

Abstract
MicroRNAs have been implicated in the modulation of gene expression programs important for normal and cancer cell development. miR-155 is known to play a role in B-cell development and is upregulated in various B-cell lymphomas, including several that are latently infected with Epstein-Barr virus (EBV). We show here that EBV infection of primary human B lymphocytes leads to the sustained elevation of miR-155 and its precursor RNA, BIC. The EBV-encoded latency membrane protein 1 (LMP1) can partially reconstitute BIC activation in B lymphocytes but not in epithelial cell cultures. LMP1 is a potent activator of NF-κB signaling pathways and is essential for EBV immortalization of B lymphocytes. An inhibitor to miR-155 further stimulated NF-κB responsive gene transcription, and IKKɛ was identified as a potential target of miR-155 translational repression. Remarkably, miR-155 inhibitor reduced EBNA1 mRNA and the EBV copy number in latently infected cells. This suggests that miR-155 contributes to EBV immortalization by modulation of NF-κB signaling and the suppression of host innate immunity to latent viral infection.
Epstein-Barr virus (EBV) is a ubiquitous human herpesvirus that has oncogenic potential, especially among immunocompromised individuals (14, 24). EBV DNA and gene products have been identified in the majority of endemic forms of Burkitt's lymphomas and nasopharyngeal carcinomas, as well as in gastric carcinomas, T-cell lymphomas, Hodgkin's disease Reed-Sternberg cells, and the majority of central nervous system lymphoma found in human immunodeficiency virus-associated AIDS. EBV can infect B lymphocytes and epithelial cells in the oropharynx and establishes long-term latent infection in memory B lymphocytes. Primary infection of B lymphocytes with EBV leads to a germinal-center-like reaction and the continuous proliferation and expansion of the latently infected B cells (32, 38). This continuous proliferation successfully immortalized B lymphocytes if T cells have been eliminated or suppressed by drugs such as cyclosporine. The efficiency of EBV immortalization of primary B cells in vitro has been a useful tool for cell culture and evidence for its oncogenic potential during immune suppression.
Immortalization of primary B cells by EBV occurs with relatively high efficiency in the absence of an antiviral T-cell response (38). During the immortalization process, EBV expresses several viral genes that have known growth-transforming activity. The EBNA2 mimics intracellular Notch by activating CBF1 (also known as CSL/RBPjk)-bound genes, while EBNA3C disrupts cell cycle checkpoints. Latency membrane protein 1 (LMP1) and LMP2 mimic constitutively active CD40 coreceptor and B-cell receptor, respectively. LMP1 binds members of the TRAF family through its intracellular domain to activate the NF-κB pathway (13, 29). However, the EBV activation and signaling mechanisms are complex and distinct from the endogenous receptors, and many of the details of gene reprogramming necessary for B-cell immortalization remain undefined.
MicroRNAs (miRNAs) are an abundant class of noncoding small RNAs ∼21 nucleotides that can affect gene expression by posttranscriptional regulation of mRNA (6, 11). miRNAs typically base pair with sequences in the 3′ untranslated region (UTR) of multiple mRNAs to inhibit mRNA translation or promote their degradation. miRNAs have been shown to play important roles in various cellular and pathogenic processes, including cellular development, immunological response, and carcinogenesis (2, 7, 39). Viruses, including EBV and Kaposi's sarcoma-associated herpesvirus (KSHV), encode multiple miRNAs, referred to as viral miRNAs, which may mimic cellular miRNAs and modulate cellular and viral gene expression (4, 9, 23). Virus infection can also affect cellular miRNA expression (20, 36).
Among the known miRNAs, miR-155 has been strongly implicated in the normal and malignant cell development of B lymphocytes. miR-155 is derived from the B-cell integration cluster (BIC)-associated noncoding RNA precursor (30). BIC and miR-155 were found to be overexpressed in a variety of B-cell malignancies, including Hodgkin's lymphomas, diffuse large cell lymphoma (16), and EBV-positive tumors, including those found in posttransplant lymphoproliferative disease (15, 36). Transgenic mice expressing high levels of miR-155 demonstrate pre-B-cell proliferation and develop lymphoblastic leukemia and high-grade lymphomas (3). miR-155 is induced by inflammatory cytokine response and by B-cell receptor activation (17, 21, 33, 37). miR-155 has been implicated in the normal B-cell developmental processes, including generation of immunoglobulin class-switched plasma cells (34) and germinal center reactions (31).
A recent study showed that that miR-155 target seed sequences exists in the 3′ UTR of IKKɛ (8). IKKɛ, the gene product of IKBKE, is a multifunctional IKB kinase protein. IKKɛ had been shown to phosphorylate IRF-3 and IRF-7 to activate interferon (IFN) response to viral infection (27). Recent studies also showed that IKKɛ regulated NF-κB activation directly by phosphorylating the C-terminal domain of RelA and c-Rel through which IKKɛ exerts its oncogenic function (1, 12). In the present study, we present evidence that EBV immortalization of B lymphocytes correlates with induction of miR-155. We show that miR-155 attenuates NF-κB signaling in EBV-positive B lymphocytes and that miR-155 is important for the stable maintenance of EBV genomes during latent infection.
MATERIALS AND METHODS
Cells and plasmids.
Raji is a type III latency B-cell line derived from Burkitt's lymphoma. LCL3473 (LCL) is a type III latency B-cell line derived from primary lymphoblasts transformed with EBV strain B95-8. DG-75 is an EBV-negative, B-cell line derived from Burkitt's lymphoma. These cell lines were maintained in RPMI supplemented with 10% fetal bovine serum, glutamine, penicillin, and streptomycin sulfate. 293T, EBV-positive 293-ZKO, and D98 cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum, glutamine, penicillin, and streptomycin sulfate. 5x κB-L reporter plasmid that contained five copies of an NF-κB-responsive element upstream of luciferase was kindly provided by R. Kaufman, The Wistar Institute, Philadelphia, PA. miR-155 sensor plasmid was kindly provided by R. Renne (University of Florida, Gainesville). pGL4.74[hRluc/TK] was from Promega. pCMV-Flag-LMP1 was constructed by PCR amplification of LMP1 with primers introducing a HinDIII-XbaI site cloned in frame to pFLAG-CMV2 (Sigma). pCMV-FLAG-EBNA2 was constructed by PCR amplification of EBNA2 with primers introducing a Asp718-HinDIII site cloned in frame to pFLAG-CMV2. pFLAG-CMV2 was used as vector control. miRIDIAN miR-155 mimic (Dharmacon old number C-300138-01/new number C-300647-05), inhibitor (Dharmacon old number I-300138-01/new number IH-300647-06), and negative control of mimic or inhibitor were purchased from Dharmacon. The miR-155 inhibitor is a single-stranded chemically enhanced oligonucleotide designed antisense to the mature miR-155 (UUAAUGCUAAUCGUGAUAGGGG), and the miR-155 mimic is a double-stranded RNA oligonucleotide miRNA with an effective mimic of endogenous mature miR-155 function (18).
Primary B-cell infection.
Infectious particles of EBV were isolated from the supernatants of B95-8 cell cultures. Supernatants were passed through a 0.45-μm-pore-size filter and then used to infect primary human B cells. Human B cells were derived from Hypaque fractionation of peripheral blood from anonymous donors.
miRNA array analysis.
RNA was isolated from human B-cell cultures by using RNazol isolation reagent. Total RNA was isolated from B-cell cultures prior to EBV infection, and at 10 days postinfection or at 10 days in culture after mock infection. RNA was then labeled by reverse transcription (RT) and assayed for miRNA by hybridization on glass slide arrays using an oligonucleotide array for 245 miRNAs as described previously (19). Microarray images were analyzed by using Genepix Pro 6.0 (Axon Instruments). Average values of the replicate spots of each miRNA were background subtracted, normalized, and further analyzed. Normalization was performed by using global median method calculated using R (www.r-project.org) with Bioconductor (www.bioconductor.org). Finally, we selected the miRNAs measured as present in at least as many samples as the smallest class in the data set (50%). Absent calls were thresholded to 4.5 (log2 scale) before statistical analysis, representing the average minimum intensity level detectable in the system. miRNAs that are differentially expressed in different tissues were identified by using the Significance Analysis of Microarrays (SAM; version 3.0) application with a threshold difference in expression set to 2, the s0 percentile set to 0.05 (default), and the number of permutations set to 100 (default). The SAM application calculates a score for each gene on the basis of the change of expression relative to the standard deviation of all measurements. Only mature miRNAs that are differentially expressed are reported. A tree cluster was generated by the hierarchical cluster analysis showing the separation of each tissue. For hierarchical clustering, we used average linkage metrics and centered Pearson correlation (Cluster 3.0). Java Treeview 1.1.1 was used for tree visualization.
Luciferase assays.
293T cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's specifications. A total of 2 × 105 293T cells were transfected with 20 μM miRNA mimic and 200 ng of reporter DNA with Renilla internal control vector pGL4.74 (Promega) and then harvested at 48 h posttransfection. For luciferase assay of DG75 and LCL cells, 3 × 106 cells were centrifuged and resuspended in 100 μl of T solution (Amaxa) containing 60 μM miRNA mimic or inhibitor and 2 μg of reporter DNA with Renilla internal control vector pGL4.74 and then electroporated on an Amaxa Nucleofector using program A-23. After nucleofection, the cells were transferred to a flask containing 10 ml of fresh, complete RPMI 1640 medium and incubated at 37°C for 48 h. The relative luciferase activity was assayed by using Promega Dual-Luciferase reporter assay system. All data points were the average of at least three independent transfections.
Transfection of miR-155 mimic and inhibitor.
Nucleofection was used for transfection of all B lymphocytes including, DG75, Raji, and LCL cells. Briefly, 5 × 106 cells were centrifuged and resuspended in 100 μl of T solution (Amaxa) containing 150 μM miRNA mimic or inhibitor and 20 μM siGLO (Dharmacon) and electroporated on an Amaxa Nucleofector using the program A-23. Mimic or inhibitor negative controls with siGLO were transfected in the same way. After nucleofection, the cells were transferred to a flask containing 20 ml of fresh, complete RPMI 1640 medium and incubated at 37°C for 48 h. The fluorescein isothiocyanate-positive cells were sorted and used in RT-PCR and Western blotting. For genome copy number assays, cells were transfected twice, with the second transfection at 48 h later. The cells were then selected by fluorescence-activated cell sorting for siGlo at 96 h after original transfection and subjected to genomic DNA purification (Promega genome DNA isolation kit).
Quantitative RT-PCR and Northern blotting.
Total RNA was prepared in TRIzol reagents (Invitrogen) in accordance with the manufacturer's instructions. RT-PCR was done as previously described. Real-time PCR was performed with Sybr green probe in an ABI Prism 7000 according to the manufacturer's specified parameters. Primer sequences for RT-PCRs were as follows: BIC, 5′-TCAAGAACAACCTACCAGAGACCTT-3′ and 5′-TCCTGGTTTGTGCCACCAT-3′; LMP-1, 5′-CCCTCAACAAGCTACCGATGAT-3′ and 5′-TCTGCCCTCGTTGGAGTTAGA-3′; EBNA-2, 5′-GGGATGCCTGGACACAAGA and 5′-GTCCATGCCCGACGTCATA-3′; β-actin, 5′-GCCATGGTTGTGCCATTACA-3′ and 5′-GGCCAGGTTCTCTTTTTATTTCTG-3′; IKKɛ, 5′-TGCGTGCAGAAGTATCAAGC-3′ and TACAGGCAGCCACAGAACAG-3′; BCL2, 5′-GAGGATTGTGGCCTTCTTTG-3′ and 5′-ACAGTTCCACAAAGGCATCC-3′; CCL5, 5′-CCATATTCCTCGGACACCAC-3′ and 5′-TGTACTCCCGAACCCATTTC-3′; and EBNA1, 5′-GGTCGTGGACGTGGAGAAAA-3′ and 5′-GGTGGAGACCCGGATGATG-3′.
For Northern blot analysis, 20 μg of total RNA was resolved on a 15% denaturing polyacrylamide gel and electrotransferred to Hybond N+ nylon membrane (Amersham). The membranes were cross-linked under UV. Prehybridization and hybridization were performed at 42°C in 10× Denhardt solution, 6× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), and 0.1% sodium dodecyl sulfate (SDS). Oligonucleotides complementary to miRNA-155 or U6 snRNA were end labeled with [γ-32P]ATP and used as probes for Northern analysis. The sequences of the oligonucleotides were 5′-CCCCTATCACGATTAGCATTAA-3′ (miR-155) and 5′-GCAGGGGCCATGCTAATCTTCTCTGTATCG-3′ (U6 snRNA). All of the probes were washed twice for 5 min at 25°C in 6× SSC-0.1% SDS.
Western blotting and antibodies.
The Western blot was preblocked with 5% dried milk in Tris-saline (20 mM Tris [pH 8.0], 200 mM NaCl), incubated with antibody in the blocking agent (diluted from 1:1,000 to 1:2,000), followed by four washes with Tris-saline containing 0.05% IPEGAL. Secondary antibody was diluted 1:2,000 in blocking agent, washed as described above, and then detected by enhanced chemiluminescence (Amersham). The following mouse monoclonal antibodies were used anti-IKKɛ (Imgenex) and anti-PCNA (Santa Cruz).
Genome copy number assay.
Cells (ca. 106 cells per sample) were collected and resuspended in 100 μl of SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris [pH 8.0]). After brief sonication, immunoprecipitation dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris [pH 8.0], 167 mM NaCl) was added to 1 ml, followed by incubation with proteinase K for 2 to 3 h at 50°C. Then, 300 μl was removed and subjected to phenol-chloroform extraction and ethanol precipitation. Precipitated DNA was then assayed by real-time PCR using primers for the DS region of EBV and normalized by the cellular DNA signal at the actin gene locus. The primers for DS were 5′-ATGTAAATAAAACCGTGACAGCTCAT-3′ and 5′-TTACCCAACGGGAAGCATATG-3′ and for actin were 5′-GCCATGGTTGTGCCATTACA-3′ and 5′-GGCCAGGTTCTCTTTTTATTTCTG-3′.
Southern blotting.
Purified genomic DNA was digested with BamHI and analyzed by 0.7% agarose gel electrophoresis and Southern blotting. EBV DNA was identified by using the OriLyt sequence amplified by the primers 5′-GCCCGTTGGGTTTCATTAAG-3′ and 5′-CCAGGAAGTGGCGAGCAT-3′. PCR products were labeled with digoxigenin using a PCR DIG probe synthesis kit (Roche) according to the manufacturer's protocol. Oligonucleotide probes for telomere repeats (6x TTAGGG) were labeled with a DIG oligonucleotide 3′ end-labeling kit (Roche).
RESULTS
EBV primary infection induces miR-155 and BIC RNA.
To determine whether primary infection of EBV induces cellular miRNAs, we assayed human peripheral blood cells before and after EBV infection for changes in miRNA abundance. RNA was isolated from peripheral blood cells before infection and at 10 days postinfection. As a secondary control for days in cell culture, we also assayed miRNA levels in peripheral blood cells after 10 days in culture after mock infection. A comparison of seven different donor samples revealed a clustering of at least two cellular miRNAs, miR-155 and miR-210, that were consistently induced by EBV infection relative to preinfection and mock infection controls (Fig. 1A). miR-338, miR-346, and miR-369 were induced by EBV infection relative to preinfection but not compared to mock infections (data not shown). No miRNAs were found to be consistently repressed by EBV infection in this analysis.
FIG. 1.

miR-155 and BIC are induced by EBV immortalization of primary B lymphocytes. (A) Total RNA was isolated from human peripheral blood cells from seven different donors at 10 days postinfection with EBV (EBV), at 10 days after mock infection (Mock), or immediately prior to EBV infection (Pre). RNA was analyzed for miRNA expression by using differential hybridization. miRNA expression was grouped by hierarchical cluster analysis with RNA samples in the vertical axis and by relevant miRNAs in the horizontal axis. (B) RNA was isolated from preinfected peripheral blood cell samples or from peripheral blood cell samples (1 to 7) at 10 days postinfection or at 10 days after mock infection and assayed by RT-PCR for the miR-155 precursor BIC RNA relative to cellular actin RNA. (C) Northern blot analysis of total RNA isolated from samples 4, 5, and 6 from preinfection (D0), mock-infected (−), or EBV (+)-infected peripheral blood samples (10 days postinfection) and analyzed with a probe specific for miR-155 (top panel) or control U6 snRNA (lower panel). (D) BIC RNA relative to actin RNA was measured by RT-PCR at various days (as indicated) after EBV infection or after mock infection of peripheral blood cells. (E) EBV-encoded LMP1 and EBNA2, along with BIC RNA, were measured at 10 days posttransfection relative to actin RNA using RT-PCR for EBV-infected samples from donors 4, 5, and 6.
BIC RNA is elevated in EBV positive B-cell lines.
To begin to validate the findings from the array analysis, we focused on miR-155, which has been previously implicated in B-cell malignancies. miR-155 is known to be derived from the noncoding BIC RNA, which can be readily detected by RT-PCR analysis. RNA from the seven peripheral blood cell samples were assayed for BIC RNA levels relative to actin after EBV infection or in preinfected and mock-infected controls (Fig. 1B). EBV infection induced BIC RNA relative to actin in all seven samples, with no detectable activation in the preinfected or mock-infected controls. BIC activation levels varied for each donor sample, with samples 1, 5, and 6 showing greater than 10- to 200-fold induction by EBV. The detection of mature miR-155 was also verified by Northern blotting of total cellular RNA (Fig. 1C, upper panel). Mature miR-155 was readily detected in EBV-infected samples from donors 6 and 5, and to a lesser extent donor 4, reflecting the pattern observed for the BIC precursor RNA. EBV infection had no effect on control U6 snRNA (Fig. 1C, lower panel). The time course of BIC induction after EBV infection was assayed with a new donor sample by using RT-PCR for BIC relative to actin (Fig. 1D). We found that BIC was strongly activated by 19 days postinfection but could be detected by 8 days postinfection relative to mock-infected controls. The expression of EBV growth-transforming genes LMP1 and EBNA2 was also assayed in donor samples 4, 5, and 6 (Fig. 1E). BIC RNA levels correlated with expression levels of EBNA2 and LMP1, with donor sample 6 showing the highest levels of all three RNAs relative to actin controls. These findings suggest that EBV infection and gene products contribute to the elevated expression of BIC RNA.
BIC RNA is elevated in EBV positive B-cell lines.
EBV infects both B lymphocytes and epithelial cells. To determine whether all EBV-positive cells have elevated BIC, we assayed EBV-positive and -negative cell lines for BIC levels, relative to cellular actin (Fig. 2A). We found that EBV-positive lymphoblastoid cell lines had the highest levels of BIC expression. The EBV-positive Burkitt's lymphoma cell line Raji had a modest increase in BIC levels relative to an EBV-negative Burkitt's lymphoma cell line DG75. In contrast, BIC levels were not significantly elevated relative to actin in EBV-positive or -negative epithelial cell lines derived from 293 HEK cells (293T or ZKO-293) or a HeLa-derived cell line (D98/HR1). The levels of EBV gene products LMP1 and EBNA2 were also assayed (Fig. 2B and C). LMP1 and EBNA2 mRNA were expressed in LCL, Raji, and ZKO-293 cells, while D98/HR1 cells express LMP1 only, and 293T and DG75 cells do not express either EBV gene product. These findings suggest that BIC activation by EBV depends on cooperating B-cell-specific factors.
FIG. 2.
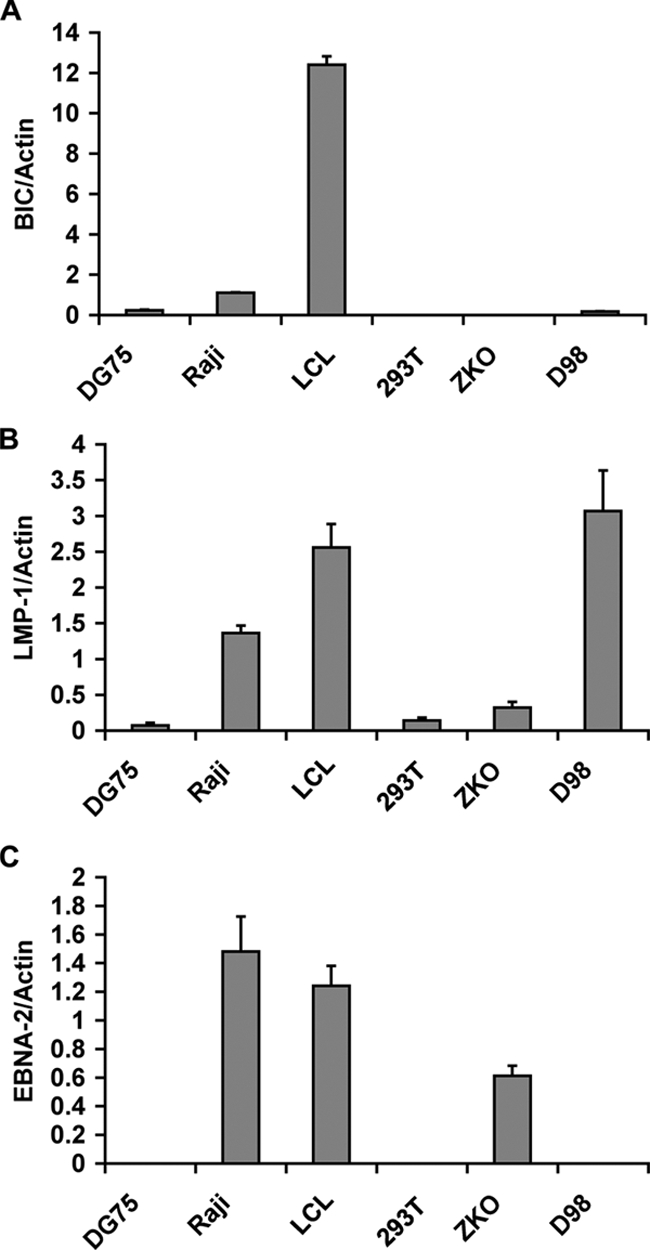
BIC RNA levels are highest in EBV-positive LCL cells. (A) BIC RNA levels were assayed by RT-PCR and normalized to actin RNA levels for various cell lines (DG75 is an EBV-negative BL cell line, Raji is EBV-positive BL cell line, LCL is an EBV-positive lymphoblastoid cell line, 293T is an EBV-negative HEK cell line, ZKO is a Zta knockout EBV bacmid-positive 293 cell line, and D98 is an EBV P3HR1 strain-positive HeLa cell line). (B and C) Same as in panel A, except that LMP1 (B) and EBNA-2 (C) mRNA were quantified relative to actin for each cell line.
LMP1 induces BIC RNA in B lymphocytes.
To further explore the mechanism of BIC activation by EBV, we assayed the ability of the EBV gene products EBNA2 or LMP1 to activate BIC mRNA in EBV-negative cell lines derived from B lymphocytes or epithelial cells. We found that BIC levels were induced ∼7-fold by transfection of LMP1 in B-cell line DG75. These levels were similar to that observed for BIC RNA in EBV-positive Raji cells. EBNA2 also induced BIC, but to a lesser extent (∼3-fold). Combining EBNA2 and LMP1 did not lead to a greater induction of BIC than when LMP1 was transfected alone. Mock transfection had no effect on BIC levels. LMP1 and EBNA2 expression levels were also monitored in transfected cells (Fig. 3B and C). Although LMP1 and EBNA2 levels were readily detected in transfected cells, the levels of expression were not as high as those observed in EBV-positive Raji cells. We also assayed the ability of LMP1 and EBNA2 to activate BIC RNA in 293T cells (Fig. 3D). In contrast to the B-cell line DG75, BIC levels were not activated above basal levels in 293T cell lines. EBNA2 and LMP1 mRNA was expressed to higher levels in 293T cells than were observed in Raji cells (Fig. 3E and F). These data indicate that LMP1, and to a lesser extent EBNA2, can activate BIC RNA in DG75 B cells but not in 293T cells.
FIG. 3.

B-cell-specific induction of BIC RNA by LMP1. (A to C) EBV-negative BL cell DG75 (5 × 106 cells) was transfected with 5 μg of expression vectors for LMP1, EBNA2, LMP1+EBNA2, or control vector (mock) and compared to Raji cell RNA for expression of BIC (A), EBNA-2 (B), or LMP1 (C) relative to actin RNA. (D to F) EBV-negative 293T cells were transfected and analyzed as described for panels A to C.
miR-155 downmodulates NF-κB signaling.
To explore the functional significance of BIC and miR-155 activation in EBV-infected B cells, we assayed the effects of an miR-155 inhibitor in EBV-positive LCL cell lines. The miR-155 inhibitor was demonstrated to efficiently activate the expression of a luciferase construct containing the miR-155 target sequence at its 3′ UTR (Fig. 4A). The miR-155 inhibitor, but not a scrambled control inhibitor, efficiently activated luciferase in LCL cell lines that are known to express high endogenous levels of miR-155 (see Fig. S1 in the supplemental material). We then tested the ability of the miR-155 inhibitor to affect several cell functions, including cell cycle profile, cellular proliferation, and apoptosis induction (data not shown). We did not detect any significant effect of miR-155 inhibitor with these functional assays. We therefore reasoned that miR-155 may play a modulatory effect on the LMP1 signaling pathways. The major LMP1 signaling target is through NF-κB. We therefore tested whether miR-155 inhibitor affected NF-κB activity in LCL cells by using a luciferase reporter construct that contains five copies of NF-κB response elements (5x κB-L). We found that miR-155 inhibitor activated 5x κB-L ∼2-fold in LCL cells (Fig. 4B). To further explore a role of miR-155 in NF-κB signaling, we assayed a miR-155 mimic in EBV-negative DG75 B cells (Fig. 4C). The miR-155 mimic reduced 5x κB-L activity by ∼5-fold, while a control miRNA mimic had no detectable effect. The miR-155 mimic had no significant effect on 5x κB-L activity relative to the control in EBV-negative 293T cell lines (Fig. 4D). This suggests that miR-155 downmodulates the relatively high levels of NF-κB activity in EBV-positive B cells but has no significant effect on low or constitutive levels of NF-κB in 293T cells.
FIG. 4.

miR-155 modulates NF-κB signaling in B lymphocytes. (A) miR-155 inhibitor or control inhibitor (0 to 100 μM) were assayed for their ability to relieve repression of the miR-155 target sequence in LCL cells (3 × 106) using a luciferase indicator plasmid (2 μg). (B) LCL cells were transfected with 5x κB-L reporter plasmid and either control or miR-155 inhibitor and assayed for luciferase activity. (C) EBV-negative DG75 BL cells were transfected with pGL3-Basic or 5x κB-L reporter plasmids and either miR-155 mimic or control and then assayed for luciferase activity. (D) Same as in panel C, except in 293T cells.
Computational studies and luciferase indicator assays have implicated IKKɛ mRNA as a potential target for translational modulation by miR-155 (8, 25, 33). To test whether miR-155 inhibitor affected IKKɛ protein levels, we assayed LCL cells by Western blotting for expression of IKKɛ relative to control protein PCNA (Fig. 5A). We found that miR-155 inhibitor elevated IKKɛ protein levels ∼2-fold in LCL cells, with little detectable effect on the levels of PCNA. We also found that the miR-155 mimic in EBV-negative DG75 cells caused an ∼2-fold reduction in IKKɛ, with no detectable effect on PCNA. Given the fact of IKKɛ being involved in both NF-κB signaling and IFN antiviral response, we further investigated the expression of NF-κB-regulated BCL2 and IFN-regulated gene CCL5 (Fig. 5B and C). We examined the mRNA levels for IKKɛ, BCL2, and CCL5 in EBV-positive LCLs (Fig. 5B) or Raji BL cells (Fig. 5C) transfected with control or miR-155 inhibitor. RT-PCR was used to measure mRNA levels relative to cellular actin. We found that miR-155 inhibitor activated BCL2 and CCL5 mRNA levels relative to actin in both cell types, while IKKɛ mRNA levels were highly activated in Raji cells but not LCL cells. These findings are consistent with a role of miR-155 in the translational inhibition of IKKɛ and suggest that miR-155 modulates both NF-κB and IFN-responsive genes through downmodulation of IKKɛ.
FIG. 5.
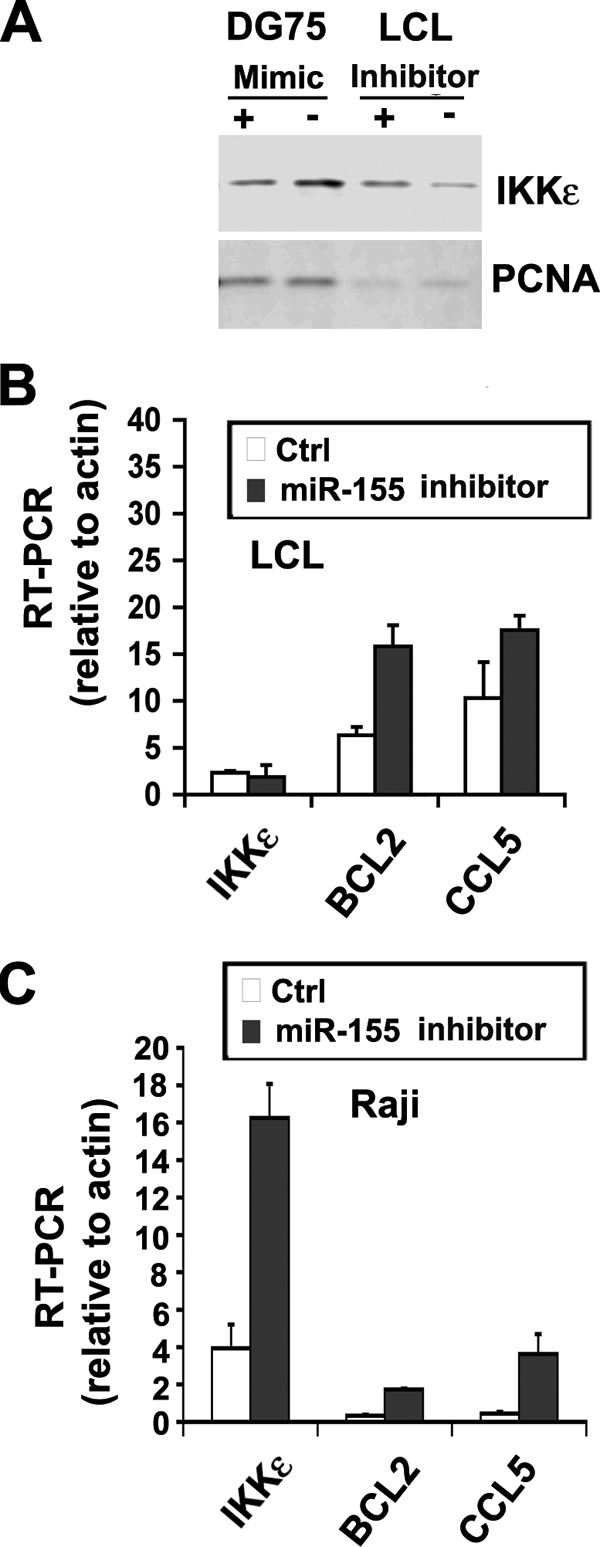
Suppression of the IKKɛ pathway by miR-155. (A) Western blot analysis of IKKɛ protein (top panel) or PCNA protein control (lower panel) in either DG75 cells (left two lanes) transfected with miR-155 mimic (+) or control (−), or EBV-positive LCL cells (right two lanes) transfected with miR-155 inhibitor (+) or control (−). (B) IKKɛ, BCL2, or CCL5 mRNA was measured relative to actin mRNA in LCL cells transfected with miR-155 inhibitor (▪) or control (□). (C) Same as in panel B, except in Raji cells.
The IFN-associated antiviral response may also play a role in the stable maintenance of the viral genome during latency. To explore this possibility, we first measured the effect of miR-155 inhibitor on the EBV genome copy number (Fig. 6A and B). We found that miR-155 inhibitor caused an ∼4-fold reduction in the EBV genome copy number in both Raji and LCL cells as measured by quantitative PCR of viral relative to cellular DNA. EBV genome maintenance during latency requires sufficient expression of the virus-encoded EBNA1 protein. We therefore examined whether miR-155 inhibitor reduced EBNA1 mRNA expression (Fig. 6C and D). We found that miR-155 inhibitor modestly reduced EBNA1 mRNA ∼2-fold in Raji (Fig. 6C) but only ∼1.3-fold in LCL cells (Fig. 6D) relative to control inhibitor. The loss of EBV DNA was further measured by Southern blotting after transfection of miR-155 inhibitor in Raji cells (Fig. 6E). We found an ∼2-fold loss of EBV genomes relative to telomere repeat DNA. These findings indicate that miR-155 contributes to the stable maintenance of EBV genomes during latent infection and may exert this effect, partly, through regulation of EBNA1 mRNA levels.
FIG. 6.

miR-155 stabilizes EBV genome copy number during latent infection. (A) EBV DNA copy number was assayed by real-time PCR of EBV dyad symmetry (DS) DNA relative to cellular actin in Raji cells transfected with miR-155 inhibitor or control. (B) Same as in panel A, except in LCL cells. (C) Raji cells were transfected with miR-155 inhibitor or control and then assayed for EBNA1 mRNA expression relative to actin using RT-PCR. (D) Same as in panel C, except in LCL cells. (E) Southern blot analysis of EBV genomic DNA (upper panel) and cellular telomere repeat DNA (Telo, lower panel) in control (Ctrl) or miR-155 inhibitor-transfected Raji cells. (F) Summary of genetic interactions of miR-155 relevant to EBV immortalization and stable latent infection. EBV LMP1 induces BIC/miR-155, which in turn attenuates NF-κB signaling and IKKɛ protein expression. miR-155 also contributes to EBV genome maintenance, in part by stimulating EBNA1 mRNA expression.
DISCUSSION
Immortalization of B lymphocytes by EBV primary infection shares several features with B-cell proliferation and maturation that occurs during the germinal center reaction (32). miR-155 has been implicated in the germinal center reaction and memory B-cell formation, as well as in lymphomagenesis in transgenic mice (2, 3, 31, 34). We present evidence from array, RT-PCR, and Northern blotting analyses that miR-155 is induced during the EBV immortalization process (Fig. 1). miR-155 expression has been detected in EBV-immortalized B-cell lines, but its expression has been variable in EBV-positive Burkitt's lymphoma cell lines (15, 16). Recent studies have found that miR-155 expression is elevated in EBV type III latency but not in matched cells with a type I latency (20, 36). This finding is consistent with our findings that EBV type III latency gene product LMP1 is capable of inducing the miR-155 precursor RNA, BIC, in EBV-negative BL cell lines (Fig. 3). LMP1 did not induce BIC in EBV-negative 293T cells, despite its ability to activate NF-κB signaling in both cell types. These findings suggest that EBV-encoded LMP1 can stimulate BIC transcription in a B-cell-dependent manner and is likely to be involved in the generation of miR-155 during EBV immortalization process.
LMP1 is essential for EBV immortalization of B lymphocytes, where it functions as a constitutive CD40-like receptor that can stimulate NF-κB signaling (5, 13, 29). Our data suggest that LMP1 also stimulates miR-155 expression. miR-155 can be induced by several B-cell stimulatory processes, including B-cell receptor engagement, lipopolysaccharide-mediated Toll receptor activation, and tumor necrosis factor alpha-associated macrophage inflammatory response (21, 33, 37). NF-κB signaling plays a central but complex role in these pathways. Our data using inhibitors and miRNA mimics suggest that miR-155 attenuates NF-κB signaling (Fig. 4). This suggests that LMP1 induces miR-155 to buffer persistent stimulation of NF-κB. Inhibitor kinases, such as IkBα, can provide temporal control over NF-κB signaling through a negative-feedback mechanism (35). Like IkBα, miR-155 may attenuate NF-κB signaling during the persistent stimulation that occurs upon EBV transformation and germinal center reaction. Signal attenuation may be necessary for cell survival (Fig. 6F).
LMP1, like the B-cell coreceptor CD40, activates NF-κB through the noncanonical signaling pathway (22). LMP1 is known to activate TRAFs and NIK, which in turn phosphorylate and activate IKKα and IKKβ (29). IKKɛ was identified as a candidate for miR-155 translational suppression because of a consensus seed sequence in the 3′ UTR of the IKKɛ mRNA (8, 33). We found that miR-155 inhibitor modestly increased IKKɛ protein levels in EBV-positive LCL cells, while a miR-155 mimic reduced IKKɛ protein levels in EBV-negative BL cells (Fig. 5). The role of IKKɛ in the NF-κB signaling pathway is not completely understood, but IKKɛ is known to regulate IFN signaling by phosphorylation and activation of IRF3 and IRF7 (10, 27). Increased IKKɛ levels in miR-155 inhibitor-treated cells correlated with an increase in NF-κB responsive BCL2 and the IFN-responsive CCL5 mRNA. These findings are consistent with a role of miR-155 in regulating the IKKɛ-dependent pathway involving both NF-κB and IFN signaling. However, we were unable to deplete IKKɛ by siRNA (data not shown) and therefore cannot rigorously conclude that IKKɛ is a functional target of miR-155 in modulating the NF-κB pathway. Nevertheless, it is possible that miR-155 depletion of IKKɛ may skew the NF-κB signaling pathway toward B-cell proliferation and antiapoptosis, as well as limit the innate immune response to viral infection.
Our data also indicate that miR-155 activity contributes to the stable maintenance of latent viral genomes. miR-155 inhibitor caused a loss of EBV genome copy number in LCL and Raji cells (Fig. 5) and a partial reduction in EBNA1 mRNA levels. This suggests that the miR-155 may suppress cellular factors, in addition to EBNA1, that may regulate viral genome stability during latency. The related gammaherpesvirus, KSHV, which also persists as a multicopy episome in human B cells, encodes one miRNA with seed sequence specificity identical to miR-155 (8, 28). Since KSHV does not immortalize primary B cells, the common function of miR-155 during gammaherpesvirus latency may be maintaining latent viral genomes in proliferating B cells. Although miR-155 levels are not elevated in EBV-positive type I Burkitt's lymphoma cell lines, other cellular changes may compensate for miR-155 function. Interestingly, elevated levels of c-myc have been shown to suppress the NF-κB and IFN signaling, similar to what we found for miR-155 (26). Thus, downmodulation of NF-κB and IFN signaling by viral or cellular miRNAs, or by elevated c-myc, may be necessary to suppress host cell innate immune response and permit viral genome persistence during latency.
Supplementary Material
Acknowledgments
We thank Rolf Renne and Russel Kaufman for generously providing plasmids. We acknowledge the valuable contributions of the Wistar Institute Cancer Center Core Facilities for Flow Cytometry and Genomics.
This study was supported by National Institutes of Health grant CA085678 to P.M.L.
Footnotes
Published ahead of print on 27 August 2008.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Agami, R. 2007. All roads lead to IKKɛ. Cell 1291043-1045. [DOI] [PubMed] [Google Scholar]
- 2.Calame, K. 2007. microRNA-155 function in B cells. Immunity 27825-827. [DOI] [PubMed] [Google Scholar]
- 3.Costinean, S., N. Zanesi, Y. Pekarsky, E. Tili, S. Volinia, N. Heerema, and C. M. Croce. 2006. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc. Natl. Acad. Sci. USA 1037024-7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cullen, B. R. 2006. Viruses and microRNAs. Nat. Genet. 38(Suppl.)S25-S30. [DOI] [PubMed] [Google Scholar]
- 5.Dirmeier, U., R. Hoffmann, E. Kilger, U. Schultheiss, C. Briseno, O. Gires, A. Kieser, D. Eick, B. Sugden, and W. Hammerschmidt. 2005. Latent membrane protein 1 of Epstein-Barr virus coordinately regulates proliferation with control of apoptosis. Oncogene 241711-1717. [DOI] [PubMed] [Google Scholar]
- 6.Engels, B. M., and G. Hutvagner. 2006. Principles and effects of microRNA-mediated posttranscriptional gene regulation. Oncogene 256163-6169. [DOI] [PubMed] [Google Scholar]
- 7.Esquela-Kerscher, A., and F. J. Slack. 2006. Oncomirs: microRNAs with a role in cancer. Nat. Rev. Cancer 6259-269. [DOI] [PubMed] [Google Scholar]
- 8.Gottwein, E., N. Mukherjee, C. Sachse, C. Frenzel, W. H. Majoros, J. T. Chi, R. Braich, M. Manoharan, J. Soutschek, U. Ohler, and B. R. Cullen. 2007. A viral microRNA functions as an orthologue of cellular miR-155. Nature 4501096-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grey, F., L. Hook, and J. Nelson. 2007. The functions of herpesvirus-encoded microRNAs. Med. Microbiol. Immunol. 197:261-267. [DOI] [PMC free article] [PubMed]
- 10.Hacker, H., and M. Karin. 2006. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006re13. [DOI] [PubMed]
- 11.Hannon, G. J., F. V. Rivas, E. P. Murchison, and J. A. Steitz. 2006. The expanding universe of noncoding RNAs. Cold Spring Harbor Symp. Quant. Biol. 71551-564. [DOI] [PubMed] [Google Scholar]
- 12.Hiscott, J. 2007. Convergence of the NF-κB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 18483-490. [DOI] [PubMed] [Google Scholar]
- 13.Izumi, K. M. 2004. Epstein-Barr virus signal transduction and B-lymphocyte growth transformation. Prog. Mol. Subcell. Biol. 36269-288. [DOI] [PubMed] [Google Scholar]
- 14.Kieff, E. 2007. Epstein-Barr virus and its replication, 5th ed. Wolters Kluwer Health/Lippincott/The Williams & Wilkins Co., Philadelphia, PA.
- 15.Kluiver, J., E. Haralambieva, D. de Jong, T. Blokzijl, S. Jacobs, B. J. Kroesen, S. Poppema, and A. van den Berg. 2006. Lack of BIC and microRNA miR-155 expression in primary cases of Burkitt lymphoma. Genes Chromosomes Cancer 45147-153. [DOI] [PubMed] [Google Scholar]
- 16.Kluiver, J., S. Poppema, D. de Jong, T. Blokzijl, G. Harms, S. Jacobs, B. J. Kroesen, and A. van den Berg. 2005. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B-cell lymphomas. J. Pathol. 207243-249. [DOI] [PubMed] [Google Scholar]
- 17.Kluiver, J., A. van den Berg, D. de Jong, T. Blokzijl, G. Harms, E. Bouwman, S. Jacobs, S. Poppema, and B. J. Kroesen. 2007. Regulation of pri- microRNA BIC transcription and processing in Burkitt lymphoma. Oncogene 263769-3776. [DOI] [PubMed] [Google Scholar]
- 18.Krutzfeldt, J., N. Rajewsky, R. Braich, K. G. Rajeev, T. Tuschl, M. Manoharan, and M. Stoffel. 2005. Silencing of microRNAs in vivo with “antagomirs.” Nature 438685-689. [DOI] [PubMed] [Google Scholar]
- 19.Liu, C. G., G. A. Calin, B. Meloon, N. Gamliel, C. Sevignani, M. Ferracin, C. D. Dumitru, M. Shimizu, S. Zupo, M. Dono, H. Alder, F. Bullrich, M. Negrini, and C. M. Croce. 2004. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc. Natl. Acad. Sci. USA 1019740-9744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motsch, N., T. Pfuhl, J. Mrazek, S. Barth, and F. A. Grasser. 2007. Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) induces the expression of the cellular microRNA miR-146a. RNA Biol. 4131-137. [DOI] [PubMed] [Google Scholar]
- 21.O'Connell, R. M., K. D. Taganov, M. P. Boldin, G. Cheng, and D. Baltimore. 2007. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 1041604-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perkins, N. D. 2007. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell. Biol. 849-62. [DOI] [PubMed] [Google Scholar]
- 23.Pfeffer, S., and O. Voinnet. 2006. Viruses, microRNAs and cancer. Oncogene 256211-6219. [DOI] [PubMed] [Google Scholar]
- 24.Rickinson, A. B., and E. Kieff. 2007. Epstein-Barr virus, 5th ed. Wolters Kluwer Health/Lippincott/The Williams & Wilkins Co., Philadelphia, PA.
- 25.Rodriguez, A., E. Vigorito, S. Clare, M. V. Warren, P. Couttet, D. R. Soond, S. van Dongen, R. J. Grocock, P. P. Das, E. A. Miska, D. Vetrie, K. Okkenhaug, A. J. Enright, G. Dougan, M. Turner, and A. Bradley. 2007. Requirement of bic/microRNA-155 for normal immune function. Science 316608-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlee, M., M. Holzel, S. Bernard, R. Mailhammer, M. Schuhmacher, J. Reschke, D. Eick, D. Marinkovic, T. Wirth, A. Rosenwald, L. M. Staudt, M. Eilers, F. Baran-Marszak, R. Fagard, J. Feuillard, G. Laux, and G. W. Bornkamm. 2007. c-myc activation impairs the NF-κB and the interferon response: implications for the pathogenesis of Burkitt's lymphoma. Int. J. Cancer 1201387-1395. [DOI] [PubMed] [Google Scholar]
- 27.Sharma, S., B. R. tenOever, N. Grandvaux, G. P. Zhou, R. Lin, and J. Hiscott. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 3001148-1151. [DOI] [PubMed] [Google Scholar]
- 28.Skalsky, R. L., M. A. Samols, K. B. Plaisance, I. W. Boss, A. Riva, M. C. Lopez, H. V. Baker, and R. Renne. 2007. Kaposi's sarcoma-associated herpesvirus encodes an ortholog of miR-155. J. Virol. 8112836-12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soni, V., E. Cahir-McFarland, and E. Kieff. 2007. LMP1 TRAFficking activates growth and survival pathways. Adv. Exp. Med. Biol. 597173-187. [DOI] [PubMed] [Google Scholar]
- 30.Tam, W., D. Ben-Yehuda, and W. S. Hayward. 1997. bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol. Cell. Biol. 171490-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thai, T. H., D. P. Calado, S. Casola, K. M. Ansel, C. Xiao, Y. Xue, A. Murphy, D. Frendewey, D. Valenzuela, J. L. Kutok, M. Schmidt-Supprian, N. Rajewsky, G. Yancopoulos, A. Rao, and K. Rajewsky. 2007. Regulation of the germinal center response by microRNA-155. Science 316604-608. [DOI] [PubMed] [Google Scholar]
- 32.Thorley-Lawson, D. A. 2001. Epstein-Barr virus: exploiting the immune system. Nat. Rev. Immunol. 175-82. [DOI] [PubMed] [Google Scholar]
- 33.Tili, E., J. J. Michaille, A. Cimino, S. Costinean, C. D. Dumitru, B. Adair, M. Fabbri, H. Alder, C. G. Liu, G. A. Calin, and C. M. Croce. 2007. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 1795082-5089. [DOI] [PubMed] [Google Scholar]
- 34.Vigorito, E., K. L. Perks, C. Abreu-Goodger, S. Bunting, Z. Xiang, S. Kohlhaas, P. P. Das, E. A. Miska, A. Rodriguez, A. Bradley, K. G. Smith, C. Rada, A. J. Enright, K. M. Toellner, I. C. Maclennan, and M. Turner. 2007. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity 27847-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Werner, S. L., D. Barken, and A. Hoffmann. 2005. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science 3091857-1861. [DOI] [PubMed] [Google Scholar]
- 36.Yin, Q., J. McBride, C. Fewell, M. Lacey, X. Wang, Z. Lin, J. Cameron, and E. K. Flemington. 2008. microRNA-155 is an Epstein-Barr virus induced gene that modulates Epstein Barr virus regulated gene expression pathways. J. Virol. [DOI] [PMC free article] [PubMed]
- 37.Yin, Q., X. Wang, J. McBride, C. Fewell, and E. Flemington. 2008. B-cell receptor activation induces BIC/miR-155 expression through a conserved AP-1 element. J. Biol. Chem. 2832654-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young, L. S., and A. B. Rickinson. 2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4757-768. [DOI] [PubMed] [Google Scholar]
- 39.Zhao, Y., and D. Srivastava. 2007. A developmental view of microRNA function. Trends Biochem. Sci. 32189-197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.