

Abstract
The current view holds that chronic autoimmune diseases are driven by the continuous activation of autoreactive B and T lymphocytes. However, despite the use of potent immunosuppressive drugs designed to interfere with this activation the production of autoantibodies often persists and contributes to progression of the immunopathology. In the present study, we analyzed the life span of (auto)antibody-secreting cells in the spleens of NZB × NZW F1 (NZB/W) mice, a murine model of systemic lupus erythematosus. The number of splenic ASCs increased in mice aged 1–5 mo and became stable thereafter. Less than 60% of the splenic (auto)antibody-secreting cells were short-lived plasmablasts, whereas 40% were nondividing, long-lived plasma cells with a half-life of >6 mo. In NZB/W mice and D42 Ig heavy chain knock-in mice, a fraction of DNA-specific plasma cells were also long-lived. Although antiproliferative immunosuppressive therapy depleted short-lived plasmablasts, long-lived plasma cells survived and continued to produce (auto)antibodies. Thus, long-lived, autoreactive plasma cells are a relevant target for researchers aiming to develop curative therapies for autoimmune diseases.
Keywords: plasma cell, autoimmunity, SLE, antibodies, anti-DNA
Introduction
Autoimmune diseases are characterized by the activation of autoantigen-specific T and B lymphocytes and by their differentiation into autoreactive effector cells. Antibodies secreted by autoreactive plasmablasts and plasma cells can contribute significantly to autoimmune pathogenesis (1, 2). Antibodies binding to double-stranded DNA characterize human and murine systemic lupus erythematosus (SLE), and their titers are an excellent measure of disease activity. Some subgroups of these autoantibodies cause severe manifestations such as lupus nephritis and vascular injury. (3). It is assumed that chronic autoantibody production is driven by the continuous activation of autoreactive T and B lymphocytes (4–6). The latter cells differentiate into proliferating, autoantibody-secreting plasmablasts, which in turn differentiate into nonproliferating, short-lived plasma cells. However, suppression of T and B lymphocyte activation with immunosuppressants often does not eliminate the production of autoantibodies (7, 8). This could be due to the presence of resistant autoreactive T and B lymphocytes with strengthened survival capabilities and a lowered threshold for activation (9, 10). Alternatively, long-lived, autoreactive plasma cells might continue to secrete autoantibodies in the absence of chronic activation of autoreactive T and B lymphocytes.
It was shown recently that plasma cells can survive in the BM for extended periods of time, presumably from several months to years (11). These long-lived plasma cells do not divide and are resistant to irradiation and mitomycin C (11–13). They no longer react to antigen or antigen-antibody complexes (14). Their survival in the BM might be dependent on a complex molecular microenvironment called the “plasma cell survival niche.” IL-5, IL-6, TNF-α, SDF-1, and CD44-mediated signaling have been identified as survival factors for plasma cells (15–17). In NZB/W mice, a murine model of SLE (18), inflamed kidneys may provide additional survival niches for long-lived plasma cells (19).
Like their short-lived counterparts, long-lived plasma cells are generated in secondary lymphoid organs (14) such as the LNs and spleen, which apparently provide only few survival niches for the cells (20). Most plasma cells either die within these organs (21) or leave the organs in search of survival niches elsewhere (13). After immunization, <5% of antigen-specific plasma cells persist in the spleen compared with 95% in the BM (14, 22, 23).
In the present study, we showed that long-lived plasma cells, including autoreactive plasma cells, constitute a major population in the spleen of NZB/W mice. Since these cells are resistant to cyclophosphamide, selective elimination of autospecific long-lived plasma cells is a provocative challenge to researchers aiming to develop new autoimmune disease treatment strategies.
Materials and Methods
Mice and Immunizations.
Female NZB/W mice were bred at the animal facility of the Deutsches Rheumaforschungszentrum Berlin under defined, pathogen-free conditions. Mice aged 20–24 wk were used in the experiments. Selected mice were immunized with OVA as described previously (14). D42/NZB/W females (24) were bred in the animal facility of the Hebrew University Medical School. To assess the effect of cyclophosphamide, we fed the mice bromodeoxyuridine (BrdU) for 14–21 d before treating them with 35 mg/kg body weight (BW) cyclophosphamide i.p. at various time intervals. Single-cell suspensions of spleen were analyzed by FACS® and ELISPOT 7 d after the initial cyclophosphamide treatment.
FACS® Analysis of BrdU Incorporation.
These mice received drinking water containing BrdU (1 mg/ml; Sigma-Aldrich)/1% glucose, which was carefully protected from light and changed every 3 d. Single-cell suspensions of spleen were filtered through a 70-μm cell strainer (BD Falcon) and washed twice with PBS/0.5% BSA. The cells were first incubated with anti-CD16/CD32 (clone 2.4G2; BD Biosciences) and rat Ig (Biotrend) for 15 min on ice and then with anti-CD138–PE (clone 281–2; BD Biosciences) for 15 min. They were then washed and subsequently fixed and stained for BrdU (BrdU-Flow-Kit; BD Biosciences) according to the manufacturer's protocol. Anti-BrdU–FITC (clone 3D4; BD Biosciences), anti-D42 idiotype rabbit IgG (provided by D. Eilat, Hadassah Medical School, Jerusalem, Israel), and Cy5-conjugated OVA (Sigma-Aldrich) were used for intracellular staining. Anti–Dig-Cy5 (Roche), SA-Cy5 (Amersham Biosciences), and goat anti–rabbit IgG-Cy5 were used as secondary reagents. Cytometric analysis was performed using a FACSCalibur cytometer (BD Biosciences) with CellQuest software (BD Biosciences). Debris and RBCs were excluded by electronic gating. Absolute cell numbers were calculated based on population frequencies and total cell numbers per organ. To evaluate annexin V binding to plasma cells and plasmablasts, single-cell suspensions of spleen were incubated with annexin V (Annexin V staining kit; BD Biosciences) according to the manufacturer's protocol. The samples were then counterstained with CD138-PE and propidium iodide and run on the FACSCalibur with CellQuest software.
Quantification of Antibody-secreting Cells by ELISPOT.
Single-cell suspensions of spleen were washed and resuspended in RPMI 1640 medium supplemented with 10% FCS (Invitrogen), penicillin, streptomycin, and glutamine (complete medium). We used a modified standard ELISPOT technique as described previously (19). Briefly, ELISPOT plates were coated with methyl-BSA (1 μg/ml; Serva electrophoresis, 2 h at 37°C) and then washed with PBS/0.01% Tween 20 (Merck); they were subsequently coated with calf thymus DNA (1 μg/ml; Serva Electrophoresis) and incubated overnight at 4°C in PBS. The cells were then pipetted onto the plates and analyzed for DNA-specific antibody-secreting cells (ASCs).
Fluorescence Microscopy.
CD138-positive spleen cells were sorted by FACS® (Moflo-Sorter), placed on slides (2 × 10 5 cells/ slide), and cytospun. The cells were then fixed with ice-cold acetone and counterstained for kappa light chain (clone 187.1; BD Biosciences). Cells for anti-BrdU staining analysis were first treated with 2 N HCl for 1 h and then washed to yield a pH of 7 and incubated for 1 h with 600 μg DNase I (Sigma-Aldrich) in 1 ml buffer (1 ml in 150 mM NaCl, 5 mM MgCl2, 10 μM HCl). BrdU incorporation was visualized by staining the cells with anti-BrdU–Alexa 488 antibody (clone PRB-1; Molecular Probes) before examining them by confocal laser scanning microscopy (DM IRE2; Leica).
Results
Proliferation of ASCs in the Spleens of NZB/W Mice.
In NZB × NZW F1 mice (NZB/W) at least 4–5 mo of age with full-blown murine SLE (18), the total number of ASCs is normal in the BM but severely increased in the spleen (19), the organ where B cells are induced to form plasma cells. In the spleens of healthy mice, ASCs do not make up >0.1% of the total number of mononuclear cells even after immunization (14). The vast majority of these ASCs are short-lived plasmablasts; only a few are long-lived (20). The frequency of splenic plasma cells increases about threefold (to 1–2%) in NZB/W mice between the ages of 1–5 mo and before the development of proteinuria. The numbers of ASCs in NZB/W mice 5 mo of age are ∼20-fold higher than in healthy mice (19). Thereafter, the frequencies and absolute numbers of splenic plasma cells remain stable (Fig. 1 A).
Figure 1.
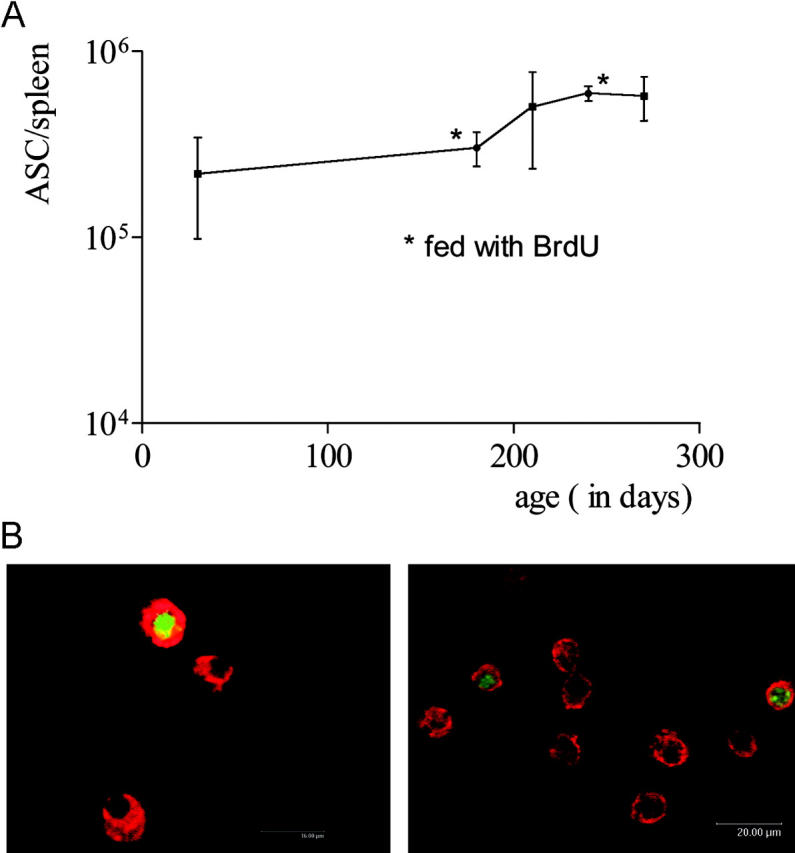
Kinetics and incorporation of BrdU in the splenic plasma cell compartment of NZB/W mice. (A) Total number of ASCs in spleen, as determined by ELISPOT, at the times indicated. Some groups were fed BrdU continuously (B). Splenic CD138-positive cells consist of dividing and nondividing cells. NZB/W mice with confirmed SLE were continuously fed BrdU for 2 wk. CD138-positive cells were then FACS® sorted and stained for intracellular immunoglobulin (red) and BrdU (green/yellow); original magnification, 160 × (left) and 80 × (right). Plasma cells negative for BrdU had not undergone DNA synthesis or replication during the period of BrdU treatment. Dividing plasmablasts and plasma cells recently derived from proliferating precursors are BrdU positive.
Here, plasma cell turnover in the spleens of NZB/W mice was determined based on BrdU incorporation. Mice 4–5 mo of age were continuously fed BrdU for variable periods of up to 12 wk. Consequently, cells that replicated their DNA during this period were labeled and could be identified using antibodies recognizing DNA-containing BrdU (25). Potentially, long-term feeding with BrdU could have mutagenic effects that might influence the formation or persistence of plasma cells. To exclude this possibility, the absolute numbers and frequencies of splenic ASCs were determined by both ELISPOT and FACS® (Fig. 1 A and Fig. 2 C). There were no detectable differences between mice fed BrdU for up to 12 wk and age-matched controls that did not receive the drug. Particularly spleen size and absolute numbers of ASCs (Fig. 1 A) were not altered by feeding with BrdU. This suggests that in these experiments BrdU did not significantly modulate the induction or persistence of plasma cells.
Figure 2.
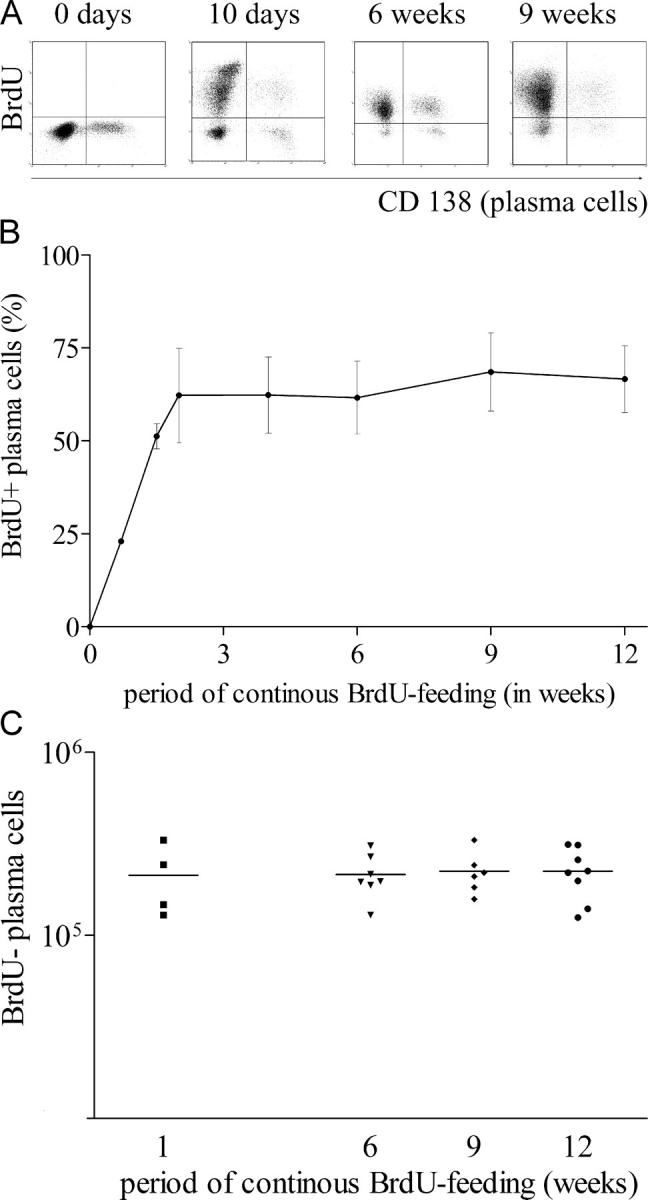
The splenic plasma cell compartment consists of continuously dividing cells and a stable population of nondividing cells. NZB/W mice with confirmed SLE were continuously fed BrdU. Splenic plasma cells (CD138-positive) were enumerated by flow cytometry at various sampling times. (A) FACS® analysis. Plasma cells were detected by CD138 staining. Debris was excluded from the analysis by electronic gating. (B) Kinetics of BrdU incorporation in splenic plasma cells based on FACS® analysis. Mean values for five mice are shown. The results are those from one run of two performed. (C) The absolute number of BrdU-negative, nondividing plasma cells was calculated based on their frequency and the absolute number of splenic cells. Each dot resembles an individual mouse.
CD138 (syndecan-1) was used for cytometric identification of ASCs (21). More than 95% of CD138+ cells in tissue sections or isolated from spleens by FACS® expressed intracellular kappa light chains and exhibited either a lymphoblastoid or plasma cell morphology (Fig. 1 B) identifying them as plasmablasts or plasma cells. Many of these cells remained BrdU negative even after 2–9 wk of continuous BrdU feeding (Fig. 1 B). This indicates that they had not undergone DNA synthesis or replication during the treatment period.
Short-lived Plasmablasts and Long-lived Plasma Cells in the NZB/W Spleen.
The kinetics of BrdU incorporation into CD138+ antibody-secreting spleen cells was analyzed by cytometry (Fig. 2 A). About 60% of CD138+ cells became BrdU positive within 10 d of BrdU feeding (Fig. 2 B). The other 40% were not labeled and remained BrdU negative for the entire period of BrdU feeding, i.e., 12 wk. The total number of BrdU-negative CD138+ plasma cells did not change during these 3 mo (Fig. 2 C), suggesting that these cells are long-lived, nondividing plasma cells.
To exclude the possibility that dividing plasmablasts might not have been labeled due to blockage of BrdU uptake and/or incorporation into the DNA of those cells, we also assessed the efficiency of BrdU incorporation in OVA-specific plasmablasts and plasma cells that develop in the immune response to this antigen. BrdU feeding was started at the time point of immunization to allow labeling of all plasmablasts regardless whether they are precursors of short-lived or long-lived plasma cells. In accordance with our earlier observations, the majority of OVA-specific plasma cells were located in the BM several weeks (here, 6, 9 and 12 wk) after immunization. All OVA-specific plasmablasts and plasma cells in the spleen and BM of NZB/W mice became and remained BrdU positive during 6–12 wk of continuous BrdU feeding (Fig. 3). Thus, all newly formed plasma cells took up BrdU during BrdU feeding. As determined by ELISOT, the frequencies and numbers of OVA-specific ASCs in mice fed with BrdU did not differ from those in control animals. In conclusion, the BrdU-negative plasma cells and their precursors observed in the spleens of NZB/W mice fed BrdU did not synthesize DNA during the period of BrdU feeding (Fig. 2 A).
Figure 3.

BrdU efficiently labels all newly formed plasma cells. Mice were continuously fed BrdU and immunized and boosted with OVA 3–4 wk later. After fixation, intracellular staining with fluorochrome-labeled OVA was performed to identify plasma cells secreting OVA-specific antibodies. OVA-specific plasma cells in the spleen (top) and BM (bottom) were enumerated 6, 9, and 12 wk after boost immunization. During the whole period between boost immunization and analysis, mice were continuously fed with BrdU. Representative data, as shown, are obtained 6 wk after boost immunization when BrdU incorporation into OVA-specific plasma cells was already complete (n = 5). Similar results were obtained 9 and 12 wk after immunization.
Despite the presence of large numbers of BrdU-positive, dividing plasmablasts, the numbers of nondividing, BrdU-negative plasma cells in the spleens of NZB/W mice did not significantly increase over time, i.e., they were not significantly replaced by newly generated plasma cells. Therefore, we investigated the possibility that the newly generated plasmablasts might be short-lived themselves or that they might differentiate into short-lived plasma cells. MHCII is expressed on early ASCs but is absent on mature plasma cells (14). Expression of this marker correlates well with the expansion phase of the ASC population and could therefore be used as a surrogate marker to distinguish proliferating plasmablasts from noncycling plasma cells (14). This is in accordance with the observation that all long-lived BrdU-negative CD138-positive plasma cells expressed little MHCII (Fig. 4). Roughly half of the BrdU-positive, CD138-positive cells expressed high levels of this molecule; the other half expressed low levels. This finding suggests that during the period of BrdU feeding, a fraction of the dividing plasmablasts differentiated into more mature plasma cells, expressing little MHCII. Compared with CD138-negative splenic cells and CD138-positive/MHCII-low plasma cells, binding of annexin V, a marker for early apoptosis, was increased on CD138-positive/MHCII-positive early plasma cells (Fig. 4).
Figure 4.

Increased binding of early MHCII-positive plasma cells to annexin V. Fixed cells from mice fed with BrdU for 2 wk were stained for CD138, BrdU, and MHCII. Result is shown on cells gated for CD138 expression (top). Frequencies of BrdU-positive, MHCII-negative CD138-positive cells were 32.5 ± 9% (n = 5). Living early plasma cells were distinguished from mature plasma cells by MHCII expression (below) and were stained for annexin V. Dead cells and debris were excluded according to propidium iodide staining and forward scatter profile. Histogram plots were additionally gated on CD138-negative, CD138-positive/MHCII-negative or CD138-positive/MHCII-positive cells.
Collectively, these results show that the splenic ASC population in NZB/W mice with an established murine SLE consists of roughly 60% dividing plasmablasts, at least a fraction of which are short-lived cells that die before differentiating into mature MHCII-low plasma cells. The remaining 40% of splenic ASC are nondividing, long-lived MHCII-low plasma cells. They constitute a stable population of ∼105 cells, which is over 10 times higher than the total ASC population in the spleens of nonautoimmune CB20 and BALB/c mice (19).
Unlike their short-lived counterparts, long-lived plasma cells are refractory to cyclophosphamide. Long-lived lymphocytes are considered to be resistant to immunosuppressive treatment (26). Our finding that roughly half of the splenic ASCs in NZB/W mice are long-lived plasma cells made us wonder whether these cells are responsible for treatment-resistant antibody titers and autoantibody production. To clarify these points, we analyzed the plasma cell compartment of NZB/W mice treated experimentally with cyclophosphamide, a drug also used to treat severe SLE in humans (Fig. 5). Three doses of 35 mg/kg BW, higher than the amount normally used for high dose human therapy, deleted short-lived BrdU-positive plasmablasts nearly completely while affecting long-lived BrdU-negative plasma cells to a much lesser extent. Higher doses (up to 140 mg/kg) depleted larger numbers of long-lived plasma cells, but a fraction of these cells survived even under these conditions. These results show that cyclophosphamide therapy prevents ongoing immune activation and formation of short-lived plasmablasts but does not delete long-lived plasma cells efficiently.
Figure 5.

Long-lived plasma cells in NZB/W mice are resistant to conventional immunotherapy. Mice were fed BrdU for 2–4 wk and received the total cyclophosphamide dose indicated (divided over 3 d) 1 wk before sampling. BrdU-positive (short-lived) and -negative (long-lived) plasma cells were identified by FACS®. Representative data for 25 untreated and 12 treated mice are shown.
Short-lived and Long-lived Plasma Cell Populations Contain Anti-DNA Cells.
To determine whether ASCs were present in the short- or long-lived plasma cell compartments, we took advantage of the preferential deletion of short-lived cells by cyclophosphamide and used ELISPOT to determine whether anti-DNA ASCs were among the remaining cells. Although BrdU-positive short-lived plasmablasts were eliminated by high doses (three to four times 35 mg/kg) of cyclophosphamide (Fig. 5), an average of ∼20% of the total IgG/IgM anti-DNA ASC survived this extreme treatment (Table I). Although these findings suggest that a fraction of the autoreactive cells is contained within the therapy-resistant long-lived plasma cell compartment, it could not formally be excluded that the remaining BrdU-positive cells are those secreting anti-DNA antibodies. To further investigate this point, we analyzed the plasma cell compartment in knock-in NZB/W mice transgenic for the anti-DNA D42 heavy chain. With this autoimmunogenic background, the D42 heavy chain transgene combines with the appropriate light chain to produce high affinity anti-DNA IgM and IgG autoantibodies (24). With this model, plasma cells producing DNA-binding antibodies can also be monitored by staining intracellular immunoglobulins with an VH11-specific rabbit anti-idiotypic antibody. On the NZB/W background, ∼90% of cells detected with that reagent bind to DNA with high affinity (24). Based on BrdU incorporation, the frequencies of short-lived plasmablasts and long-lived plasma cells in the spleens of D42H transgenic NZB/W mice were very similar to those observed in nontransgenic NZB/W mice (Fig. 6). About 20% of the D42 transgene–expressing plasma cells were contained within the BrdU-negative long-lived plasma cell compartment after 12 wk of BrdU feeding. This is a direct indication that autoreactive cells can enter the compartment of long-lived plasma cells. These results show that a considerable proportion of the persistent autoantibody production in lupus can be mediated by long-lived plasma cells.
Table I.
Number of Splenic ASCs in Cyclophosphamide-treated NZB/W Mice
| Samples | CY | Number of IgG−
ASC/spleen |
Number of IgM−
ASC/spleen |
Number of DNA-IgG−
ASC/spleen |
Number of DNA-IgM−
ASC/spleen |
|---|---|---|---|---|---|
| mg/kg | |||||
| 1 | 4 × 35 | 414 ± 76 | 18,675 ± 3,297 | 5 ± 0 | 130 ± 23 |
| 2 | 4 × 35 | 4,680 ± 1,464 | 24,150 ± 687 | 94 ± 19 | 496 ± 120 |
| 3 | 3 × 35 | 616 ± 140 | 20,538 ± 6,865 | 49 ± 6 | 344 ± 40 |
| 4 | 3 × 35 | 2,093 ± 217 | 23,625 ± 11,137 | 58 ± 6 | 351 ± 38 |
| 5 | 3 × 35 | 1,485 ± 63 | 9,788 ± 997 | 72 ± 12 | 432 ± 114 |
| 6 | — | 1,3050 ± 3,307 | 45,000 ± 3,182 | 155 ± 23 | 783 ± 90 |
| 7 | — | 68,625 ± 14,319 | 69,750 ± 6,364 | 1,141 ± 108 | 2,653 ± 259 |
| 8 | — | 81,000 ± 6,364 | 73,125 ± 4,773 | 1,782 ± 187 | 2,025 ± 147 |
| 9 | — | 10,575 ± 7,319 | 20,250 ± 0 | 1,215 ± 343 | 1,823 ± 192 |
Animals 4–5 mo of age with confirmed SLE received the cyclophosphamide (CY) dose indicated. The spleens of the mice were analyzed by ELISPOT 7 days after initiation of treatment.
Figure 6.

Autoreactive cells can enter the long-lived plasma cell compartment. D42 knock-in mice with an NZB/W genetic background were fed BrdU for 12 wk. The spleens of the mice were then analyzed by FACS® for the presence of BrdU-negative plasma cells (left). D42 transgene-bearing plasma cells were detected by CD138 and intracellular staining with an VH11-specific anti-idiotypic antibody. On the NZB/W background, ∼90% of cells detected with that reagent bind to DNA with high affinity (24) (middle). Gating on D42-positive/CD138-positive plasma cells as shown allowed the identification of BrdU-positive and BrdU-negative plasma cells expressing the D42 transgene (right). Results are representative data from four mice.
Discussion
Autoantibodies can contribute significantly to the immunopathology of autoimmune diseases. There is impressive evidence for this, e.g., the spontaneous development of lupus-like disease in MRL/lpr mice. In this mouse strain, disease-related mortality is reduced by ∼50% by rendering the mice deficient in the production of serum antibodies (27). During flares of autoimmune diseases like SLE, serum titers of pathogenic autoantibodies, e.g., those directed against dsDNA, nucleosomes, or SmD1, increase dramatically (3, 28, 29). In patients with sustained disease activity, e.g., in those who are consequently included in experimental therapeutic approaches like autologous stem cell transplantation, these antibodies can persist despite aggressive cyclophosphamide therapy (30, 31). Furthermore, antibodies to several other autoantigens observed in SLE and other autoimmune disorders are known to remain stable after immunosuppressive therapy (7, 8, 32). Traditionally, the persistent secretion of autoantibodies and of serum antibodies in general is considered to be the result of the continuous activation of specific B lymphocytes and their differentiation into short-lived plasma cells (33). However, it recently became clear that long-lived plasma cells residing in the BM can maintain the humoral memory provided by secreted serum antibodies (11, 12, 13, 14). Since these plasma cells do not divide, they are neither sensitive to irradiation nor to cell division inhibitors. The murine model of SLE studied here shows that such long-lived plasma cells can be part of an autoimmune response; hence, they provide a provocative target for novel immunosuppressive therapies.
An earlier study suggested that the production of autoantibodies can be mediated by two independent mechanisms which have not been characterized in detail (34). Here, we showed that murine SLE autoantibodies are secreted by short-lived plasmablasts and long-lived plasma cells. Equal numbers of both types of ASCs coexist in the spleens of NZB/W mice with manifest disease. High doses of cyclophosphamide (three to four times 35 mg/kg BW) completely suppressed the formation of short-lived plasmablasts but were less effective in suppressing the long-lived plasma cells. The numerical reduction in long-lived plasma cells may well be an indirect effect, since the overall size of the spleen is reduced at these concentrations. This could indicate the destruction of survival niches for long-lived plasma cells by the treatment. We and other investigators have shown that survival of long-lived plasma cells is dependent on a supportive environment, i.e., specific survival niches, which can be found in BM, inflamed tissue, and to a limited extent in the normal spleen (13, 16, 20, 35). It is remarkable that the spleens of NZB/W mice provide such niches for ∼10 times more long-lived plasma cells than those of normal mice. A complete molecular definition of plasma cell survival niches is still lacking (15, 16). The present work makes it clear that a significant increase in the number of such niches occurs in NZB/W mice before the age of 5 mo. Our finding that long-lived plasma cells in NZB/W spleens secrete mostly IgM (Table I and unpublished data), an isotype dominating the early autoantibody response, supports the idea that the cells in question had been generated in an initial flare of the disease. Between the ages of 5 and 8 mo, the number of long-lived plasma cells remained constant. Annexin V staining showed that a fraction of the splenic plasmablasts were apoptotic (Fig. 4), suggesting that these cells are short-lived. The stable numbers of splenic long-lived plasma cells enumerated during the observation period indicate that these short-lived plasmablasts do not contribute significantly to the long-lived splenic plasma cell compartment. Whether some of these cells leave the spleen and differentiate into long-lived plasma cells in other organs remains unclear.
It should be noted that the number of plasma cells in the BM of diseased NZB/W mice was not significantly higher than in normal mice (19). Less than 4 × 103 DNA-specific plasma cells were detected in the BM of 7-mo-old NZB/W mice (unpublished data). This is low compared with the number of NZB/W plasma cells generated in an antigenic response against a potent immunogen. 50 d after secondary immunization, the BM of NZB/W mice immunized with OVA contains ∼2 × 105 OVA-specific plasma cells. Roughly 2 × 104 OVA-specific plasma cells could be detected in the spleens of these mice at that time (unpublished data).
The spleen normally is not a major site of persistent antibody secretion (23). The dramatic pathologic increase in plasma cells in the NZB/W spleen might be due to expression of the inflammatory chemokine IP10 (CXCL10) in that organ (36). IP10 attracts early plasmablasts that express CXCR3 and is presumably a major physiological factor in the relocation of plasmablasts to inflamed tissue (18, 35). It remains unclear why and how plasmablasts generated in T cell–dependent immune responses to the antigen OVA leave the spleen and home in on the BM since they also are attracted by ligands for CXCR3 (36). One could speculate that plasmablasts of T cell–dependent responses against nonself antigens such as OVA and of autoimmune responses are generated in distinct areas of the spleen, e.g., in germinal centers versus extrafollicular areas. For MRL/lpr mice, it has been demonstrated conclusively that splenic autoreactive B cells can proliferate, undergo somatic hypermutation and differentiate into plasmablasts at the T zone red pulp border (37). In such a scenario, local inflammatory chemokine production might retain site-specific autoreactive plasmablasts. On the other hand, these sites do not appear to support the long-term survival of the plasmablasts in light of their short half-life. This scenario must be different in the early stages of the disease, when large numbers of survival niches for plasma cells apparently are generated in the spleen, giving autoreactive plasmablasts a chance to form a prominent population of long-lived splenic plasma cells. Based on the kinetics of BrdU incorporation, their half-life is estimated to be in the range of at least 6–9 mo. This is roughly equivalent to the half-life of long-lived plasma cells formed in a protective immune response to lymphocytic choriomeningitis virus (12). It should be noted that these estimates represent minimum values. Long-lived plasma cells in humans could well survive for a half-life of several years or decades as long as they are maintained in a functional survival niche. The present results suggest that reversion of the inflammatory pathophysiology of the spleen in lupus-prone NZB/W mice may be one way to target autoreactive long-lived plasma cells residing primarily in the spleen as opposed to protective long-lived plasma cells residing in the BM. In any case, the elimination of autospecific, long-lived plasma cells is a challenge to researchers developing novel immunosuppressive treatment strategies.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 421-C4 and MA 2273). B.F. Hoyer received a student grant from the Faculty of Medicine, Charité University Hospital.
F. Hiepe and R.A. Manz are senior authors and contributed equally to this work.
Abbreviations used in this paper: ASC, antibody-secreting cell; BrdU, bromodeoxyuridine; BW, body weight; SLE, systemic lupus erythematosus.
References
- 1.Tan, E.M. 1991. Autoantibodies in pathology and cell biology. Cell. 67:841–842. [DOI] [PubMed] [Google Scholar]
- 2.Ji, H., A.S. Korganow, S. Mangialaio, P. Hoglund, I. Andre, F. Luhder, A. Gonzalez, L. Poirot, C. Benoist, and D. Mathis. 1999. Different modes of pathogenesis in T-cell-dependent autoimmunity: clues from two TCR transgenic systems. Immunol. Rev. 169:139–146. [DOI] [PubMed] [Google Scholar]
- 3.Hahn, B.H. 1998. Antibodies to DNA. N. Engl. J. Med. 338:1359–1368. [DOI] [PubMed] [Google Scholar]
- 4.Lipsky, P.E. 2001. Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nat. Immunol. 2:764–766. [DOI] [PubMed] [Google Scholar]
- 5.Duke-Cohan, J.S., A. Rubinow, R. Hirt, and D. Naor. 1990. The reaction against autologous lymphoblasts as an indicator of lymphocyte hyperreactivity in rheumatoid arthritis. Clin. Immunol. Immunopathol. 54:298–308. [DOI] [PubMed] [Google Scholar]
- 6.Moutsopoulos, H.M., and M.N. Manoussakis. 1989. Immunopathogenesis of Sjogren's syndrome: facts and fancy. Autoimmunity. 5:17–24. [DOI] [PubMed] [Google Scholar]
- 7.Wahren, M., P. Tengner, I. Gunnarsson, I. Lundberg, E. Hedfors, N.R. Ringertz, and I. Pettersson. 1998. Ro/SS-A and La/SS-B antibody level variation in patients with Sjogren's syndrome and systemic lupus erythematosus. J. Autoimmun. 11:29–38. [DOI] [PubMed] [Google Scholar]
- 8.De Block, C.E., I.H. De Leeuw, R.P. Rooman, F. Winnock, M.V. Du Caju, and L.F. Van Gaal. 2000. Gastric parietal cell antibodies are associated with glutamic acid decarboxylase-65 antibodies and the HLA DQA1*0501-DQB1*0301 haplotype in type 1 diabetes mellitus. Belgian Diabetes Registry. Diabet. Med. 17:618–622. [DOI] [PubMed] [Google Scholar]
- 9.Liu, Y.J., C. Barthelemy, O. de Bouteiller, C. Arpin, I. Durand, and J. Banchereau. 1995. Memory B cells from human tonsils colonize mucosal epithelium and directly present antigen to T cells by rapid up-regulation of B7-1 and B7-2. Immunity. 2:239–248. [DOI] [PubMed] [Google Scholar]
- 10.Martin, S.W., and C.C. Goodnow. 2002. Burst-enhancing role of the IgG membrane tail as a molecular determinant of memory. Nat. Immunol. 3:182–188. [DOI] [PubMed] [Google Scholar]
- 11.Manz, R.A., A. Thiel, and A. Radbruch. 1997. Lifetime of plasma cells in the bone marrow. Nature. 388:133–134. [DOI] [PubMed] [Google Scholar]
- 12.Slifka, M.K., R. Antia, J.K. Whitmire, and R. Ahmed. 1998. Humoral immunity due to long-lived plasma cells. Immunity. 8:363–372. [DOI] [PubMed] [Google Scholar]
- 13.Manz, R.A., S. Arce, G. Cassese, A.E. Hauser, F. Hiepe, and A. Radbruch. 2002. Humoral immunity and long-lived plasma cells. Curr. Opin. Immunol. 14:517–521. [DOI] [PubMed] [Google Scholar]
- 14.Manz, R.A., M. Lohning, G. Cassese, A. Thiel, and A. Radbruch. 1998. Survival of long-lived plasma cells is independent of antigen. Int. Immunol. 10:1703–1711. [DOI] [PubMed] [Google Scholar]
- 15.Cassese, G., S. Arce, A.E. Hauser, K. Lehnert, B. Moewes, M. Mostarac, G. Muehlinghaus, M. Szyska, A. Radbruch, and R.A. Manz. 2003. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J. Immunol. 171:1684–1690. [DOI] [PubMed] [Google Scholar]
- 16.Minges Wols, H.A., G.H. Underhill, G.S. Kansas, and P.L. Witte. 2002. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J. Immunol. 169:4213–4221. [DOI] [PubMed] [Google Scholar]
- 17.O'Connor, B.P., V.S. Raman, L.D. Erickson, W.J. Cook, L.K. Weaver, C. Ahonen, L.L. Lin, G.T. Mantchev, R.J. Bram, and R.J. Noelle. 2004. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 199:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Theofilopoulos, A.N., and F.J. Dixon. 1985. Murine models of systemic lupus erythematosus. Adv. Immunol. 37:269–390. [DOI] [PubMed] [Google Scholar]
- 19.Cassese, G., S. Lindenau, B. de Boer, S. Arce, A. Hauser, G. Riemekasten, C. Berek, F. Hiepe, V. Krenn, A. Radbruch, et al. 2001. Inflamed kidneys of NZB/W mice are a major site for the homeostasis of plasma cells. Eur. J. Immunol. 31:2726–2732. [DOI] [PubMed] [Google Scholar]
- 20.Sze, D.M., K.M. Toellner, C. Garcia de Vinuesa, D.R. Taylor, and I.C. MacLennan. 2000. Intrinsic constraint on plasmablast growth and extrinsic limits of plasma cell survival. J. Exp. Med. 192:813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith, K.G., T.D. Hewitson, G.J. Nossal, and D.M. Tarlinton. 1996. The phenotype and fate of the antibody-forming cells of the splenic foci. Eur. J. Immunol. 26:444–448. [DOI] [PubMed] [Google Scholar]
- 22.Slifka, M.K., M. Matloubian, and R. Ahmed. 1995. Bone marrow is a major site of long-term antibody production after acute viral infection. J. Virol. 69:1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benner, R., W. Hijmans, and J.J. Haaijman. 1981. The bone marrow: the major source of serum immunoglobulins, but still a neglected site of antibody formation. Clin. Exp. Immunol. 46:1–8. [PMC free article] [PubMed] [Google Scholar]
- 24.Friedmann, D., N. Yachimovich, G. Mostoslavsky, Y. Pewzner-Jung, A. Ben-Yehuda, K. Rajewsky, and D. Eilat. 1999. Production of high affinity autoantibodies in autoimmune New Zealand Black/New Zealand white F1 mice targeted with an anti-DNA heavy chain. J. Immunol. 162:4406–4416. [PubMed] [Google Scholar]
- 25.Schittek, B., K. Rajewsky, and I. Forster. 1991. Dividing cells in bone marrow and spleen incorporate bromodeoxyuridine with high efficiency. Eur. J. Immunol. 21:235–238. [DOI] [PubMed] [Google Scholar]
- 26.Miller, J.J., III, and L.J. Cole. 1967. Resistance of long-lived lymphocytes and plasma cells in rat lymph nodes to treatment with prednisone, cyclophosphamide, 6-mercaptopurine, and actinomycin D. J. Exp. Med. 126:109–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan, O.T., L.G. Hannum, A.M. Haberman, M.P. Madaio, and M.J. Shlomchik. 1999. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J. Exp. Med. 189:1639–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruns, A., S. Blass, G. Hausdorf, G.R. Burmester, and F. Hiepe. 2000. Nucleosomes are major T and B cell autoantigens in systemic lupus erythematosus. Arthritis Rheum. 43:2307–2315. [DOI] [PubMed] [Google Scholar]
- 29.Riemekasten, G., J. Marell, G. Trebeljahr, R. Klein, G. Hausdorf, T. Haupl, J. Schneider-Mergener, G.R. Burmester, and F. Hiepe. 1998. A novel epitope on the C-terminus of SmD1 is recognized by the majority of sera from patients with systemic lupus erythematosus. J. Clin. Invest. 102:754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosen, O., A. Thiel, G. Massenkeil, F. Hiepe, T. Haupl, H. Radtke, G.R. Burmester, E. Gromnica-Ihle, A. Radbruch, and R. Arnold. 2000. Autologous stem-cell transplantation in refractory autoimmune diseases after in vivo immunoablation and ex vivo depletion of mononuclear cells. Arthritis Res. 2:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Traynor, A.E., J. Schroeder, R.M. Rosa, D. Cheng, J. Stefka, S. Mujais, S. Baker, and R.K. Burt. 2000. Treatment of severe systemic lupus erythematosus with high-dose chemotherapy and haemopoietic stem-cell transplantation: a phase I study. Lancet. 356:701–707. [DOI] [PubMed] [Google Scholar]
- 32.Decochez, K., J. Tits, J.L. Coolens, L. Van Gaal, G. Krzentowski, F. Winnock, E. Anckaert, I. Weets, D.G. Pipeleers, and F.K. Gorus. 2000. High frequency of persisting or increasing islet-specific autoantibody levels after diagnosis of type 1 diabetes presenting before 40 years of age. The Belgian Diabetes Registry. Diabetes Care. 23:838–844. [DOI] [PubMed] [Google Scholar]
- 33.Ahmed, R., and D. Gray. 1996. Immunological memory and protective immunity: understanding their relation. Science. 272:54–60. [DOI] [PubMed] [Google Scholar]
- 34.Richards, H.B., M. Satoh, M. Shaw, C. Libert, V. Poli, and W.H. Reeves. 1998. Interleukin 6 dependence of anti-DNA antibody production: evidence for two pathways of autoantibody formation in pristane-induced lupus. J. Exp. Med. 188:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manz, R.A., and A. Radbruch. 2002. Plasma cells for a lifetime? Eur. J. Immunol. 32:923–927. [DOI] [PubMed] [Google Scholar]
- 36.Hauser, A.E., G.F. Debes, S. Arce, G. Cassese, A. Hamann, A. Radbruch, and R.A. Manz. 2002. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J. Immunol. 169:1277–1282. [DOI] [PubMed] [Google Scholar]
- 37.William, J., C. Euler, S. Christensen, and M.J. Shlomchik. 2002. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 297:2066–2070. [DOI] [PubMed] [Google Scholar]