

Abstract
This review focuses on recent developments in our understanding of neurodegeneration at the mammalian neuromuscular junction. We provide evidence to support a hypothesis of compartmental neurodegeneration, whereby synaptic degeneration occurs by a separate, distinct mechanism from cell body and axonal degeneration. Studies of the spontaneous mutant Wlds mouse, in which Wallerian degeneration is characteristically slow, provide key evidence in support of this hypothesis. Some features of synaptic degeneration in the absence of Wallerian degeneration resemble synapse elimination in neonatal muscle. This and other forms of synaptic plasticity may be accessible to further investigations, exploiting advantages afforded by the Wlds mutant, or transgenic mice that express the Wlds gene.
Orthograde degeneration in the distal segment of severed axons was first described by Augustus Waller in 1850, when he examined lesioned hypoglossal and glossopharyngeal nerves in the frog. Waller noted that the axon disintegrated and the remaining debris was subsequently removed within a few days of axotomy. However, our present knowledge and understanding of the underlying mechanisms of Wallerian degeneration (WD) remain sketchy, despite the advent and improvement of physiological, immunocytochemical and molecular techniques.
Our aim here is fourfold. First, to briefly review what is known about WD in wild-type animals. Second, to discuss the characteristic phenotype of the spontaneous mutant Wlds mouse, and the opportunities this mutant offers to gain insights into the molecular mechanisms of WD. Third, to appraise the evidence that WD is one of several distinctive, compartmentalised degeneration mechanisms in neurones, whereby survival of cell bodies and dendrites, axons, and synaptic terminals may be regulated independently. Finally, we argue for the utility of the Wlds mouse as a paradigm for studying other issues in neurobiology, such as mechanisms responsible for plasticity of synaptic structure and function.
(1) Wallerian degeneration of axons and motor nerve terminals
Axons
The primary event in WD is axonal fragmentation and degeneration (Vial, 1958; Allt, 1976; Hallpike, 1976; Nicholls et al. 1992). Subsequent breakdown and removal of the myelin sheath occurs by phagocytosis involving the invasion of myelomonocytic cells after the onset of axonal degeneration (Beuche & Friede, 1984). Once the lesioned axon has begun to fragment, the myelin sheath retracts from the nodes of Ranvier creating enlarged nodal spaces (Fig. 1). These then segregate the nerve into ‘digestive chambers’ or ‘ellipsoids’ (Allt, 1976). Within each of these compartments, the axon fragments proceed to a state of complete degradation. Electron microscopy has demonstrated that the major early axonal changes include fragmentation of endoplasmic reticulum and dissolution of neurofilaments and microtubules within 48 h (Vial, 1958; Honjin et al. 1959; Ballin & Thomas, 1969; Donat & Wisniewski, 1973). These changes have been attributed by Schlaepfer (1974) to the influx of calcium ions at the lesion site. Soon after the onset of these events it is also possible to detect a more distinctive degenerative marker, the swelling and lysis of axonal mitochondria.
Figure 1. Schematic representation of Wallerian degeneration.
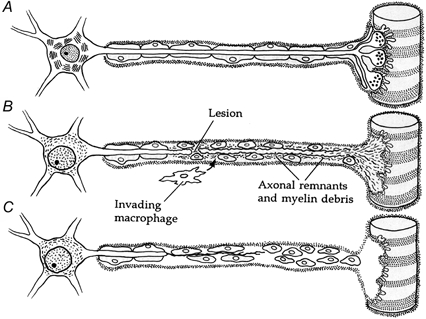
A, the cellular organisation of a motoneurone, skeletal muscle fibre and Schwann cells. B, following an axonal lesion, the distal stump of the axon and its motor nerve terminal degenerate. The resident Schwann cells dedifferentiate and proliferate. The axonal, nerve terminal and myelin debris are removed by phagocytosing Schwann cells as well as invading macrophages. The cell body undergoes chromatolysis and the nucleus translocates. C, after the removal of all debris and the formation of bands of Büngner by Schwann cells, the proximal nerve stump regenerates back to the denervated muscle fibre. (Adapted from Nicholls et al. (1992) with permission.)
As degradation of the axon continues, the ellipsoids are removed by phagocytosing Schwann cells and invading macrophages. At the same time, Schwann cells proliferate: in lesioned rabbit sciatic nerve after 25 days there are up to 13 times the number present before injury (Abercrombie & Johnson, 1946). These Schwann cells join together, tip to tip, forming longitudinal bands known as ‘bands of Büngner’. The Schwann cell bands may play a role in guiding the regenerating proximal nerve stump axons back to the denervated site. Schwann cells secrete many different growth and adhesive factors such as nerve growth factor (NGF; Heumann et al. 1987), the neural cell adhesion molecule (N-CAM; Nieke & Schachner, 1985) and cytokines including members of the interleukin (IL-x) family (Rutkowski et al. 1999; for review see Fu & Gordon, 1997).
Cell body reaction
In adults the proximal portion of an axotomised motoneurone does not normally degenerate alongside the distal stump following axotomy, at least in the short term (Romanes, 1941; Johnson & Duberley, 1998). However, marked changes occur to both the cell body and its nucleus (Nicholls et al. 1992). The cell body swells, the nucleus translocates and the Nissl substance (endoplasmic reticulum) becomes dispersed.
In contrast, survival of neonatal motoneurones is strongly dependent upon their maintaining synaptic contact with their target muscles. Thus neonatal motoneurones normally die by apoptosis within a few days of nerve section. In this case, motoneurone death is mitigated by neurotrophic factors originating in the target muscles signalling through transmembrane receptors to the Bax-Bcl-2-Bcl-X system, activating calcium-dependent proteases (Martinou et al. 1994; Knudson et al. 1995; Arce et al. 1998; Villa et al. 1998; reviewed by Pettmann & Henderson, 1998).
Immediate early genes (IEGs; see Morgan & Curran, 1991), which control the cell body response following axotomy of adult motoneurones, are activated by transcription factors including c-Jun and JunD, which are selectively expressed in axotomised peripheral and central neurones within 10-15 h of nerve lesion (for review see Herdegen & Leah, 1998). By contrast, c-Fos, FosB, JunB and Fras expression do not occur at all (Leah et al. 1991; Jenkins & Hunt, 1991; Herdegen & Zimmermann, 1994). Induction of c-Jun expression precedes the activation of several regeneration-associated genes such as GAP-43, tubulins and other cytoskeletal proteins required for axon regeneration (Mikucki & Oblinger, 1991; Tetzlaff et al. 1991; Herdegen & Zimmermann, 1994).
Neuromuscular junction
The progress of WD at the mammalian neuromuscular junction (NMJ) is well documented (Miledi & Slater, 1968, 1970; Manalov, 1974; Winlow & Usherwood, 1975). Axotomy induces degeneration of nerve terminals before the degeneration of their motor axons (Birks et al. 1960): within 24-26 h after axotomy in rodents, depending upon the length of the remaining distal nerve stump. Initial degenerative changes in the nerve terminal include (after a lag period of 3-8 h): (i) swelling, disruption of the cristae and destruction of mitochondrial membranes; (ii) a reduction in the number of and clustering of synaptic vesicles; and (iii) active invasion of terminal Schwann cell processes into the synaptic cleft. More advanced degenerative changes involve extensive fragmentation of nerve terminal membranes, accompanied by axoplasmic autolysis and phagocytic engulfment of the nerve terminal by the terminal Schwann cell. These sometimes remain apposed to the postsynaptic folds for up to 3 weeks after phagocytosis is complete. Retention of Schwann cells in the period following denervation is significant, because they promote reinnervation by extending processes that guide sprouts and regenerating axons back to denervated endplates (Son & Thompson, 1995a,b).
In summary, axotomy induces distinct cellular responses on either side of an axonal lesion. Distal axons and synaptic terminals degenerate. In neonates, axotomised neurones also die, by apoptosis. Adult neurones normally survive axotomy, however. Thus these distinct reactions of normal neurones to interruption of their axons suggest that different cellular mechanisms regulate cell survival; and these mechanisms may, to a certain extent, be compartmentalised.
(2) The Wlds mouse
Discovery of the Wlds mouse
During their investigations into the role of recruited myelomonocytic cells in WD of mouse peripheral nerve, M. C. Brown, V. H. Perry and their colleagues discovered, quite serendipitously, a spontaneous mutation in the C57Bl/6 line of mice supplied originally by Harlan-Olac (Lunn et al. 1989). The mutant mice show no readily discernible phenotype and they breed easily. What distinguishes these mice is that WD is significantly delayed and protracted after axotomy. Thus, the distal portion of cut axons and their motor nerve terminals remain morphologically intact for as long as 2 weeks. Remarkably, the isolated distal axons are still capable of conducting action potentials, and neuromuscular synapses continue to release neurotransmitter and recycle synaptic vesicles for several days, despite being disconnected from their cell bodies (Tsao et al. 1994; Ribchester et al. 1995). Both sensory and motor axons are delayed in their degeneration, and the mutation also delays WD in the central nervous system, for example following section of the optic nerve (Perry et al. 1990a; Ludwin & Bisby, 1992). The Wlds phenotype appears to be an age-dependent phenomenon, however; mice over 4 months of age appear to revert to the WD pattern of a wild-type (Perry et al. 1992; Tsao et al. 1994; Ribchester et al. 1995; Gillingwater et al. 2000; T. H. Gillingwater, D. Thompson, M. P. Coleman & R. R. Ribchester, manuscript in preparation; but see Crawford et al. 1995).
Molecular genetics of the Wlds mouse
Genomic analysis of the mutant subsequently led to its redesignation as Wlds (Lyon et al. 1993). The nature of the Wlds mutation has now also been unequivocally demonstrated.
Perry et al. (1990c) produced genetic outcrosses and backcrosses with the Wlds mouse and as a result showed that the mutation is controlled by a single autosomal dominant gene. Further studies mapped the Wld locus to the distal end of chromosome 4, the region homologous with the human chromosomal region 1p34-1p36 (Lyon et al. 1993). The Wld locus was subsequently identified to be an 85 kb tandem triplication within the candidate region, although in some mice there is a tandem duplication indicating that there may be some instability in the repeat copy number (Coleman et al. 1998).
Recent studies have identified exons of three genes located within the 85 kb repeat sequence that could account for the Wlds phenotype (Conforti et al. 2000). Two genes, ubiquitin fusion degradation protein 2 (Ufd2) and a novel gene, D4Cole1e, were reported to span the proximal and distal boundaries of the repeat unit, forming a chimeric gene with an open reading frame coding for a 43 kDa fusion protein (Fig. 2). The Wlds gene and its protein product are strongly expressed in the nervous system of Wlds mice. The third exon, coding for Rbp7, is not expressed in the nervous system (Conforti et al. 2000).
Figure 2. Genetics of the Wlds mouse.
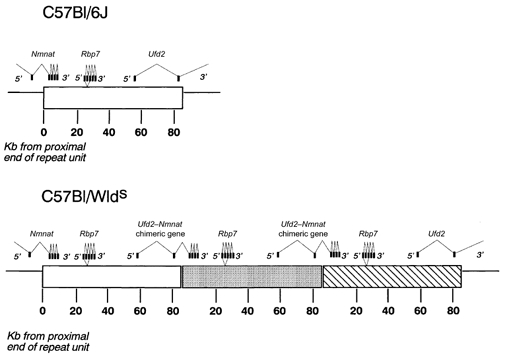
Location of exons within the 85 kb triplication repeat unit in the Wlds mutant mouse, showing the formation of the Ufd2-Nmnat chimeric gene and the triplication of the complete Rbp7 gene. (Based on Conforti et al. (2000).)
It has since been shown that the novel sequence D4Cole1e incorporates the complete sequence encoding nicotinamide mononucleotide adenylyl transferase (Nmnat), the enzyme responsible for synthesising NAD (Emanuelli et al. 2001), but incorporating an N-terminal sequence of 18 amino acids that is not normally translated in Nmnat. Transgenic mice expressing the complete chimeric protein sequence under the control of a β-actin promoter also show the Wlds phenotype (M. P. Coleman, personal communication).
Slow WD in the Wlds mouse is an intrinsic property of the nerve
Despite the recent demonstration of the genetic characteristics of the Wlds mutant, the exact function of the gene and the mechanisms by which it confers protection from the effects of axonal injury are not known. In their initial paper describing the Wlds mouse, Lunn et al. (1989) postulated that the reason for the slow progression of WD was a problem with the recruitment of myelomonocytic cells to the distal portion of the axon. They subsequently showed in transplant experiments that the phenotype was not due to a defect in the circulating monocyte population (Perry et al. 1990b). They concluded that the mutation therefore alters intrinsic properties of axons. These findings were supported by Glass et al. (1993) who showed that Wlds axons still degenerate slowly within grafted peripheral nerve sheaths containing wild-type Schwann cells, while axotomised wild-type axons degenerate normally within an environment containing Wlds Schwann cells. Likewise, axotomy of Wlds axons demyelinated by intraneural injection of lysophosphatidyl choline resulted in slow degeneration of the distal stump (Hall, 1993). Axotomised Wlds neurites also persist in tissue culture. For example, Deckwerth & Johnson (1994) studied Wlds sympathetic neurones from the superior cervical ganglion. In wild-type neurones the cell body and axons degenerate concurrently following the removal of NGF (Edwards & Tolkovsky, 1994). However, in cultures of Wlds neurones where NGF was absent, the axons survived whilst cell bodies underwent apoptosis. Distal neurites also persist following physical axotomy in culture (Glass et al. 1993), although the survival of axons depends on how long the neurones are cultured before their axons are cut (Buckmaster et al. 1995). From these experiments it was suggested that the cell body of a neurone may provide a maintenance factor which is transported to the extremities of the cell via axoplasmic transport and inhibits the initiation of WD. The Wlds mutation may either cause a stabilisation of such a factor, or impair the axonal machinery responsible for its breakdown, thereby prolonging the initiation of WD following separation from the cell body.
The hypothesis that the Wlds phenotype may be due to the impairment or failure of axonal transport was examined by Glass and colleagues (Glass & Griffin, 1991, 1994; Watson et al. 1993), but refuted with the demonstration that bi-directional transport of neurofilaments continues at a normal rate for up to 14 days post-axotomy. Tsao et al. (1994) also found no abnormalities in neurofilament phosphorylation and stability. A number of studies have addressed the possibility that an altered regulation of Ca2+ ions within the nerve causes the Wlds phenotype. For instance, both Glass et al. (1994) and Buckmaster et al. (1995) demonstrated that degeneration in Wlds axons, as in wild-type axons, is calcium dependent. Their data also suggest that calcium-dependent WD-associated proteases are present in Wlds axons, but that these may require higher levels of calcium for their activation than in normal axons. However, Tsao et al. (1994) found that the levels of calcium-activated proteases in Wlds axons were normal and Glass et al. (1998) found no evidence for a defect in the m-calpain 80 kDa subunit. The calpain system of neurofilament degradation therefore appears to be functioning normally, but the possibility that calpain activation by calcium is impaired in the Wlds mouse has not been ruled out.
Recent evidence in support of the hypothesis that calcium ions may play a pivotal role in the regulation of the Wlds phenotype is provided by experiments examining the role of NAD in intracellular Ca2+ regulation. Both cyclic ADP-ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP), derivatives of NAD and NADP, respectively, initiate release of Ca2+ from intracellular stores (Lee, 1999; Ziegler, 2000; Podesta et al. 2000). This has led to the suggestion that NAD derivatives are endogenous modulators of intracellular Ca2+ (White et al. 2000) and may also have a role in the modulation of neurotransmitter release from pre-synaptic nerve terminals (Mothet et al. 1998). This hypothesis is noteworthy given the identification of Nmnat in the Wlds genotype (Conforti et al. 2000).
Intriguingly, a recent study by Benavides et al. (2000) suggests that the elevation of intracellular Ca2+ in Wlds hippocampal neurones following depolarisation is significantly less than in control preparations. They suggest that this is evidence for abnormal calcium ion entry into Wlds neurones. However, if the mutation produces abnormal calcium buffering, perhaps via its effects on NAD levels, this could perhaps explain their findings.
Finally, proteins targeted by ubiquitin (Laney & Hochstrasser, 1999) may play a critical role in the Wlds phenotype. It is possible that the fragment of Ufd2 expressed in the chimeric Wlds protein may act to competitively inhibit the action of native Ufd2 (a ‘dominant-negative’ effect).
Effect of axotomy on Wlds NMJs
Axotomised Wlds motor nerve terminals, as well as axons, undergo a process remarkably distinct from classical WD. Ribchester et al. (1995) showed that Wlds NMJs are also preserved and retain their ability to release neurotransmitter and recycle synaptic vesicle membrane for at least 3 days, and in a few instances as long as 2 weeks, following nerve lesion.
Evidence for piecemeal withdrawal of axotomised Wlds nerve terminals and retraction of axons has since been presented, based on studies using vital dye labelling, immunocytochemistry, electrophysiology and nerve/ muscle cultures (Fig. 3; Mattison et al. 1996; Parson et al. 1998a,b; Ribchester et al. 1999). These studies suggest that nerve terminals remove themselves from the endplate, bouton by bouton, until they form a characteristic bulbous swelling at the distal end of the axon, detached from the endplate (Mattison, 1999; D. Thomson, T. H. Gillingwater & R. R. Ribchester, manuscript in preparation). Physiological changes at these synapses include a reduced quantal content and the occasional appearance of ‘giant’ miniature endplate potentials (MEPPs). The progressive nature of this synapse withdrawal is quite distinct from the synchronous degeneration of terminals observed at wild-type denervated NMJs (Miledi & Slater, 1968, 1970; Winlow & Usherwood, 1975).
Figure 3. Piecemeal withdrawal of motor nerve terminals at axotomised Wlds NMJs.
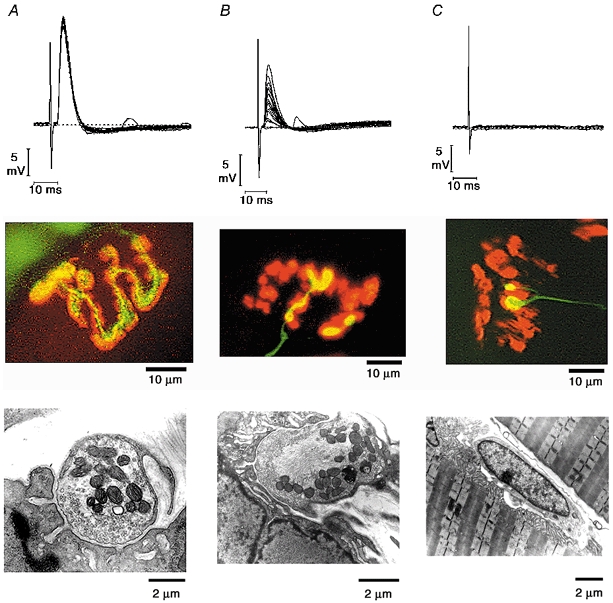
Electrophysiological, immunocytochemical and ultrastructural correlates of control, withdrawing and vacant NMJs. Control preparations (A) have strong endplate potentials, exact matching of pre- and postsynaptic components and a healthy distribution of mitochondria and clear 50 nm synaptic vesicles within terminal boutons. Withdrawing terminals (B) show a weakening of synaptic transmission as well as piecemeal retraction of boutons. Ultrastructurally, withdrawing boutons appear normal (with intact mitochondria and normal vesicle numbers) except for the accumulation of neurofilaments within the terminal. Vacant and nearly vacant endplates (C) show no electrophysiological activity and nerve terminals are sometimes replaced at the synaptic site by Schwann cells. The axon shown stained with neurofilament and SV2 antibodies ends in two ‘retraction bulbs’ resembling structures seen during neonatal synapse elimination. (Authors’ unpublished data.)
The ultrastructure of withdrawing terminals in axotomised Wlds neurones is also distinctive (Fig. 3; Ribchester et al. 1995; Gillingwater et al. 2000). They appear morphologically intact and show none of the classical signs of degeneration such as mitochondrial disruption and swelling. Morphological alterations include dense packing of synaptic vesicles towards the pre-synaptic membrane, with some vesicles as large as 130 nm in diameter. The appearance of large synaptic vesicles suggests that the synaptic vesicle retrieval mechanism may be impaired during withdrawal of Wlds nerve terminals. Neurofilaments also accumulate in the centre of withdrawing nerve terminal boutons.
Like axon degeneration, the degeneration of denervated NMJs in the Wlds mouse is also age dependent. In mice older than 3-4 months, both the time course and morphology of degeneration revert to that seen in wild-type mice (Gillingwater et al. 2000, 2001).
Secondary characteristics associated with the Wlds phenotype
Although the Wlds mouse appears indistinguishable in appearance and behaviour from the C57Bl/6J mouse, both operated and unoperated Wlds mice show some subtle, as yet unexplained, phenotypic features, suggesting that slow nerve degeneration is not the only effect of the Wlds mutation.
Brown et al. (1991a) found that normal, unoperated Wlds soleus muscles express greater intrinsic tension, fewer macrophages per muscle fibre and lower basal levels of acetylcholine sensitivity than their wild-type counterparts. Using high resolution 1H magnetic resonance spectroscopy, Tsao et al. (1999) detected altered cerebral metabolism (indicated by decreased levels of glutamate and phosphocholine relative to total N-acetyl aspartate content) in Wlds mice compared to wild-type controls.
Other studies describe specific axotomy-related secondary characteristics associated with the Wlds phenotype. For example, axotomy-induced motoneurone death in neonates (Lapper et al. 1994) and retrograde degeneration of cell bodies in axotomised adult retinal ganglion cells (Perry et al. 1990a) are also significantly delayed. In denervated Wlds muscles, the initiation of muscle atrophy and development of acetylcholine sensitivity have a slower onset, and the rise in serum creatine kinase levels is also delayed (Brown et al. 1991a).
Axon-glia signalling plays an important role in the maintenance and control of both cell types in vivo. Sprouting responses of non-myelinating terminal Schwann cells are delayed by several days (both in vivo and in vitro) following denervation in the Wlds mouse (Barry et al. 1997; Parson et al. 1998b). The resident Schwann cells in peripheral nerve produce potential maintenance factors that are taken up by the axon: for example, ciliary neurotrophic factor (CNTF). Following axotomy in the Wlds mouse, both mRNA and protein levels of CNTF remain normal for up to 4 days, whereas in wild-type animals they both decline rapidly and synchronously (Subang et al. 1997). However, by 10 days after axotomy, CNTF mRNA levels in the Wlds mice have decreased to wild-type levels, but levels of CNTF protein remain unchanged. This suggests that the CNTF protein is relatively stable in axotomised Wlds axons. Its retention could contribute some protection against degeneration.
The failure of macrophage recruitment after axotomy (see above) may be due to a failure of the nerve to produce a chemotactic signal, or there may be some form of blockade that prevents the myelomonocytic cells from entering the nerve (Perry et al. 1990b). Some candidates for this chemotactic signal are the monocyte chemoattractant protein-1 (JE), which fails to be induced in Wlds mice following axotomy (Carroll & Frohnert, 1998), and granulocyte macrophage colony-stimulating factor (GM-CSF), whose levels are also deficient following axotomy in Wlds mice. It is also possible that persisting distal axons produce a factor that inhibits macrophage recruitment, whereas production of this normally ceases in axotomised wild-type axons (Ludwin & Bisby, 1992). Whether any or all of these features are related to Ufd2-Nmnat overexpression in Wlds mice remains to be tested.
Nerve regeneration in the Wlds mouse
Following axotomy in the PNS, immediate-early gene expression is initiated within a few hours (see above), and after an initial ‘dying back’, the proximal axon stump is primed for regeneration, which begins 5-48 h after the lesion (Ramon y Cajal, 1928; Brecknell & Fawcett, 1996). In mice, this process gains momentum and proceeds over a number of days before achieving a reinnervation of skeletal muscle after about 2-3 weeks.
Previously WD was thought to play a functional role in generating an environment conducive to nerve regeneration (Ramon y Cajal, 1928). It was therefore hypothesised that in the Wlds mouse, intact distal nerve stumps would be equivalent in their obstructive effect to intact nerves. Brown et al. (1991b) showed that severed axons would not grow into a completely undegenerated portion of nerve. Remarkably, however, regeneration of motor axons after a nerve crush injury in the Wlds mouse is not prevented by the presence of axons in the remaining distal nerve stump (Lunn et al. 1989). This suggests that even though Wlds axons persist after axotomy and remain capable of functioning normally, they may undergo some conformational changes that allow regenerating nerves to progress along the distal stump.
Other studies suggest that whilst motor axons appear able to regenerate almost as well as in wild-type mice, sensory nerve regeneration in Wlds mice is significantly impaired (Bisby & Chen, 1990; Brown et al. 1992). Sommer & Schafers (1998) showed that the delay in sensory nerve regeneration leads to prolonged mechanical allodynia. One possible explanation for the disparity between the regeneration of motor and sensory nerves is that axotomised sensory nerve axons degenerate and regenerate more slowly because they are responsive to NGF levels (Bisby & Chen, 1990) and NGF levels do not increase as rapidly after nerve section in Wlds mice compared to wild-type mice (Brown et al. 1991b). Motor axons do not respond to NGF. They also appear to be more opportunistic, using myelinating and non-myelinating Schwann cells to guide regeneration (Brown et al. 1992, 1993). Tenascin-C is upregulated selectively in Schwann cells of axotomised muscle nerves of Wlds mice, and axons regenerate more rapidly in these nerves than in cutaneous nerves, where there is delayed expression of tenascin-C (Fruttiger et al. 1995). Conversely, myelin-associated glycoprotein (MAG) normally inhibits axon regeneration, but the rate of axon regeneration in Wlds mice was partially restored when these mice were cross-bred with MAG knockout mice (Schafer et al. 1996).
Shi & Stanfield (1996) also reported differences in sprouting and regeneration patterns in the CNS of the Wlds mouse. In wild-type mice, sprouting responses are detected in the septohippocampal and hippocampal commissural projections within 3 days of a lesion to the perforant path to the dentate gyrus. However, in Wlds mice septohippocampal axonal sprouting was only observed 5-7 days after a perforant pathway lesion and changes in the distribution of commissural axons in the dentate gyrus were not observed before 12 days. It would clearly be of interest to establish the fate of axotomised synapses, and their associations with astrocytic neuroglia following axonal lesions in the brains of Wlds mice.
Alterations in the rate and extent of both motor and sensory reinnervation occurring as a result of the Wlds mutation are not due to problems with the cell body reaction to axotomy. The time course of expression of transcription factors such as c-Jun is very similar in Wlds mice to that in BALB/c controls (Brown et al. 1993).
(3) Compartmental neurodegeneration
Whilst the major structural and functional compartments of the neurone are well defined (the cell body and dendrites, the axon and the synaptic terminals), there has been little debate on a possible compartmentalisation of the neurone with regard to pathophysiology. The distinctive nature of neuronal cell death by apoptosis (for reviews see Nijhawan et al. 2000; Yuan & Yanker, 2000), and the WD of distal axons suggests that different mechanisms are embedded in neurones for executing these processes. Taken together with observations of slow synapse withdrawal at axotomised Wlds NMJs, and other recent findings (see below), it appears the degeneration of synapses may also occur by a distinct, compartmentalised mechanism.
Neuronal apoptosis
The term ‘apoptosis’ was coined by Kerr et al. (1972), to describe a process involved in the normal turnover of hepatocytes: intrinsic cell suicide. There are three main regulators of apoptosis in neurones: the Bcl-2 family of proteins, an adaptor protein known as apoptotic protease-activating factor 1 (Apaf-1) and the cysteine protease caspase family (Yuan & Yanker, 2000). Neuronal apoptosis is inhibited by growth factors and by over-expression of genes such as Bcl-2. By contrast, WD appears to be independent of the same growth factors from the cell bodies, and is unaffected by the overexpression of Bcl-2 (Dubois-Dauphin et al. 1994; Sagot et al. 1995; Burne et al. 1996).
‘Cytoplastic apoptosis’ of axons
Whilst some of the cellular processes that occur during WD (for example ellipsoid body formation) are remarkably similar to those seen during apoptosis, WD obviously cannot involve all apoptotic cellular processes (eg. DNA degradation) because axotomised distal stumps do not include motoneurone cell bodies. However, it has previously been shown that apoptosis can occur in the absence of RNA or protein synthesis (Martin, 1993) and in cells lacking nuclei (Jacobson et al. 1994; Ellerby et al. 1997). Thus, a number of authors have suggested that axonal degeneration might be viewed as a form of ‘cytoplastic apoptosis’ (Ribchester et al. 1995; Buckmaster et al. 1995; Alvarez et al. 2000). To test this hypothesis, however, it will be necessary to discover the enzymes and signalling molecules that are involved.
Studies in which the role of Bcl-2 in axonal degeneration was examined also concluded that the molecular mechanisms involved are distinct from those activated during apoptosis. Burne et al. (1996) showed that overexpression of the human Bcl-2 protein in retinal ganglion cells protects the cell body as expected. However, the axons were not protected from WD. Similar findings were reported in a study by Sagot et al. (1995) who examined the fate of cell bodies and axons in a mouse model of motoneurone disease with Bcl-2 overexpression. The increased level of Bcl-2 rescued facial motoneurones and restored their soma size and choline acetyltransferase expression. However, there was no effect on the rate of axonal degeneration in facial and phrenic motoneurones. Thus it seems unlikely that Bcl-2 itself plays a significant role in axon degeneration.
Finn et al. (2000) recently examined whether the molecular machinery of WD depends upon the caspase family of cysteine proteases. They found that caspase-3 (which is thought to be important for apoptosis in neurones) was not activated in the axon during WD. Thus, they argued that WD is molecularly distinct from the classical caspase-dependent apoptotic process implicated in axonal degeneration.
‘Synaptosis’
The slow withdrawal of axotomised nerve terminals in Wlds mice suggests a novel form of synapse-specific neurodegeneration that is unmasked when axonal degeneration (WD) is delayed. We tentatively designate this process ‘synaptosis’. Interestingly, nerve terminals withdraw in a similar fashion during synapse elimination, an essential step in the formation or reformation of normal neuromuscular innervation patterns, which takes place in postnatal development or following reinnervation (Sanes & Lichtman, 1999; Lichtman & Colman, 2000; Ribchester, 2001). Thus, nerve terminals undergoing withdrawal during synapse elimination are removed from the endplate bouton by bouton, ending with the formation of a characteristic ‘retraction bulb’ (Brown et al. 1976; Riley, 1977, 1981; Balice-Gordon et al. 1993; Gan & Lichtman, 1998). These events resemble those at axotomised Wlds NMJs in both morphology and time course (Ribchester et al. 1995, 1999; Mattison et al. 1996; Parson et al. 1998a; Costanzo et al. 2000; Gillingwater et al. 2000).
Developmental synapse elimination takes place at a normal rate in Wlds mice, although axotomy in the neonate still delays degeneration (Parson et al. 1997). Thus we are not suggesting that the mechanisms of synapse withdrawal are in any way abnormal in Wlds mice. However, it is noteworthy that the role of ‘degeneration’ in synapse elimination was a matter of debate during the 1970s. For instance, ultrastructural analysis of neonatal synapse elimination by Rosenthal & Taraskevich (1977) led to their suggestion that the retraction of supernumerary inputs from developing motor endplates occurs by a mechanism similar to WD. Subsequent electron microscopic studies repudiated these findings, however, and provided evidence that the removal of excess inputs occurs by a distinctive process of retraction (Korneliussen & Jansen, 1976; Riley, 1977, 1981; Bixby, 1981). Since the withdrawal of nerve terminals in axotomised Wlds and normal development appears to be morphologically similar, and yet distinct from WD, this raises the possibility of a common underlying mechanism of nerve terminal retraction. One way of rationalising these findings is to suggest that synaptic maintenance depends upon the supply of essential and specific maintenance factors or molecules. If the supply of these factors is compromised, then synapses withdraw at a rate that varies inversely with the concentration of such factors. During normal development, an adequate supply may depend on their trafficking into motor nerve terminals. In Wlds mice disruption of this trafficking is synchronised by axotomy. Synapse withdrawal (rather than degeneration) is observed because WD is absent (or profoundly delayed). In wild-type animals axotomy induces additional, rapid degenerative mechanisms in axons, which therefore mask the slower process of synapse withdrawal. In normal development there is no axotomy as such, but disruptive trafficking of the same molecules could lead to selective loss of synaptic inputs to muscle fibres. Such a mechanism is consistent with earlier proposals of ‘sibling neurite bias’ or ‘intrinsic withdrawal’ as important processes in establishing the normal motor innervation pattern of skeletal muscle fibres (Brown et al. 1976; Thompson & Jansen, 1977; Smalheiser & Crain, 1984; Fladby & Jansen, 1987).
Disruption of axonal transport under experimental conditions also triggers synapse withdrawal. For instance, blocking fast axonal transport with batrachotoxin causes nerve terminals to withdraw from endplates within 18 h; but they then grow back (Hudson et al. 1984). Nerve terminals also retract in a ‘non-Wallerian’ fashion following a single subcutaneous injection of the organophosphate sarin (Kawabuchi et al. 1991). Rich & Lichtman (1989) observed comparable, reversible nerve terminal withdrawal from degenerating muscle fibres. Nerve terminals also retract in piecemeal fashion in a mouse model of myasthenia gravis (Rich et al. 1994).
The molecular mechanisms of synapse withdrawal, either during synapse elimination in development or following axotomy in Wlds mice, remain unknown. However, further evidence in support of the hypothesis that the ‘degeneration’ of synapses is regulated independently from that of cell bodies or axons has been obtained in recent studies of CNS neurons. For example, Ivins et al. (1998) have shown that activation of caspases plays a crucial role in neurite degeneration in cultured hippocampal neurones exposed to an apoptotic stimulus (amyloid β-peptide). Using the same cell-death stimulus, Mattson et al. (1998b) found no caspase activation in axons, but instead reported that caspases were activated in cortical synaptosomes. Their data also provide evidence that apoptotic biochemical cascades (such as caspase activation) are selectively triggered at synaptic sites following exposure to staurosporine and Fe2+. The term ‘synaptic apoptosis’ was proposed (Mattson et al. 1998a,b; Mattson, 2000; Guo & Mattson, 2000).
In summary, analysis of the reactions of cell bodies, axons and synaptic terminals to axotomy in Wlds mice suggests that mechanisms of degeneration are highly compartmentalised in neurones (Fig. 4). Neuronal cell bodies react to certain pathophysiological stimuli, or deprivation of growth factors by changing their pattern of gene expression, leading - at some stages in development, or after prolonged periods of deprivation in adults - to programmed cell death by apoptosis. Under normal circumstances, the death of the cell body is followed by rapid degeneration of the other neuronal compartments: dendrites, axons and synaptic terminals. Nerve injury disconnects axons and synapses from their cell bodies, and this normally triggers an independent mechanism - WD - in the isolated distal axon. The dissociation between neuronal cell body apoptosis and WD is revealed when WD is absent or delayed, as in the Wlds mutant mouse. The slow pace of axon degeneration in this mutant (and in transgenic mice expressing the Wlds chimeric protein) also reveals that disconnection of synapses from cell bodies can induce at least one additional mechanism of degeneration, namely withdrawal of synaptic terminals.
Figure 4. Compartmental organisation of degeneration mechanisms in the neurone based on the present review.
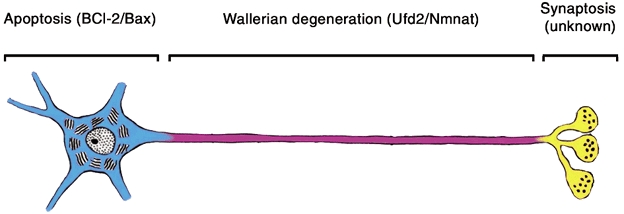
Genes regulating cell body degeneration by apoptosis (blue) and axon degeneration (magenta) are given in parentheses. The genes controlling synaptic degeneration (Synaptosis; yellow) have not yet been defined.
(4) Future research and additional utilities of the Wlds mouse
With the full characterisation of the chimeric Wlds gene, it should become possible to gain deeper insights into the functions of this gene and mechanisms by which it mitigates axon degeneration. One possibility is that the Ufd2-Nmnat chimeric protein upregulates the production of NAD, which in turn acts as a maintenance factor or indirectly as an axonal calcium buffer. To discover the subcellular location and action of such a maintenance or buffering mechanism within the axon would be of great importance in uncovering the mechanisms and fine control of calcium-mediated degeneration in both the peripheral and central nervous systems. An alternative hypothesis is that the mutant gene destabilises the proteolytic system involving ubiquitin. As ubiquitin is crucial in the selection process of which proteins are to be degraded (Laney & Hochstrasser, 1999), it will be interesting to find out whether the mutation in the Wlds mouse affects the degradation of all, some or none of the constituent proteins of the axon. This could perhaps be examined by creating transgenic animals in which the Ufd2 fragment or Nmnat are independently expressed.
Further research into the underlying mechanisms of synapse withdrawal at axotomised Wlds NMJs may also provide insights into the mechanisms responsible for synapse elimination during development and following reinnervation after nerve injury. Since the phenomenon of synapse elimination is not restricted to the PNS, study of axotomy-induced synaptic withdrawal in the CNS of these mice could provide a greater understanding of the mechanisms of remodelling central connections, for example in the developmental organisation of the visual system and cerebellum (Lohof et al. 1996). Understanding the response to brain or spinal cord injury, especially the role of collateral secondary degeneration (Dirnagl et al. 1999), might also be facilitated by experiments using Wlds mice.
Finally, the persistence of functional synapses in isolated preparations of neural tissue from Wlds mice - or mice transgenically expressing the Wlds gene - beyond the period when axotomy normally induces axons and synaptic terminals to degenerate, suggests that these mice may be of considerable value for studies aimed towards improved understanding of mechanisms of functional synaptic plasticity. It may be possible to use the absence of axon degeneration in brain slice preparations from Wlds mice to track synaptic strengths over many more hours than is presently feasible. For example, this could prove valuable in studies of the induction and expression of ‘late’ long term potentiation of synaptic transmission (Frey & Morris, 1998). The mechanisms of other activity-dependent changes in the physiological responses of synapses could also now become more accessible, by utilising in vitro preparations in which classical neurodegeneration of synapses and axons remains absent for more than 48 h after axonal injury.
Acknowledgments
We are grateful to the MRC and the Wellcome Trust for support, Derek Thomson for expert technical assistance and for providing the electrophysiology records used in Fig. 3 and Dr M. P. Coleman for discussions and helpful comments on this review.
References
- Abercrombie M, Johnson ML. Quantitative histology of Wallerian degeneration, I. Nuclear population in rabbit sciatic nerve. Journal of Anatomy. 1946;80:37–50. [PubMed] [Google Scholar]
- Allt G. Pathology of the peripheral nerve. In: Landon DN, editor. The Peripheral Nerve. London: Chapman & Hall; 1976. pp. 666–739. [Google Scholar]
- Alvarez J, Giuditta A, Koenig E. Protein synthesis in axons and terminals: Significance for maintenance, plasticity and regulation of phenotype with a critique of slow transport theory. Progress in Neurobiology. 2000;62:1–62. doi: 10.1016/s0301-0082(99)00062-3. [DOI] [PubMed] [Google Scholar]
- Arce V, Pollock RA, Philippe JM, Pennica D, Henderson CE, De Apeyrie'Re O. Synergistic effects of Schwann- and muscle-derived factors on motoneuron survival include GDNF and cardiotrophin-1 (CT-1) Journal of Neuroscience. 1998;18:1440–1448. doi: 10.1523/JNEUROSCI.18-04-01440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Chua CK, Nelson CC, Lichtman JW. Gradual loss of synaptic cartels precedes axon withdrawal at developing neuromuscular junctions. Neuron. 1993;11:801–815. doi: 10.1016/0896-6273(93)90110-d. [DOI] [PubMed] [Google Scholar]
- Ballin RHM, Thomas PK. Changes at the nodes of Ranvier during Wallerian degeneration: an electron microscope study. Acta Neuropathologica (Berlin) 1969;14:237–249. doi: 10.1007/BF00685303. [DOI] [PubMed] [Google Scholar]
- Barry JA, Mattison RJ, Nelson C, Lichtman JW, Ribchester RR. Schwann cell sprouting in a mutant mouse with slowed Wallerian degeneration. Society for Neuroscience Abstracts. 1997;23:P244.10. [Google Scholar]
- Benavides E, Miranda M, Alvarez J. Cytosolic calcium pulses in Wlds mouse neurones. Journal of Physiology. 2000;523.P:28P. [Google Scholar]
- Beuche W, Friede RL. The role of non-resident cells in Wallerian degeneration. Journal of Neurocytology. 1984;13:767–796. doi: 10.1007/BF01148493. [DOI] [PubMed] [Google Scholar]
- Birks R, Katz B, Miledi R. Physiological and structural changes at the amphibian myoneural junction, in the course of nerve degeneration. Journal of Physiology. 1960;150:145–168. doi: 10.1113/jphysiol.1960.sp006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisby MA, Chen S. Delayed Wallerian degeneration in sciatic nerves of C57Bl/Ola mice is associated with impaired regeneration of sensory axons. Brain Research. 1990;530:117–120. doi: 10.1016/0006-8993(90)90666-y. [DOI] [PubMed] [Google Scholar]
- Bixby JL. Ultrastructural observations on synapse elimination in neonatal rabbit skeletal muscle. Journal of Neurocytology. 1981;10:81–100. doi: 10.1007/BF01181746. [DOI] [PubMed] [Google Scholar]
- Brecknell JE, Fawcett JW. Axonal regeneration. Biological Reviews of the Cambridge Philosophical Society. 1996;71:227–255. doi: 10.1111/j.1469-185x.1996.tb00748.x. [DOI] [PubMed] [Google Scholar]
- Brown MC, Booth CM, Lunn ER, Perry VH. Delayed response to denervation in muscles of C57Bl/Ola mice. Neuroscience. 1991a;43:279–283. doi: 10.1016/0306-4522(91)90435-q. [DOI] [PubMed] [Google Scholar]
- Brown MC, Jansen JK, Van Essen D. Polyneuronal innervation of skeletal muscle in new-born rats and its elimination during maturation. Journal of Physiology. 1976;261:387–422. doi: 10.1113/jphysiol.1976.sp011565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Lunn ER, Perry VH. Poor growth of mammalian motor and sensory axons into intact proximal nerve stumps. European Journal of Neuroscience. 1991b;3:1366–1369. doi: 10.1111/j.1460-9568.1991.tb00069.x. [DOI] [PubMed] [Google Scholar]
- Brown MC, Lunn ER, Perry VH. Consequences of slow Wallerian degeneration for regenerating motor and sensory axons. Journal of Neurobiology. 1992;23:521–536. doi: 10.1002/neu.480230507. [DOI] [PubMed] [Google Scholar]
- Brown MC, Perry VH, Hunt SP, Lapper SR. Further studies on motor and sensory nerve regeneration in mice with delayed Wallerian degeneration. European Journal of Neuroscience. 1993;6:420–428. doi: 10.1111/j.1460-9568.1994.tb00285.x. [DOI] [PubMed] [Google Scholar]
- Brown MC, Perry VH, Lunn ER, Gordon S, Heumann R. Macrophage dependence of peripheral sensory nerve regeneration - Possible involvement of nerve growth-factor. Neuron. 1991;6:359–370. doi: 10.1016/0896-6273(91)90245-u. [DOI] [PubMed] [Google Scholar]
- Buckmaster EA, Perry VH, Brown MC. The rate of Wallerian degeneration in cultured neurons from wild type and C57Bl/Wlds mice depends on time in culture and may be extended in the presence of elevated K+ levels. European Journal of Neuroscience. 1995;7:1596–1602. doi: 10.1111/j.1460-9568.1995.tb01155.x. [DOI] [PubMed] [Google Scholar]
- Burne JF, Staple JK, Raff MC. Glial cells are increased proportionally in transgenic optic nerves with increased numbers of axons. Journal of Neuroscience. 1996;16:2064–2073. doi: 10.1523/JNEUROSCI.16-06-02064.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SL, Frohnert PW. Expression of JE (monocyte chemoattractant protein-1) is induced by sciatic axotomy in wild type rodents but not in C57Bl/Wlds mice. Journal of Neuropathology and Experimental Neurology. 1998;57:915–930. doi: 10.1097/00005072-199810000-00004. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proceedings of the National Academy of Sciences of the USA. 1998;95:9985–9990. doi: 10.1073/pnas.95.17.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TGA, Weiqian M, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration mouse. Proceedings of the National Academy of Sciences of the USA. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo EM, Barry JA, Ribchester RR. Competition at silent synapses in reinnervated skeletal muscle. Nature Neuroscience. 2000;3:694–700. doi: 10.1038/76649. [DOI] [PubMed] [Google Scholar]
- Crawford TO, Hsieh ST, Schryer BL, Glass JD. Prolonged axonal survival in transacted nerves of C57Bl/Ola mice is independent of age. Journal of Neurocytology. 1995;24:333–340. doi: 10.1007/BF01189060. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM. Neurites can remain viable after the destruction of the neuronal soma by programmed cell death. Developmental Biology. 1994;165:63–72. doi: 10.1006/dbio.1994.1234. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: An integrated view. Trends in Neurosciences. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Donat JR, Wisniewski HM. The spatio-temporal pattern of Wallerian degeneration in mammalian peripheral nerves. Brain Research. 1973;53:41–53. doi: 10.1016/0006-8993(73)90765-8. [DOI] [PubMed] [Google Scholar]
- Dubois-Dauphin M, Frankowski H, Tsujimoto Y, Huarte J, Martinou J-C. Neonatal motoneurons overexpressing the Bcl-2 protooncogene in transgenic mice are protected from axotomy-induced cell death. Proceedings of the National Academy of Sciences of the USA. 1994;91:3309–3313. doi: 10.1073/pnas.91.8.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SN, Tolkovsky AM. Characterization of apoptosis in cultured rat sympathetic neurons after nerve growth factor withdrawal. Journal of Cell Biology. 1994;124:537–546. doi: 10.1083/jcb.124.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellerby HM, Martin SJ, Ellerby LM, Naiem SS, Rabizadeh S, Salvesen GS, Casiano CA, Cahsman NR, Green DR, Bredesen DE. Establishment of a cell-free system of neuronal apoptosis: comparison of premitochondrial, mitochondrial and postmitochondrial phases. Journal of Neuroscience. 1997;17:6165–6178. [PMC free article] [PubMed] [Google Scholar]
- Emanuelli M, Carnevali F, Saccucci F, Pierella F, Amici A, Raffaelli N, Magni G. Molecular cloning, chromosomal localization, tissue mRNA levels, bacterial expression, and enzymatic properties of human NMN adenylyltransferase. Journal of Biological Chemistry. 2001;276:406–412. doi: 10.1074/jbc.M008700200. [DOI] [PubMed] [Google Scholar]
- Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. Journal of Neuroscience. 2000;20:1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fladby T, Jansen JK. Postnatal loss of synaptic terminals in the partially denervated mouse soleus muscle. Acta Physiologica Scandinavica. 1987;129:239–246. doi: 10.1111/j.1748-1716.1987.tb08064.x. [DOI] [PubMed] [Google Scholar]
- Frey E, Morris RGM. Synaptic tagging: Implications for late maintenance of hippocampal long-term potentiation. Trends in Neurosciences. 1998;21:181–188. doi: 10.1016/s0166-2236(97)01189-2. [DOI] [PubMed] [Google Scholar]
- Fruttiger M, Schachner M, Martini R. Tenascin-C expression during Wallerian degeneration in C57Bl/Wlds mice: Possible implications for axonal regeneration. Journal of Neurocytology. 1995;24:1–14. doi: 10.1007/BF01370156. [DOI] [PubMed] [Google Scholar]
- Fu SY, Gordon T. The cellular and molecular basis of peripheral nerve regeneration. Molecular Neurobiology. 1997;14:67–116. doi: 10.1007/BF02740621. [DOI] [PubMed] [Google Scholar]
- Gan WB, Lichtman JW. Synaptic segregation at the developing neuromuscular junction. Science. 1998;282:1508–1511. doi: 10.1126/science.282.5393.1508. [DOI] [PubMed] [Google Scholar]
- Gillingwater TH, Koutsikou S, Barry JA, Ribchester RR. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wlds mutant mice. Journal of Physiology. 2000;523.P:53–54P. doi: 10.1113/jphysiol.2002.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater TH, Thomson D, Ribchester RR. Age-dependent transition in axotomy-induced phenotype at neuromuscular synapses of Wlds mutant mice. British Neuroscience Association Abstracts. 2001;16:P121. [Google Scholar]
- Glass JD, Brushart TM, George EB, Griffin JW. Prolonged survival of transected nerve fibres in C57Bl/Ola mice is an intrinsic characteristic of the axon. Journal of Neurocytology. 1993;22:311–321. doi: 10.1007/BF01195555. [DOI] [PubMed] [Google Scholar]
- Glass JD, Griffin JW. Neurofilament redistribution in transected nerves: Evidence for bidirectional transport of neurofilaments. Journal of Neuroscience. 1991;11:3146–3154. doi: 10.1523/JNEUROSCI.11-10-03146.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass JD, Griffin JW. Retrograde transport of radiolabeled cytoskeletal proteins in transected nerves. Journal of Neuroscience. 1994;14:3915–3921. doi: 10.1523/JNEUROSCI.14-06-03915.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass JD, Nash N, Dry I, Culver D, Levey AI, Wesselingh S. Cloning of M-Calpain 80kD subunit from the axonal degeneration-resistant Wlds mouse mutant. Journal of Neuroscience Research. 1998;52:653–660. doi: 10.1002/(SICI)1097-4547(19980615)52:6<653::AID-JNR4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Glass JD, Schryer BL, Griffin JW. Calcium-mediated degeneration of the axonal cytoskeleton in the Ola mouse. Journal of Neurochemistry. 1994;62:2472–2475. doi: 10.1046/j.1471-4159.1994.62062472.x. [DOI] [PubMed] [Google Scholar]
- Guo ZH, Mattson MP. In vivo 2-deoxyglucose administration preserves glucose and glutamate transport and mitochondrial function in cortical synaptic terminals after exposure to amyloid β-peptide and iron: Evidence for a stress response. Experimental Neurology. 2000;166:173–179. doi: 10.1006/exnr.2000.7497. [DOI] [PubMed] [Google Scholar]
- Hall SM. Observations on the progress of Wallerian degeneration in transected peripheral nerves of C57Bl/Wld mice in the presence of recruited macrophages. Journal of Neurocytology. 1993;22:480–490. doi: 10.1007/BF01181567. [DOI] [PubMed] [Google Scholar]
- Hallpike JF. Histochemistry of peripheral nerves and nerve terminals. In: Landon DN, editor. The Peripheral Nerve. London: Chapman & Hall; 1976. pp. 605–665. [Google Scholar]
- Herdegen T, Leah JD. Inducible and constitutive transcription factors in the mammalian nervous system: Control of gene expression by jun, fos and krox, and creb/atf proteins. Brain Research Reviews. 1998;28:370–490. doi: 10.1016/s0165-0173(98)00018-6. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Zimmermann M. Expression of c-jun and jund transcription factors represent specific changes in neuronal gene expression following axotomy. Progress in Brain Research. 1994;103:153–169. doi: 10.1016/s0079-6123(08)61135-8. [DOI] [PubMed] [Google Scholar]
- Heumann R, Korsching S, Bandtlow C, Thoenen H. Changes of nerve growth factor synthesis in non-neuronal cells in response to sciatic nerve transection. Journal of Cell Biology. 1987;104:1623–1631. doi: 10.1083/jcb.104.6.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjin R, Nakamura T, Imura M. Electron microscopy of peripheral nerve fibres. III On the axoplasmic changes during Wallerian degeneration. Okajimas Folia Anatomica Japonica. 1959;33:131–156. [Google Scholar]
- Hudson CS, Deshpande SS, Albuquerque EX. Consequences of axonal transport blockade by batrachotoxin on mammalian neuromuscular junction. III. An ultrastructural study. Brain Research. 1984;296:319–332. doi: 10.1016/0006-8993(84)90068-4. [DOI] [PubMed] [Google Scholar]
- Ivins KJ, Bui ETN, Cotman CW. β-amyloid induces local neurite degeneration in cultured hippocampal neurons: Evidence for neuritic apoptosis. Neurobiology of Disease. 1998;5:365–378. doi: 10.1006/nbdi.1998.0228. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Burne JF, Raff MC. Programmed cell death and bcl-2 protection in the absence of a nucleus. EMBO Journal. 1994;13:1899–1910. doi: 10.1002/j.1460-2075.1994.tb06459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins R, Hunt SP. Long term increase in the levels of c-jun mRNA and jun protein-like immunoreactivity in motor and sensory neurons following axon damage. Neuroscience Letters. 1991;129:107–110. doi: 10.1016/0304-3940(91)90731-8. [DOI] [PubMed] [Google Scholar]
- Johnson IP, Duberley RM. Motoneuron survival and expression of neuropeptides and neurotrophic factor receptors following axotomy in adult and ageing rats. Neuroscience. 1998;84:141–150. doi: 10.1016/s0306-4522(97)00500-9. [DOI] [PubMed] [Google Scholar]
- Kawabuchi M, Cintra WM, Deshpande SS, Albuquerque EX. Morphological and electrophysiological study of distal motor nerve fiber degeneration and sprouting after irreversible cholinesterase inhibition. Synapse. 1991;8:218–228. doi: 10.1002/syn.890080308. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. British Journal of Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- Korneliussen H, Jansen JKS. Morphological aspects of the elimination of polyneuronal innervation of skeletal muscle fibres in newborn rats. Journal of Neurocytology. 1976;5:591–604. doi: 10.1007/BF01175572. [DOI] [PubMed] [Google Scholar]
- Laney JD, Hochstrasser M. Substrate targeting in the ubiquitin system. Cell. 1999;97:427–430. doi: 10.1016/s0092-8674(00)80752-7. [DOI] [PubMed] [Google Scholar]
- Lapper SR, Brown MC, Perry VH. Motor neuron death induced by axotomy in neonatal mice occurs more slowly in a mutant strain in which Wallerian degeneration is very slow. European Journal of Neuroscience. 1994;6:473–477. doi: 10.1111/j.1460-9568.1994.tb00289.x. [DOI] [PubMed] [Google Scholar]
- Leah JD, Herdegen T, Bravo R. Selective expression of jun proteins following axotomy and axonal transport block in peripheral nerves in the rat: evidence for a role in the regeneration process. Brain Research. 1991;566:198–207. doi: 10.1016/0006-8993(91)91699-2. [DOI] [PubMed] [Google Scholar]
- Lee HC. A unified mechanism of enzymatic synthesis of two calcium messengers: Cyclic ADP-ribose and NAADP. Biological Chemistry. 1999;380:785–793. doi: 10.1515/BC.1999.098. [DOI] [PubMed] [Google Scholar]
- Lichtman JW, Colman H. Synapse elimination and indelible memory. Neuron. 2000;25:269–278. doi: 10.1016/s0896-6273(00)80893-4. [DOI] [PubMed] [Google Scholar]
- Lohof AM, Delhaye-Bouchard N, Mariani J. Synapse elimination in the central nervous system: Functional significance and cellular mechanisms. Reviews in the Neurosciences. 1996;7:85–101. doi: 10.1515/revneuro.1996.7.2.85. [DOI] [PubMed] [Google Scholar]
- Ludwin SK, Bisby MA. Delayed Wallerian degeneration in the central nervous system of Ola mice: An ultrastructural study. Journal of the Neurological Sci.ences. 1992;109:140–147. doi: 10.1016/0022-510x(92)90160-m. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. European Journal of Neuroscience. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Lyon MF, Ogunkolade BW, Brown MC, Atherton DJ, Perry VH. A gene affecting Wallerian nerve degeneration maps distally on mouse chromosome 4. Proceedings of the National Academy of Sciences of the USA. 1993;90:9717–9720. doi: 10.1073/pnas.90.20.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolov S. Initial changes in the neuromuscular synapses of denervated rat diaphragm. Brain Research. 1974;65:303–316. doi: 10.1016/0006-8993(74)90042-0. [DOI] [PubMed] [Google Scholar]
- Martin SJ. Protein or RNA synthesis inhibition induces apoptosis of mature human CD4+ T cell blasts. Immunology Letters. 1993;35:125–134. doi: 10.1016/0165-2478(93)90080-l. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowsky H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Overexpression of bcl-2 in transgenic mice protects neurons from naturally occuring cell death and experimental ischaemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- Mattison RJ. University of Edinburgh; 1999. Synapse withdrawal at neuromuscular junctions in mutant mice with slow Wallerian degeneration (Wlds) PhD Thesis. [Google Scholar]
- Mattison RJ, Thomson D, Barry JA, Ribchester RR. Sudden death of axotomized motor nerve terminals at neuromuscular junctions in Wlds mice. Journal of Physiology. 1996;495.P:152–153P. [Google Scholar]
- Mattson MP. Apoptosis in neurodegenerative disorders. Nature Reviews Molecular Cellular Biology. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Experimental Neurology. 1998a;153:35–48. doi: 10.1006/exnr.1998.6863. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Partin J, Begley JG. Amyloid β-peptide induces apoptosis-related events in synapses and dendrites. Brain Research. 1998b;807:167–176. doi: 10.1016/s0006-8993(98)00763-x. [DOI] [PubMed] [Google Scholar]
- Mikucki SA, Oblinger MM. Corticospinal neurons exhibit a novel pattern of cytoskeletal gene expression after injury. Journal of Neuroscience Research. 1991;30:213–225. doi: 10.1002/jnr.490300122. [DOI] [PubMed] [Google Scholar]
- Miledi R, Slater CR. Electrophysiology and electron-microscopy of rat neuromuscular junctions after nerve degeneration. Proceedings of the Royal Society B. 1968;169:289–306. doi: 10.1098/rspb.1968.0012. [DOI] [PubMed] [Google Scholar]
- Miledi R, Slater CR. On the degeneration of rat neuromuscular junctions after nerve section. Journal of Physiology. 1970;207:507–528. doi: 10.1113/jphysiol.1970.sp009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JI, Curran T. Stimulus-transcription coupling in the nervous system: Involvement of the inducible proto-oncogenes fos and jun. Annual Review of Neuroscience. 1991;14:421–451. doi: 10.1146/annurev.ne.14.030191.002225. [DOI] [PubMed] [Google Scholar]
- Mothet J-P, Fossier P, Meunier F-M, Stinnakre J, Tauc L, Baux G. Cyclic ADP-ribose and calcium-induced cacium release regulate neurotransmitter release at a cholinergic synapse of aplysia. Journal of Physiology. 1998;507:405–414. doi: 10.1111/j.1469-7793.1998.405bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls JG, Martin AR, Wallace BG. From Neuron to Brain: A Cellular and Molecular Approach to the Function of the Nervous System. 3. MA, USA: Sinauer Associates, Inc.; 1992. [Google Scholar]
- Nieke J, Schachner M. Expression of the neural cell adhesion molecules L1 and N-CAM and their common carbohydrate epitope L2/HNK-1 during development and after transection of the mouse sciatic nerve. Differentiation. 1985;30:141–151. doi: 10.1111/j.1432-0436.1985.tb00525.x. [DOI] [PubMed] [Google Scholar]
- Nijhawan D, Honarpour N, Wang X. Apoptosis in neural development and disease. Annual Review of Neuroscience. 2000;23:73–87. doi: 10.1146/annurev.neuro.23.1.73. [DOI] [PubMed] [Google Scholar]
- Parson SH, Davie N, Ribchester RR. Synapse elimination in organ cultures of Wlds mouse skeletal muscle. Journal of Physiology. 1998a;507.P:30P. doi: 10.1002/jnr.20016. [DOI] [PubMed] [Google Scholar]
- Parson SH, Dilley J, Gandhi N, Gillingwater TH, Ribchester RR. Schwann cell responses at disconnected nerve terminals in organ cultures of Wlds mouse neuromuscular junctions. Society for Neuroscience Abstracts. 1998b;24:413.17P. [Google Scholar]
- Parson SH, Mackintosh CL, Ribchester RR. Elimination of motor nerve terminals in neonatal mice expressing a gene for slow Wallerian degeneration (C57Bl/Wlds) European Journal of Neuroscience. 1997;9:1586–1592. doi: 10.1111/j.1460-9568.1997.tb01516.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER. Very slow retrograde and Wallerian degeneration in the CNS of C57Bl/Ola mice. European Journal of Neuroscience. 1990a;3:102–105. doi: 10.1111/j.1460-9568.1991.tb00815.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER, Tree P, Gordon S. Evidence that very slow Wallerian degeneration in C57Bl/Ola mice is an intrinsic property of the peripheral nerve. European Journal of Neuroscience. 1990b;2:802–808. doi: 10.1111/j.1460-9568.1990.tb00472.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Tsao JW. The effectiveness of the gene which slows the rate of Wallerian degeneration in C57Bl/Ola mice declines with age. European Journal of Neuroscience. 1992;4:1000–1002. doi: 10.1111/j.1460-9568.1992.tb00126.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Lunn ER, Brown MC, Cahusac S, Gordon S. Evidence that the rate of Wallerian degeneration is controlled by a single autosomal dominant gene. European Journal of Neuroscience. 1990c;2:408–413. doi: 10.1111/j.1460-9568.1990.tb00433.x. [DOI] [PubMed] [Google Scholar]
- Pettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- Podesta M, Zocchi E, Pitto A, Usai C, Franco L, Bruzzone S, Guida L, Bacigalupo A, Scadden DT, Walseth TF, De Flora A, Daga A. Extracellular cyclic ADP-ribose increases intracellular free calcium concentration and stimulates proliferation of human hemopoietic progenitors. FASEB Journal. 2000;14:680–690. doi: 10.1096/fasebj.14.5.680. [DOI] [PubMed] [Google Scholar]
- Raman y Cajal S. In: Degeneration and Regeneration of the Nervous System. May RM, editor. London: Oxford University Press; 1928. [Google Scholar]
- Ribchester RR. Development and plasticity of neuromuscular connections. In: Kalverboer AF, Gramsbergen A, editors. Brain and Behaviour in Human Neural Development. Kluwer Academic Press; 2001. [Google Scholar]
- Ribchester RR, Pakiam JG, O'Carroll CM, Thomson D, Mattison RJ, Costanzo EM, Gillingwater TH, Barry JA. Impaired neuromuscular transmission preceding synapse withdrawal in axotomized adult Wlds mutant mouse skeletal muscle. Journal of Physiology. 1999;520.P:76P. [Google Scholar]
- Ribchester RR, Tsao JW, Barry JA, Asgari-Jirandeh N, Perry VH, Brown MC. Persistence of neuromuscular junctions after axotomy in mice with slow Wallerian degeneration (C57Bl/Wlds) European Journal of Neuroscience. 1995;7:1641–1650. doi: 10.1111/j.1460-9568.1995.tb01159.x. [DOI] [PubMed] [Google Scholar]
- Rich MM, Coleman H, Lichtman JW. In vivo imaging shows loss of synaptic sites from neuromuscular junctions in a model of myasthenia gravis. Neurology. 1994;44:2138–2144. doi: 10.1212/wnl.44.11.2138. [DOI] [PubMed] [Google Scholar]
- Rich MM, Lichtman JW. Motor nerve terminal loss from degenerating muscle fibres. Neuron. 1989;3:677–688. doi: 10.1016/0896-6273(89)90236-5. [DOI] [PubMed] [Google Scholar]
- Riley DA. Spontaneous elimination of nerve terminals from endplates of developing skeletal muscle fibres. Brain Research. 1977;134:279–285. doi: 10.1016/0006-8993(77)91073-3. [DOI] [PubMed] [Google Scholar]
- Riley DA. Ultrastructural evidence for axon retraction during the spontaneous elimination of polyneuronal innervation of the rat soleus muscle. Journal of Neurocytology. 1981;10:425–440. doi: 10.1007/BF01262414. [DOI] [PubMed] [Google Scholar]
- Romanes GJ. The development and significance of the cell columns in the ventral horn of the cervical and upper thoracic spinal cord of the rabbit. Journal of Anatomy. 1941;76:112–130. [PMC free article] [PubMed] [Google Scholar]
- Rosenthal JL, Taraskevich PS. Reduction of multiaxonal innervation at the neuromuscular junction of the rat during develoment. Journal of Physiology. 1977;270:299–310. doi: 10.1113/jphysiol.1977.sp011953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski JL, Tuite GF, Lincoln PM, Boyer PJ, Tennekoon GI, Kunkel SL. Signals for proinflammatory cytokine secretion by human Schwann cells. Journal of Neuroimmunology. 1999;101:47–60. doi: 10.1016/s0165-5728(99)00132-0. [DOI] [PubMed] [Google Scholar]
- Sagot Y, Dubois-Dauphin M, Tan SA, De Bilbao F, Aebischer P, Martinou JC, Kato AC. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. Journal of Neuroscience. 1995;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annual Review of Neuroscience. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Schafer M, Fruttiger M, Montag D, Schachner M, Martini R. Disruption of the gene for the myelin-associated glycoprotein improves axonal regrowth along myelin in C57Bl/Wlds mice. Neuron. 1996;16:1107–1113. doi: 10.1016/s0896-6273(00)80137-3. [DOI] [PubMed] [Google Scholar]
- Schlaepfer WW. Calcium-induced degeneration of axoplasm in isolated segments of rat peripheral nerve. Brain Research. 1974;69:203–215. doi: 10.1016/0006-8993(74)90002-x. [DOI] [PubMed] [Google Scholar]
- Shi B, Stanfield BB. Differential sprouting responses in axonal fiber systems in the dentate gyrus following lesions of the perforant path in Wlds mutant mice. Brain Research. 1996;740:89–101. doi: 10.1016/s0006-8993(96)00849-9. [DOI] [PubMed] [Google Scholar]
- Smalheiser NR, Crain SM. The possible role of ‘sibling neurite bias’ in the coordination of neurite extension, branching, and survival. Journal of Neurobiology. 1984;15:517–29. doi: 10.1002/neu.480150609. [DOI] [PubMed] [Google Scholar]
- Sommer C, Schafers M. Painful mononeuropathy in C57Bl/Wld mice with delayed Wallerian degeneration: Differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Research. 1998;784:154–162. doi: 10.1016/s0006-8993(97)01327-9. [DOI] [PubMed] [Google Scholar]
- Son Y-J, Thompson WJ. Schwann cell processes guide regeneration of peripheral axons. Neuron. 1995a;14:125–132. doi: 10.1016/0896-6273(95)90246-5. [DOI] [PubMed] [Google Scholar]
- Son Y-J, Thompson WJ. Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron. 1995b;14:133–141. doi: 10.1016/0896-6273(95)90247-3. [DOI] [PubMed] [Google Scholar]
- Subang MC, Bisby MA, Richardson PM. Delay of CNTF decrease following peripheral nerve injury in C57Bl/Wld mice. Journal of Neuroscience Research. 1997;49:563–568. doi: 10.1002/(SICI)1097-4547(19970901)49:5<563::AID-JNR6>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Tetzlaff W, Alexander SW, Miller FD, Bisby MA. Response of facial and rubrospinal neurons to axotomy: Changes in mRNA expression for cytoskeletal proteins and GAP-43. Journal of Neuroscience. 1991;11:2528–2544. doi: 10.1523/JNEUROSCI.11-08-02528.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson W, Jansen JK. The extent of sprouting of remaining motor units in partly denervated immature and adult rat soleus muscle. Neuroscience. 1977;2:523–535. doi: 10.1016/0306-4522(77)90049-5. [DOI] [PubMed] [Google Scholar]
- Tsao JW, Brown MC, Carden MJ, McLean WG, Perry VH. Loss of the compound action potential: An electrophysiological, biochemical and morphological study of early events in axonal degeneration in the C57Bl/Ola mouse. European Journal of Neuroscience. 1994;6:516–524. doi: 10.1111/j.1460-9568.1994.tb00295.x. [DOI] [PubMed] [Google Scholar]
- Tsao JW, Paramananthan N, Parkes HG, Dunn JF. Altered brain metabolism in the C57Bl/Wld mouse strain detected by magnetic resonance spectroscopy: association with delayed Wallerian degeneration? Journal of the Neurological Sciences. 1999;168:1–12. doi: 10.1016/s0022-510x(99)00161-6. [DOI] [PubMed] [Google Scholar]
- Vial JD. The early changes in the axoplasm during Wallerian degeneration. Journal of Biophysical and Biochemical Cytology. 1958;4:551–555. doi: 10.1083/jcb.4.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa PG, Henzel WJ, Sensenbrenner M, Henderson CE, Pettmann B. Calpain inhibitors, but not caspase inhibitors, prevent actin proteolysis and DNA fragmentation during apoptosis. Journal of Cell Science. 1998;111:713–722. doi: 10.1242/jcs.111.6.713. [DOI] [PubMed] [Google Scholar]
- Waller A. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philisophical Transactions of the Royal Society of London. 1850;140:423–429. [Google Scholar]
- Watson DF, Glass JD, Griffin JW. Redistribution of cytoskeletal proteins in mammalian axons disconnected from their cell bodies. Journal of Neuroscience. 1993;13:4354–4360. doi: 10.1523/JNEUROSCI.13-10-04354.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TA, Johnson S, Walseth TF, Lee HC, Graeff RM, Munshi CB, Prakesh YS, Sieck GC, Kannan MS. Subcellular localization of cyclic ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities in porcine airway smooth muscle. Biochimica et Biophysica Acta. 2000;1498:64–71. doi: 10.1016/s0167-4889(00)00077-x. [DOI] [PubMed] [Google Scholar]
- Winlow W, Usherwood PNR. Ultrastructural studies of normal and degenerating mouse neuromuscular junctions. Journal of Neurocytology. 1975;4:377–394. doi: 10.1007/BF01261371. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yanker BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Ziegler M. New functions of a long-known molecule - Emerging roles of NAD in cellular signalling. European Journal of Biochemistry. 2000;267:1550–1564. doi: 10.1046/j.1432-1327.2000.01187.x. [DOI] [PubMed] [Google Scholar]