind to GTRAP3-18, retained the interaction with EAAC1, indicating this trans-membrane domain was not involved in EAAC1 binding (Fig. 2, B, and C, lane 12). Under a closer examination, 90% of EAAC1 was present in the mature form (open arrowhead) in the total cell lysates, whereas in the precipitates, this percentage decreased to 40%, indicating RTN2B bound to both mature and immature forms (filled arrowhead) of EAAC1 with a preference to the immature one enriched in the ER (Fig. 2C, comparing lanes 1 and 6 with 7 and 12).
The results of the truncation studies were summarized in Table 1. Taken together, it showed that the first transmembrane domain of RTN2B was required and sufficient to bind to GTRAP3-18. For EAAC1 association, the NH2 terminus of RTN2B, more specifically the first hydrophilic peak was required. Because EAAC1 and GTRAP3-18 interacted with different domains of RTN2B, they did not compete for the binding sites. Thus, it was likely that all three proteins interacted in one complex.
TABLE 1. Summary of RTN2B truncation studies.
In “expression/stability,” ++++ represents the highest and + the lowest level. In the other sections, + indicates the ability to bind to EAAC1 or GTRAP3-18, whereas − means no ability. −/? indicates a very low level of RTN2B-ΔC was expressed and recovered from the IP, but no clear conclusion can be drawn regarding the interaction between this truncation of RTN2B and EAAC1.
| FL | ΔN1 | ΔN2 | ΔN | ΔC | ΔTM1 | |
|---|---|---|---|---|---|---|
| Expression/stability | ++++ | +++ | ++ | ++ | + | +++ |
| Binding to GTRAP3-18 | + | + | + | + | + | − |
| Binding to EAAC1 | + | − | − | − | −/? | + |
Expression and Localization of RTN2B in Neurons
Our data showed that RTN2B interacted with neuronal glutamate transporter EAAC1 and the ER protein GTRAP3-18. We expected to find RTN2B protein in neurons and associated with the ER. To further verify this hypothesis, we examined the expression profile and localization of RTN2B.
Two specific immunoreactive bands, migrating at 52 and 18 kDa, were detected in Western blot analysis of rat tissue homogenates, using a rabbit antibody against the last 19 aa on the COOH-terminal region of RTN2, where the two RTN2 isoforms shared identity. The long isoform was present in the brain and spinal cord, whereas the short one was expressed in the skeleton muscle and heart (data not shown). The distribution of the long isoform of RTN2 (i.e. RTN2B) in all regions of the brain and the spinal cord was further confirmed in a Western blot, using a chicken antibody to the NH2 terminus of RTN2B. This antibody specifically recognized the 52-kDa RTN2B. The transfected HEK sample was loaded to verify that the transfected RTN2B-V5 was recognized by this antibody as well. A little higher molecular weight of RTN2B-V5 was caused by the tag (Fig. 3A). In a primary neuron and astrocyte mixed culture from embryonic rat cortex, the anti-RTN2B antibody reacted only with neurons, which was co-labeled with neuronal specific β-tubulin III. In contrast, GTRAP3-18 had a broader distribution than RTN2B and was found in both cultured neurons and astrocytes (Fig. 3B). In addition, using reverse transcriptase-PCR, RTN2B transcripts were detected only in neuronal culture, but not in astrocyte culture (data not shown). Co-staining the primary cultured neurons for RTN2B, EAAC1, and GTARP3–18 showed that these three proteins, which interacted with each other as demonstrated in the whole brain co-immunoprecipitation (Fig. 1E), co-expressed in neurons (Fig. 3C).
FIGURE 3. Expression of RTN2B in brain and neurons.

A, Western blot analyses of RTN2B protein expression in HEK 293 cells transfected with RTN2B-V5 and multiple tissues, using a chicken anti-RTN2B against the N-terminal peptide of RTN2B. As estimated by molecular weight, the bands marked with arrows likely represented dimer and multimers of RTN2B. The band marked with * in white in the skeleton muscle lane was determined as a cross-reaction band by peptide block blots. B, immunostaining of RTN2B and GTRAP3-18 proteins in primary cortex neuron and astrocyte mixed culture from E16 rat brain. Neurons were labeled with the neuronal specific β-tubulin antibodies. Surrounding astrocytes were stained with the astrocyte marker glial fibrillary acidic protein. Scale bars represented 20 μm. C, immunofluorescence microscopy of primary cortex neuronal culture. Neurons were stained with anti-RTN2B, anti-EAAC1, and anti-GTRAP3-18 antibodies. Scale bar, 20 μm. D, immunoblots of total (T), intracellular (I), and plasma membrane (M) fractions of cell surface biotinylation of primary neurons. Actin was used as total protein and intracellular controls.
Reticulon family members were generally thought to be located in the ER (hence the name), according to the immunocytochemical localization of RTN1A in transfected COS cells (26, 30). However, it has been reported that Nogo/RTN4-A, -B, and -C are present on the cell surface of fibroblasts, cultured neurons, and muscle cells (31). In the primary neuronal cultures, using a cell surface biotinylation technique, we found that a small fraction (17 ± 4%) of RTN2B was present on the cell surface compared with total protein, similar to NogoA (23 ± 3% on the plasma membrane), whereas RTN1A was exclusively distributed in the intracellular fraction (Fig. 3D). This finding suggests that RTN2B function might not be restricted to the ER.
Consistent with the nomenclature, we observed ER localization of RTN2B in transfected COS7 cells (Fig. 4). Immunostaining showed that transfected RTN2B displayed a combined pattern of reticular filaments and punctuate perinuclear structures. The vast majority of the RTN2B staining co-localized with Calnexin, showing the typical distribution of an ER protein with the reticular network around the nuclear envelope. In addition to the prominent peripheral tubular distribution, some RTN2B exhibited a tight and punctuate perinuclear staining, suggesting a Golgi localization shown by cis-Golgi marker GM130 staining, where Calnexin was essentially absent (Fig. 4, A and B, arrows). The stable and stationary RTN2B puncta distributed on the ER membrane (Fig. 4B, circles and inset) identified the ER exit sites, specialized subdomains of ER membrane where cargo is concentrated into vesicles that bud from the ER and are transported to the Golgi (32). This distribution pattern suggested that at least a portion of RTN2B may exit out of the ER and traffic to the Golgi apparatus.
FIGURE 4. Subcellular localization of RTN2B in transfected cells.

Confocal immunofluorescence microscopy of COS7 cells transiently transfected with RTN2B-V5. A, fixed cells were stained with anti-Calnexin (ER marker), anti-GM130 (Golgi marker, indicated by arrows), and anti-V5 antibodies. B, a higher magnification view of the RTN2B ER association. The representative examples of ER exit sites were marked with dotted circles and shown in the inset panel. Arrows indicate the putative Golgi localization. Scale bars, 20 μm.
RTN2B Increases EAAC1 Trafficking from ER to the Cell Surface
Having established that RTN2B is localized in neurons and associated with EAAC1, we next investigated the biological effects of this interaction on EAAC1. We first examined the cell surface distribution of EAAC1. We used a cell membrane impermeant biotinylation reagent to selectively label cell surface protein. The densitometry analysis of the following Western blot showed that co-expression of RTN2B with EAAC1 resulted in a 26% increase in the percentage of EAAC1 protein distributed on the cell surface. Co-expression of GTRAP3-18 increased the ER form of EAAC1, and reduced its cell surface distribution (Fig. 5, A and B). In parallel, we measured the sodium-dependent glutamate transport activity of HEK cells that had been transfected either with EAAC1 alone or together with RTN2B or GTRAP3-18. Consistent with previous reports (13),4 glutamate transport activity decreased progressively with increasing expression of the GTRAP3-18 protein. As co-expression of RTN2B modestly increased the cell surface level of EAAC1, it also resulted in a concomitant 27% increase in Na+-dependent glutamate transport activity. Notably, expression of RTN2B did not alter the inhibitory effect of GTRAP3-18 on transport activity (Fig. 5C).
FIGURE 5. Effect of RTN2B on EAAC1 trafficking in transfected cells.
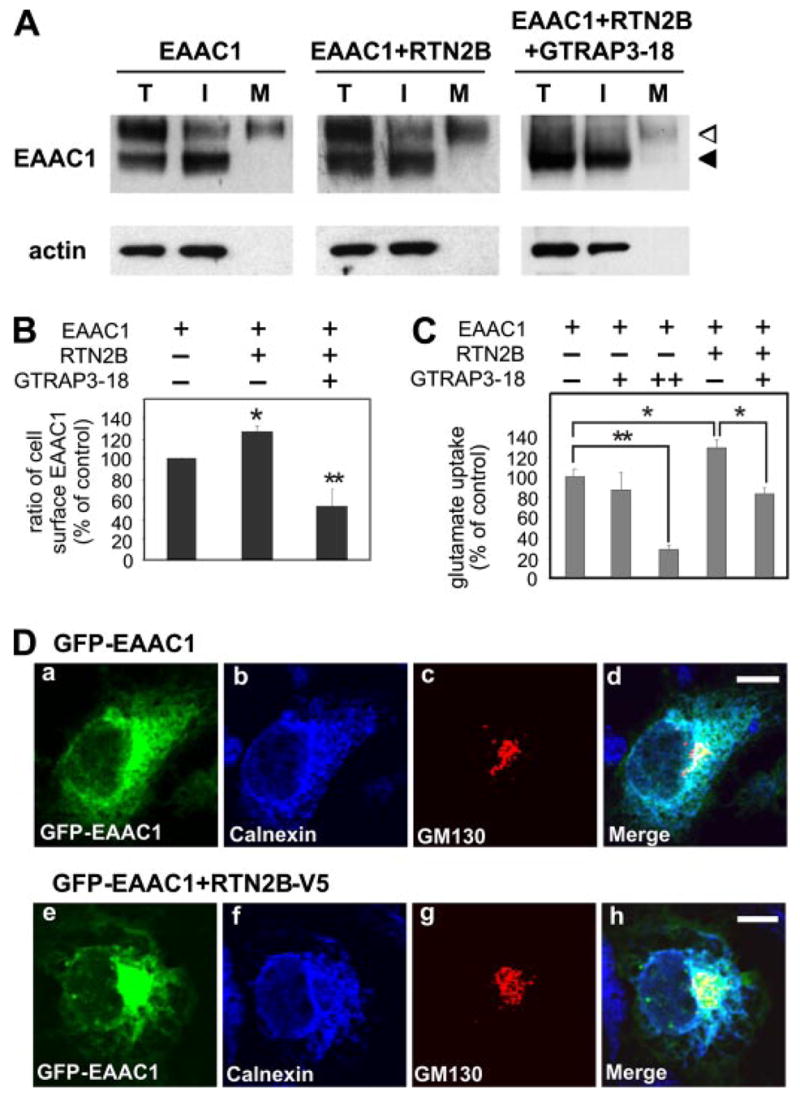
A, cell surface biotinylation of HEK 293 cells transfected with Myc-EAAC1 alone or together with RTN2B-V5 and GTRAP3-18. Total (T), intracellular (I), and biotinylated plasma membrane (M) fractions were blotted with a Myc antibody. Actin was used as total and intracellular controls. B, quantification of immunoblots for cell surface biotinylation. Ratio of cell surface EAAC1 was compared (*, p < 0.05; **, p < 0.01). Cell surface (mature form in the M fraction) EAAC1 was normalized with total EAAC1 (mature and immature forms in the T fraction) and calculated as the ratio of cell surface EAAC1. C, glutamate uptake assay in HEK 293 cells transfected with the indicated plasmids. Each + indicates equal amount, and ++ represents double amount of DNA added in the transfection reaction (*, p < 0.05; **, p < 0.001). D, immunofluorescence microscopy of COS7 cells transfected with GFP-EAAC1 alone or together with RTN2B-V5. Fixed cells were stained with anti-Calnexin (ER marker) and anti-GM130 (cis-Golgi marker) antibodies. Scale bars, 10 μm.
To determine whether the increased cell surface EAAC1 was the result of up-regulated ER exit of EAAC1, we compared the intracellular localization of GFP-EAAC1 in the presence or absence of RTN2B. As shown in Fig. 5D, we observed a strong but not complete co-localization of intracellular GFP-EAAC1 with the ER marker Calnexin in the perinuclear region and tubuloreticular network. Some GFP-EAAC1 displayed a punctuated staining pattern that overlapped with the cis-Golgi marker GM130. Because only a small amount of EAAC1 (about 25%) was delivered to the plasma membrane in transfected cells, according to surface biotinylation (Fig. 5A), a cell surface GFP signal was difficult to detect in this imaging paradigm, because of the presence of strong intracellular signals. Co-expression of RTN2B caused an apparent shift of localization of GFP-EAAC1 signals with a reduced ER co-localization and an increased punctuated Golgi staining (Fig. 5D).
In summary, these results demonstrated that RTN2B protein positively regulated EAAC1 trafficking to the plasma membrane by facilitating its ER exit. In our transient transfection conditions, RTN2B was expressed at a level 5–10 times higher than the endogenous protein, whereas EAAC1 was overexpressed more than 100-fold (data not shown). This extremely low ratio of RTN2B to EAAC1 may be the reason of the modest effects observed in the heterologous cell expression system. To justify these observations from the overexpressed heterologous system, we next studied primary cortical neurons where RTN2B and EAAC1 are endogenously expressed.
Knocking Down RTN2B Reduces EAAC1 Expression in Neurons
To test if the endogenous RTN2B affects the trafficking of EAAC1 in neurons, we used an RNA interference approach to knock-down Rtn2B mRNA in cultured primary cortical neurons. The specificity and efficacy of siRNAs were tested in HEK 293 cells. The Rtn2B siRNA almost completely abolished transfected RTN2B protein (Fig. 6A) and was used for our loss-of-function studies in rat neurons. A GFP plasmid was co-transfected with the siRNA in the primary neuronal cultures to serve as a marker of the transfected cells. Under normal conditions, RTN2B and EAAC1 were present in all cultured neurons (33). Recently, Horton and Ehlers and other groups (32) demonstrated that ER and ER exit sites were distributed throughout neurons, including the dendrites. Consistent with their reports and our immunocyto-chemistry result obtained from COS7 cells, which showed that RTN2B localized in the ER and ER exit sites (Fig. 4), RTN2B was found abundant in neurites (Fig. 6B). There was no change in RTN2B and EAAC1 staining intensity in neurons transfected with negative control siRNA (Fig. 6, B, panels a–c, and C, panels a and b). Transfection of Rtn2B siRNA resulted in a significant reduction of endogenous RTN2B staining (red) on the soma and processes of transfected neurons to 26 ± 7 and 22 ± 11%, respectively, relative to the untransfected ones (Fig. 6, B, panels d2–i2, and C, panel a). Meanwhile, the intensity of EAAC1 in the soma and process domains in these neurons was remarkably decreased to 49 ± 11 (soma) and 40 ± 15% (processes) of that in the untransfected neurons (Fig. 6, B, panels d3–f3, and C, panel b). The unaltered NogoA staining intensity indicated that Rtn2B siRNA specifically knocked down RTN2B, and did not cause nonspecific protein degradation as observed in unhealthy neurons (Fig. 6, B, panels g3–i3, and C, panel c). Corroborating our results obtained from the transfected COS cells, we conclude that the EAAC1 expression level was reduced if its trafficking from the ER was compromised through loss of RTN2B. These findings were consistent with those reported for another transporter subtype, GLT1 (23). In this case, mutation of the leucine-based ER export motif resulted in retention of GLT1 in the ER and a reduced expression level. In a different scenario, we observed the reduced expression of EAAC1, versus an accumulation/build-up in the ER when we inhibited protein trafficking from the ER in the primary cultured neurons (data not shown). This also suggested that the ER form of EAAC1 was rapidly degraded when its trafficking out of ER was compromised.
FIGURE 6. Knock down of RTN2B in neurons results in reduced total expression of EAAC1.

A, immunoblot analysis of heterologously expressed RTN2B in the HEK 293 cell co-transfected with negative (Neg) or Rtn2B siRNA for 24 h. B, Rtn2B siRNA knocks down endogenous RTN2B in neurons. Hippocampal neurons at 5 days in vitro were transfected with GFP plus negative (Neg) or RTN2B siRNA for 48 h. Neurons were fixed and stained with antibodies against RTN2B (red), EAAC1 (blue), or NogoA (blue). Soma and processes of GFP positive neurons were marked with arrows and shown with neighboring GFP negative counterparts. Scale bars, 10 μm. C, quantification of the relative immunostaining intensity for RTN2B (a), EAAC1 (b), and NogoA (c) on the soma and processes in neurons transfected with siRNA and GFP (n = 5– 8, *, p < 0.0001). The staining intensity of neighboring GFP negative neurons was used as control to calculate the percentage.
DISCUSSION
Role of RTN2B on Protein Trafficking
In this study, we show that RTN2B, a reticulon family member localized to neuronal ER and ER exit sites, interacts with neuronal glutamate transporter EAAC1. We further show that an EAAC1-RTN2B complex exists in brain and that RTN2B influences EAAC1 trafficking to its normal targets in the soma and neurites. Co-expression with RTN2B reduces the EAAC1 ER localization, increases the Golgi distribution, and eventually enhances the cell surface expression. Our results indicate that the RTN2B protein plays an important role on efficient targeting of EAAC1 to the plasma membrane by facilitating its ER exit. However, because a small fraction of RTN2B localized in the Golgi and plasma membrane, our data did not exclude the possible effects of RTN2B on other stages of the secretory pathway, such as the endocytosis for instance. Considering the localization of RTN2B in the ER, the early stage of the protein trafficking, it is very possible that other cell surface transporters or receptors may bind to RTN2B and their ER exit may be under the regulation of RTN2B. Our preliminary data show that RTN2B has the ability to increase the cell surface expression of another neuronal transporter, EAAT4, which is enriched in Purkinje cells.5 Several possible mechanisms underlying this regulation can be proposed. First, RTN2B may promote the correct folding or assembly of EAAC1 complexes. EAAC1 and other glutamate transporters form homomultimers (34, 35). Oligomer formation occurs in the ER and is a prerequisite for newly formed transporters to pass the stringent ER quality control mechanisms. Only properly assembled transporters are able to be recruited into COPII vesicles that mediate ER to Golgi trafficking (36). RTN2B may facilitate EAAC1 conformation change and complex assembly in the neurons. The second possibility is that RTN2B may serve as a particular adaptor protein coupling the ER export machinery with multimeric complex cargos. Given the widespread, neuronal localization of RTN2B, it is not unreasonable to speculate that RTN2B may function as an adaptor protein for a variety of neuron surface transporters or receptors that exit out of the ER as multimeric complexes. Third, RTN2B may accelerate the EAAC1 ER exit by binding to the export signal or shielding the retention motif and eventually making it more accessible to the COPII transport machinery. A conserved arginine-based ER retention signal and a luminal leucine motif facilitating ER exit have been identified in the GLT1 subtype of glutamate transporters (24). These two motifs are also present on the COOH-terminal domain of EAAC1. The high homology between transporters suggests that they use similar mechanisms to regulate their ER retention and forward trafficking. Further mapping the RTN2B binding site on EAAC1 will provide more information on whether RTN2B acts by masking or unmasking ER transport signals.
Although more than 300 reticulon family members have been identified in all eukaryotic organisms from yeast to human, their functions still remain unrevealed. On the basis of recent reports, several hypotheses have been formulated. RTN1 interacts with SNARE proteins (37), a medium chain of the AP-2 adaptor complex (38), RME-1 (39), Bcl-2 (40), and Spastin, an ATPase belonging to the AAA family of proteins (41). Those studies implicate that RTN1 plays a role in protein trafficking processes. A role in membrane trafficking in the early secretory pathway compartments of RTN3 also has been proposed, because overexpression of RTN3 inhibits ERGIC-53 recycling back to ER and interferes with vesicular stomatitis virus glycoprotein transport (29). Moreover, RTN3 has been found to associate with BACE1, and negatively regulates the secretion of Aβ peptide (42). Recently, the role of RTNs as “morphogenic” proteins was proposed. Rapoport and colleagues (43) show that RTN4A/NogoA is required for ER tubule formation by an in vitro ER formation assay. In yeast, deletion of the RTN1 gene results in reorganization of the cortical ER from a highly reticulated structure to a more cisternal one, pointing to involvement of RTN1p in ER structuring (44). However, yeast cells lacking the RTNs grow normally and have no secretion defects or morphological changes in the peripheral ER network. Our results on RTN2B strengthen the hypothesis that RTNs affect the trafficking of certain groups of proteins (e.g. membrane transporters) along the early secretory pathway, because knocking down RTN2B in neurons did not result in expression or distribution changes of NogoA (Fig. 6, B and C). This indicates that the general secretory machinery and ER morphology remain intact after loss of RTN2B.
Regulation of Transporter Cell Surface Expression in the Early Stages of the Secretory Pathway
Cellular localization of glutamate transporters is important for the precise control of extra-cellular glutamate levels. In particular, the polarized distribution of transporters has received substantial attention. Previous studies have documented the perisynaptic localization and enrichment of EAAC1 in hippocampus (45). However, trafficking of transporters in the early secretory pathway has been largely ignored (36). It was thought that, once properly folded and assembled, proteins exit out of the ER by default (46). As more and more ER retention and export signals were identified in transmembrane and soluble secretory cargos (36, 47, 48), unexpected complexity has been revealed in the regulation of protein trafficking from the ER to the Golgi. Moreover, the interactions of cell surface channels and neurotransmitter receptors with their binding partners have been reported to affect their ER to Golgi transport. Besides, ER exit of multimeric cell membrane proteins is regulated by other diverse mechanisms including mRNA splicing and editing, phosphorylation, and interactions with the ER quality control machinery (48, 49). All of these mechanisms controlling ER exit have a major impact on the surface abundance and function of channels and receptors. Like these proteins, glutamate transporters assemble into multimeric complexes in the ER and move along the secretory pathway to surface membranes of dendrites and soma. A number of similar mechanisms have been reported governing the regulation of transporter ER exit, such as export and retention signals and quality control of oligomerization (36). In this report, we show that GTRAP3-18, RTN2B, and EAAC1 are co-expressed in the neurons. ER to Golgi transport of EAAC1 is inhibited and facilitated by GTRAP3-18 and RTN2B, respectively. The inhibitory role of GTRAP3-18 in neurons is only observed when its expression is increased by induction (13). Based on these observations, we proposed the following model on the regulation of EAAC1 ER exit modulated by RTN2B and GTRAP3-18. In normal conditions, RTN2B facilitates EAAC1 exit from the ER, and the natural expression of GTRAP3-18 is too low to display any inhibitory effect. In some non-physiological conditions, such as induction by methyl-β-cyclodextrin (13, 50) and in some cancer lines, the increased GTRAP3-18 has a dominant inhibitory effect on EAAC1 trafficking. This regulation of ER exit governs the activity and density of EAAC1 on the cell surface. Our work demonstrates that regulation of ER exit of transporters plays a critical role in its trafficking to the cell surface. Interestingly, increased Golgi localization of EAAC1, and redistribution of the transporter between intracellular and surface membranes has been reported in a kainic acid-induced rodent epilepsy model (51). Furthermore, increased protein expression of EAAC1 has been observed in the pilocarpine model of epilepsy (52, 53). In addition, other proteins such as β3 spectrin (GTRAP41) and PDZrhoGEF (GTRAP48) have been identified that regulate cytoplasmic and plasma membrane distribution of the neuronal glutamate transporter EAAT4 (54, 55). In fact mutations of one of these, β3 spectrin, is the cause of spinal cerebellar ataxia and involves cytoplasmic/plasma membrane mishandling of transporter trafficking (14). It will be interesting to see whether the altered transporter surface composition under these pathologic conditions involves, or is caused by, changes in ER exit of the transporter, regulated by proteins such as RTN2B and/or GTRAP3-18.
Footnotes
This work was supported by National Institutes of Health Grants NS33958, NS40151, and NS52179 (to J. D. R.) and Austrian Science Foundation/FWF Grants P17076 and 18706 (to H. H. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: EAAT1, excitatory amino acid transporter 1; FRET, fluorescence resonance energy transfer; GFP, green fluorescent protein; CFP, cyan fluorescent protein; YFP, yellow fluorescent protein; HA, hemagglutinin; aa, amino acid(s); IP, immunoprecipitation; HEK, human embryonic kidney; siRNA, small interfering RNA.
Ruggiero, A. M., Liu, Y., Vidensky, S., Maier, S., Jung, E., Farhan, H., Robinson, M. B., Sitte, H., and Rothstein, J. D. (December 31, 2007) J. Biol. Chem.
Y. Liu and J. D. Rothstein, unpublished data.
References
- 1.Fonnum F. J Neurochem. 1984;42:1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- 2.Dingledine R, Borges K, Bowie D, Traynelis SF. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 3.Ozawa S, Kamiya K, Tsuzuki K. Prog Neurobiol. 1998;54:581–618. doi: 10.1016/s0301-0082(97)00085-3. [DOI] [PubMed] [Google Scholar]
- 4.Seal RP, Amara SG. Annu Rev Pharmacol Toxicol. 1999;39:431–456. doi: 10.1146/annurev.pharmtox.39.1.431. [DOI] [PubMed] [Google Scholar]
- 5.Maragakis NJ, Rothstein JD. Neurobiol Dis. 2004;15:461–473. doi: 10.1016/j.nbd.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 7.Conti F, DiBiasi S, Minelli A, Rothstein JD, Melone M. Cereb Cortex. 1998;8:108–116. doi: 10.1093/cercor/8.2.108. [DOI] [PubMed] [Google Scholar]
- 8.Diamond JS. J Neurosci. 2001;21:8328–8338. doi: 10.1523/JNEUROSCI.21-21-08328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sepkuty JP, Cohen AS, Eccles C, Rafiq A, Behar K, Ganel R, Coulter DA, Rothstein JD. J Neurosci. 2002;22:6372–6379. doi: 10.1523/JNEUROSCI.22-15-06372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathews GC, Diamond JS. J Neurosci. 2003;23:2040–2048. doi: 10.1523/JNEUROSCI.23-06-02040.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- 12.Levenson J, Weeber E, Selcher JC, Kategaya LS, Sweatt JD, Eskin A. Nat Neurosci. 2002;5:155–161. doi: 10.1038/nn791. [DOI] [PubMed] [Google Scholar]
- 13.Lin G, Orlov I, Ruggiero AM, Dykes-Hoberg M, Lee A, Jackson M, Rothstein JD. Nature. 2001;410:84–88. doi: 10.1038/35065084. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda Y, Dick KA, Weatherspoon MR, Gincel D, Armbrust KR, Dalton JC, Stevanin G, Durr A, Zuhlke C, Burk K, Clark HB, Brice A, Rothstein JD, Schut LJ, Day JW, Ranum LP. Nat Genet. 2006;38:184–190. doi: 10.1038/ng1728. [DOI] [PubMed] [Google Scholar]
- 15.Roche KW, Huganir RL. Neuroscience. 1995;69:383–393. doi: 10.1016/0306-4522(95)00253-f. [DOI] [PubMed] [Google Scholar]
- 16.Xia Z, Liu Y. Biophys J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farhan H, Korkhov VM, Paulitschke V, Freissmuth M, Sitte HH. J Biol Chem. 2004;279:28553–28563. doi: 10.1074/jbc.M307325200. [DOI] [PubMed] [Google Scholar]
- 18.Schmid JA, Scholze P, Kudlacek O, Freissmuth M, Singer EA, Sitte HH. J Biol Chem. 2001;276:3805–3810. doi: 10.1074/jbc.M007357200. [DOI] [PubMed] [Google Scholar]
- 19.Duan S, Anderson CM, Stein BA, Swanson RA. J Neurosci. 1999;19:10193–10200. doi: 10.1523/JNEUROSCI.19-23-10193.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 21.Rumbaugh G, Adams JP, Kim JH, Huganir RL. Proc Natl Acad Sci U S A. 2006;103:4344–4351. doi: 10.1073/pnas.0600084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roebroek AJ, Contreras B, Pauli IG, Van de Ven WJ. Genomics. 1998;51:98–106. doi: 10.1006/geno.1997.5175. [DOI] [PubMed] [Google Scholar]
- 23.Kalandadze A, Wu Y, Fournier K, Robinson MB. J Neurosci. 2004;24:5183–5192. doi: 10.1523/JNEUROSCI.0839-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danbolt NC. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 25.Geisler JG, Stubbs LJ, Wasseman WW, Mucenski ML. Mamm Genome. 1998;9:274–282. doi: 10.1007/s003359900748. [DOI] [PubMed] [Google Scholar]
- 26.Oertle T, Schwab ME. Trends Cell Biol. 2003;13:187–194. doi: 10.1016/s0962-8924(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 27.Oertle T, van der Haar ME, Bandtlow CE, Robeva A, Burfeind P, Buss A, Huber AB, Simonen M, Schnell L, Brosamle C. J Neurosci. 2003;23:5393–5406. doi: 10.1523/JNEUROSCI.23-13-05393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senden NH, van de Velde H, Broers JL, Timmer ED, Kuijpers HJ, Roebroek AJ, Van de Ven WJ, Ramaekers FC. Eur J Cell Biol. 1994;65:341–353. [PubMed] [Google Scholar]
- 29.Wakana Y, Koyama S, Nakajima K, Hatsuzawa K, Nagahama M, Tani K, Hauri HP, Melancon P, Tagaya M. Biochem Biophys Res Commun. 2005;334:1198–1205. doi: 10.1016/j.bbrc.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 30.Van de Velde HJ, Senden NH, Roskams TA, Broers JL, Ramaekers FC, Roebroek AJ, Van de Ven WJ. J Cell Sci. 1994;107:2403–2416. doi: 10.1242/jcs.107.9.2403. [DOI] [PubMed] [Google Scholar]
- 31.Dodd DA, Niederoest B, Bloechlinger S, Dupuis L, Loeffler JP, Schwab ME. J Biol Chem. 2005;280:12494–12502. doi: 10.1074/jbc.M411827200. [DOI] [PubMed] [Google Scholar]
- 32.Horton AC, Ehlers MD. Nat Cell Biol. 2004;6:585–591. doi: 10.1038/ncb0704-585. [DOI] [PubMed] [Google Scholar]
- 33.Coco S, Verderio C, Trotti D, Rothstein JD, Volterra A, Matteoli M. Eur J Neurosci. 1997;9:1902–1910. doi: 10.1111/j.1460-9568.1997.tb00757.x. [DOI] [PubMed] [Google Scholar]
- 34.Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, Lehre KP, Danblot NC. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- 35.Eskandari S, Kreman M, Kavanaugh MP, Wright EM, Zampighi GA. Proc Natl Acad Sci U S A. 2000;97:8641–8646. doi: 10.1073/pnas.97.15.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farhan H, Freissmuth M, Sitte HH. Handb Exp Pharmacol. 2006;175:233–249. doi: 10.1007/3-540-29784-7_12. [DOI] [PubMed] [Google Scholar]
- 37.Steiner P, Kulangara K, Sarria JC, Glauser L, Regazzi R, Hirling H. J Neurochem. 2004;89:569–580. doi: 10.1111/j.1471-4159.2004.02345.x. [DOI] [PubMed] [Google Scholar]
- 38.Iwahashi J, Hamada H. Cell Mol Biol (Noisyle-Grand) 2003;49:OL467–OL471. [PubMed] [Google Scholar]
- 39.Iwahashi J, Kawasaki I, Kohara Y, Gengyo-Ando K, Mitani S, Ohshima Y, Hamada N, Hara K, Kashiwagi T, Toyoda T. Biochem Biophys Res Commun. 2002;293:698–704. doi: 10.1016/S0006-291X(02)00282-6. [DOI] [PubMed] [Google Scholar]
- 40.Tagami S, Eguchi Y, Kinoshita M, Takeda M, Tsujimoto Y. Oncogene. 2000;19:5736–5746. doi: 10.1038/sj.onc.1203948. [DOI] [PubMed] [Google Scholar]
- 41.Mannan AU, Boehm J, Sauter SM, Rauber A, Byrne PC, Neesen J, Engel W. Neurogenetics. 2006;7:93–103. doi: 10.1007/s10048-006-0034-4. [DOI] [PubMed] [Google Scholar]
- 42.He W, Lu Y, Qahwash I, Hu XY, Chang A, Yan R. Nat Med. 2004;10:959–965. doi: 10.1038/nm1088. [DOI] [PubMed] [Google Scholar]
- 43.Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 44.De Craene JO, Coleman J, Estrada de Martin P, Pypaert M, Anderson S, Yates JR, III, Ferro-Novick S, Novick P. Mol Biol Cell. 2006;17:3009–3020. doi: 10.1091/mbc.E06-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He Y, Janssen WG, Rothstein JD, Morrison JH. J Comp Neurol. 2000;418:255–269. [PubMed] [Google Scholar]
- 46.Wieland FT, Gleason ML, Sarafini TA, Rothman JE. Cell. 1987;50:189–300. doi: 10.1016/0092-8674(87)90224-8. [DOI] [PubMed] [Google Scholar]
- 47.Barlowe C. Trends Cell Biol. 2003;13:295–300. doi: 10.1016/s0962-8924(03)00082-5. [DOI] [PubMed] [Google Scholar]
- 48.Ma D, Jan LY. Curr Opin Neurobiol. 2002;12:287–292. doi: 10.1016/s0959-4388(02)00319-7. [DOI] [PubMed] [Google Scholar]
- 49.Vandenberghe W, Bredt DS. Curr Opin Cell Biol. 2004;16:134–139. doi: 10.1016/j.ceb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 50.Butchbach ME, Guo H, Lin CL. J Neurochem. 2003;84:891–894. doi: 10.1046/j.1471-4159.2003.01588.x. [DOI] [PubMed] [Google Scholar]
- 51.Furuta A, Noda M, Suzuki SO, Goto Y, Kanahori Y, Rothstein JD, Iwaki T. Am J Pathol. 2003;163:779–787. doi: 10.1016/S0002-9440(10)63705-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crino PB, Jin H, Shumate MD, Robinson MB, Coulter DA, Brooks-Kayal A. Epilepsia. 2002;43:211–218. doi: 10.1046/j.1528-1157.2002.35001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voutsinos-Porche B, Koning E, Clement Y, Kaplan H, Ferrandon A, Motte J, Nehlig A. J Cereb Blood Flow Metab. 2006;26:1419–1430. doi: 10.1038/sj.jcbfm.9600295. [DOI] [PubMed] [Google Scholar]
- 54.Jackson M, Song W, Liu MY, Jin L, Dykes-Hoberg M, Lin CI, Bowers WJ, Federoff HJ, Sternweis PC, Rothstein JD. Nature. 2001;410:89–93. doi: 10.1038/35065091. [DOI] [PubMed] [Google Scholar]
- 55.Longhurst DM, Watanabe M, Rothstein JD, Jackson M. J Biol Chem. 2006;281:12030–12040. doi: 10.1074/jbc.M513756200. [DOI] [PubMed] [Google Scholar]