

Abstract
Wild-type (WT) and targeted-mutant mice incapable of making αβ T cells, γδ T cells, class I major histocompatibility complex (MHC), class II MHC, interferon (IFN)-γ, or inducible nitric oxide synthase (NOS2), were infected with Mycobacterium tuberculosis (Mtb) by aerosol, and monitored over time for their ability to (a) control infection, (b) develop histopathology at sites of infection, and (c) survive. WT mice acquired the ability to control and to hold infection at a stationary level from day 20 on. This was associated with the development of a macrophage-dominated alveolitis at sites of infection, with increased synthesis of IFN-γ and NOS2 mRNA, and with an median survival time (MST) of 258.5 d. In the absence of αβ T cells, Mtb grew progressively and rapidly to induce a necrotic, neutrophil-dominated lung pathology that killed mice with an MST of 48 d. In the absence of CD4-mediated immunity (class II−/− mice), progressive bacterial growth continued in the lungs and in other organs beyond day 20, resulting in an MST of 77 d. By contrast, in the absence of CD8 T cell–mediated immunity, lung infection was controlled at a 1 log higher stationary level that induced a similar histopathologic response to that of WT mice, and resulted in an MST of 232 d.
Keywords: αβ, CD8, CD4, interferon γ, nitric oxide synthase
Introduction
The rational design of an improved antituberculosis vaccine will be based, among other things, on a knowledge of the primary immune response that serves to protect most humans from the disease, and an understanding of why this immunity is not generated or expressed efficiently in the small percentage of humans who are susceptible. According to recent review articles 1 2 3, antituberculosis immunity is mediated by antigen-specific CD4 Th1 and CD8 T cells with apparent help from γδ T cells. Most of our understanding of the T cells involved in protective anti–Mycobacterium tuberculosis (Mtb) immunity is based on cause and effect evidence from studies of tuberculosis in mice in which the expression of immunity can be measured in terms of the control of infection in major organs in the absence of selected T cell subpopulations 4. Our knowledge of the T cells involved in immunity to human tuberculosis, on the other hand, is based on correlative evidence that comes from experiments designed to identify T cells that respond to appropriately presented Mtb antigens in vitro. Although there is general agreement that all of the aforementioned T cells participate in the immune response in both host species, disagreement remains about the relative importance of each in protective immunity. For example, although most evidence favors a dominant role for CD4 Th1 cell–mediated immunity, it has been argued by some that CD8 T cells are more important 5 or equally important 6 7, whereas still others claim 8 that CD8 T cells play no protective role at all. Again, in spite of numerous publications showing that Mtb-specific γδ T cells are generated in response to Mtb infections 9, and one publication showing that these T cells contribute significantly to protective immunity in mice 10, a more recent study suggests 11 that γδ T cells, like B cells 12, contribute little to protective immunity in mice. Again, although there is evidence 13 14 that class Ib (CD1)-dependent immunity might be involved in the response to Mtb infection in humans, it has recently been shown 15 that this component of immunity plays no obvious role in protective immunity in mice. This makes it unlikely that NK+CD4−CD8− αβ T cells are involved in the expression of immunity in this host species. Therefore, as far as the mouse model is concerned, it is mainly the importance of CD8 relative to CD4 T cells that needs clarification. Confusion that exists about this subject is partly a consequence of the fact that the role of these T cell subsets in anti-Mtb immunity in mice has not been investigated with a standard mouse model of infection. Instead, it has been investigated with models that have differed with regards to the mouse strain employed, the strain and number of Mtb inoculated, and route of inoculation. Moreover, in recent key studies of the role of CD8 T cells in anti-Mtb immunity 7 15, the contribution of CD4 T cells was not determined concurrently for comparison. Given the importance that now seems to be attributed to CD8 T cells in vaccine development 16 17 18 19, there is a need to determine whether these T cells are as essential to immunity as appears to be generally believed. This would require that their contribution be compared with that of CD4 T cells and other T cells in a single study that uses mice of a single strain infected via the natural route of infection with a small number of a given strain of virulent Mtb. This paper shows that when such a study is performed, CD8 T cells, in contrast to CD4 T cells, are seen not to be essential for the control of infection.
Materials and Methods
Mice.
All mice were of the C57BL/6 strain. Wild-type (WT) mice and mice with targeted disruption of the gene for IFN-γ (IFN-γ−/−), inducible nitric oxide synthase (NOS2−/−), β2-microglobulin (class I−/−), and H-2Aβ chain (class II−/−) were purchased from the Trudeau Institute Animal Breeding Facility. Mice deleted of the gene for TCR-α chain (TCR-α/β−/−) or the gene for the TCR-δ chain (TCR-γδ−/−) were purchased from The Jackson Laboratory. Mice were used in experiments when they were between 8 and 10 wk of age.
Bacteria and Infection.
Mtb, strain H37Rv (Trudeau Mycobacterial Culture collection no. 102), originally obtained from the Trudeau Mycobacterial Culture collection, was grown as a suspension culture in Proskauer and Beck medium containing 0.01% Tween 80, and harvested while in log-phase growth, as described previously 20. The culture was subjected to two 5-s bursts of ultrasound to break up clumps, and diluted in PBS containing 0.01% Tween 80 for infection via the respiratory route. This was done in an aerosol infection apparatus (Tri Instruments), and involved exposing mice to an aerosol generated by nebulizing 10 ml of a suspension of 106 bacilli/ml for 30 min. At the times indicated, mice were killed and their lungs, livers, and spleens were removed and homogenized on ice with motorized Teflon pestles in close-fitting glass tubes containing PBS-Tween. The homogenates were subjected to 10-fold serial dilution and the dilutions plated on enriched Middlebrook 7H11 agar. Colonies were counted after 2–3 wk incubation at 37°C. Survival studies were done with 10 mice per group. Significance of differences between means was determined with Student's t test, and differences between survival times determined with the Logrank test using GraphPad Prism v3 for Windows (GraphPad Software).
mAb Depletion of T Cells.
Mice were thymectomized at 3 wk of age by making a midline incision along the sternum, opening the sternum with retractors, removing the exposed thymus by suction, and closing the incision with surgical clips. The mice were infused intraperitoneally 4 and 5 wk later with 0.5 mg of monoclonal rat anti–mouse CD4 mAb (clone GK1.5), anti–mouse CD8 mAb (clone TIB 210), or with both mAbs. They were infused again with the same mAbs 2 wk later, and used in experiments after a further 2 d.
Ribonuclease Protection Assay.
The lungs of noninfected mice and mice infected by aerosol with 102 CFU of H37Rv 0, 10, 20, 30, or 50 d earlier were removed and snap frozen in liquid nitrogen. The frozen lungs were later homogenized and RNA extracted with Trizol reagent (Life Technologies). Detection of mRNA for NOS2, IL-4, IL-10, IL-13, IL-2, IL-2R-α, and IFN-γ was performed using the RiboQuant Multiprobe Ribonuclease Protection Assay System (BD PharMingen) as detailed by the manufacturer. Radiolabeling of antisense RNA probes was done using 1,250 Ci/mMol [35S]UTP (ICN Biomedicals) at a concentration of 1.5 × 106 cpm/μl. For detection of cytokine lung mRNA, 2 μl of labeled probe solution was added to 20 μg of RNA in manufacturer's hybridization buffer, heated to 90°C, and incubated overnight at 56°C. The hybridized RNA was treated with RNase for 45 min at 30°C followed by treatment with Proteinase K for 15 min at 37°C. Hybridized RNA was precipitated with ethanol and electrophoresed in a standard 5% acrylamide/8 M urea gel.
Histology and Immunocytochemistry.
Lungs were fixed in 10% neutral buffered formalin for 18 h, washed in distilled water, dehydrated in ethanol, and embedded in wax, according to standard procedures. Paraffin sections were cut on a rotary microtome, stained for acid-fast bacteria by a modified basic fuchsin stain 21, and counter stained with methylene blue or azure A and eosin. Immunocytochemistry to detect NOS2 in lung sections involved reacting deparifinized lung sections with 0.1 μg/ml of affinity-purified monospecific rabbit Ig anti–mouse NOS2 (Transduction laboratories) as the primary Ab. After washing, the sections were reacted with biotinylated goat Ig anti–rabbit Ig as the second reagent, and after washing they were incubated with avidin-coupled biotinylated horseradish peroxidase with diaminobenzidine as substrate (Vectastain ABC Kit; Novocastra Laboratories Ltd.) to produce the reaction product, according to the supplier's instructions. Photomicrographs were taken with a Microphot-Fx microscope (Nikon).
Results
Mycobacterial Growth and Host Survival in the Absence of Selected T Cell Subsets.
The ability of WT mice and mice devoid of αβ or γδ T cells to control infection initiated with ∼102 H37Rv given via the respiratory route is shown in Fig. 1. H37Rv grew in the lungs of WT mice for ∼20 d to reach ∼6.5 logs, after which infection was controlled and caused to become approximately stationary by acquired immunity. In these mice, infection was not detected in the livers and spleens until ∼day 20, and progressed very little thereafter, almost certainly because immunity had been acquired systemically. In the absence of αβ T cells, H37Rv continued to grow in all three organs beyond day 20 to reach very large numbers by day 50, at which time most animals were dead. On the other hand, the absence of γδ T cells had no effect on the growth of H37Rv in any organ during the 150-d period of the experiment.
Figure 1.
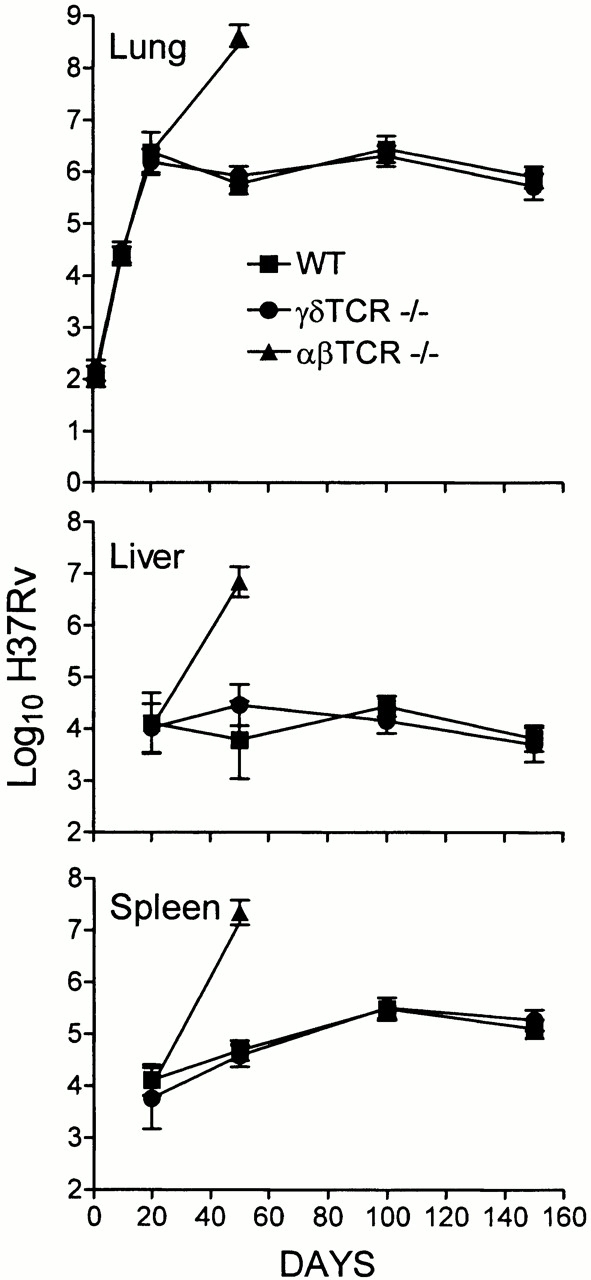
Growth of H37Rv in the lungs, livers, and spleens of WT, TCR-α/β−/−, and TCR-γ/δ−/− mice infected with 102 CFU via the respiratory route. WT and TCR-γ/δ−/− controlled infection and caused it to become stationary in all organs after day 20, whereas TCR-α/β−/− mice were unable to control infection in any organ. This is one of two experiments that gave essentially the same results. Means of five mice per time point per group ± SD.
The relative importance of CD8 versus CD4 αβ T cells in immunity is shown in Fig. 2, where it can be seen that mice incapable of generating CD4 T cell–mediated immunity (class II−/− mice) were incapable of controlling bacterial growth in their lungs and other organs after day 20, although the rate of bacterial growth was slower than in the organs of mice devoid of αβ T cells. The absence of CD8 T cell–mediated immunity (class I−/− mice) was of much less consequence, in that it resulted in Mtb growth continuing in the lungs beyond day 20 at a slower rate than in class II−/− mice. Moreover, the absence of CD8 T cells caused only a 1 log higher level of stationary lung infection than in WT mice. This was achieved sometime after day 40. Infection in the livers and spleens of class I mutant mice also persisted at about a 1 log higher level than in WT mice, thus indicating that the ability to stabilize infection was acquired in all organs.
Figure 2.
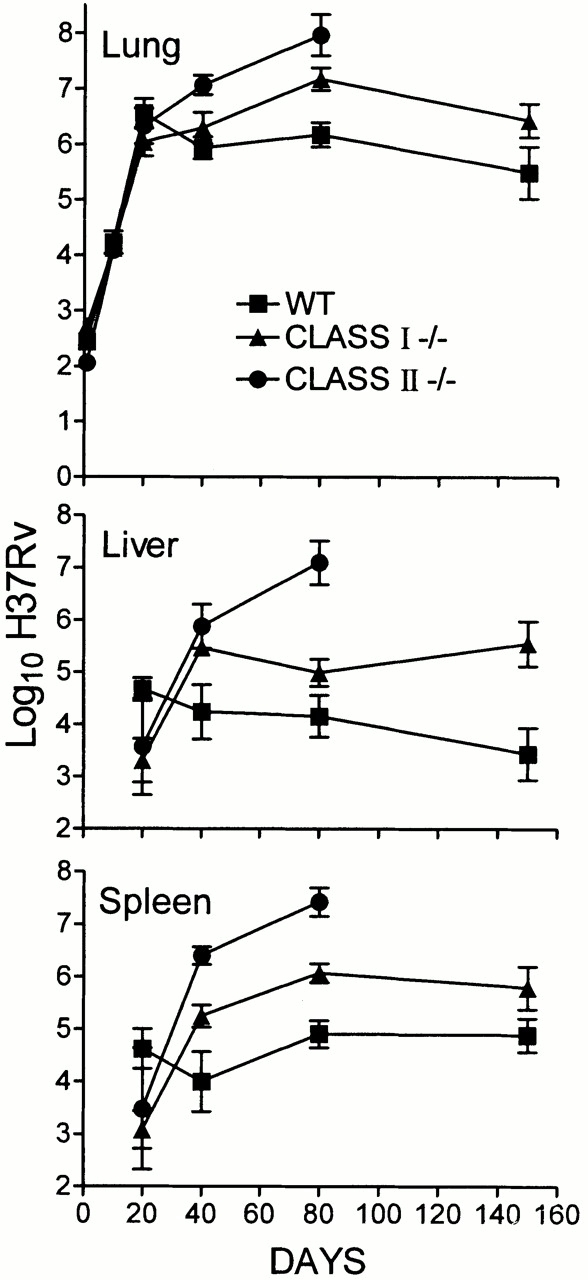
Growth of H37Rv in the lungs, livers, and spleens of WT, class I−/−, and class II−/− mice infected with 102 H37Rv via the respiratory route. Infection in class I−/− mice was stabilized in the lungs and other organs at a 1 log higher level than in WT mice. Class II−/− mice were unable to control infection in any organ. This is one of two experiments that gave essentially the same results. Means of five mice per time point per group ± SD.
The effect on host survival of the absence of different T cell subpopulations is shown in Fig. 3 and Fig. 4. Fig. 3 also shows the survival times of mice incapable of generating IFN-γ or NOS2. These mice were included in the study because it is known that in order for immunity to be expressed, NOS2-dependent NO and reactive nitrogen intermediates (RNIs) need to be generated by macrophages 22 23, and it is believed that this is dependent on IFN-γ secreted by Mtb-specific T cells 24 25. Therefore, the consequence of not being able to synthesize either molecule should be similar to that of not being able to generate αβ T cell–mediated immunity. It can be seen in Fig. 3 and Fig. 4 that, whereas WT mice died with a median survival time (MST) of 258.5 and 313.5 d, respectively, the MSTs of IFN-γ−/−, NOS2−/−, TCR-α/β−/−, class II−/−, and class I−/− mice were 32, 39, 48, 77, and 232.6 d, respectively. The differences in MST between TCR-α/β−/− and IFN-γ−/− mice, and between IFN-γ−/− and NOS2 mice, were statistically significant (P = 0.0039 and 0.0001, respectively) by Logrank test. Mice devoid of γδ T cells died with a MST of 243.5 d, which was not significantly different (P = 0.2) from WT mice.
Figure 3.

Survival times of WT, TCR-α/β−/−, TCR-γ/δ−/−, IFN-γ−/−, and NOS2−/− mice (10 mice per group) infected with 102 H37Rv via the respiratory route. IFN-γ−/− mice died earlier (MST = 32 d) than NOS2−/− mice (MST = 39 d), and NOS2−/− mice died earlier than TCR-α/β−/− mice (MST = 48). TCR-γ/δ−/− mice died with an MST of 243.5 d, which was not significantly different from the 258.5-d MST of WT mice.
Figure 4.

Survival times of WT (MST = 313.5 d), class I−/− (MST = 232 d), and class II−/− (MST = 77 d) mice (10 per group) infected with 102 H37Rv via the respiratory route.
To determine whether the susceptibility of mice without αβ T cell subsets as shown above was due entirely to the absence of CD4 and CD8 T cells, mice depleted of either, or both, subsets of T cells by treatment with mAbs were infected with 102 CFU of Mtb by aerosol, and their survival monitored. The results in Fig. 5 show that, whereas untreated mice died with an MST of 289.5 d, mice depleted of CD4 T cells or CD8 T cells died with an MST of 82 or 256 d, respectively. Mice depleted of both T cell subsets died with an MST of 50 d. Therefore, the results with T cell–depleted mice were essentially the same as those obtained with MHC mutant mice.
Figure 5.

The survival times of mice depleted of CD4, CD8, or both T cell subsets by treatment with mAbs were similar to survival times of class II−/−, class I−/−, and TCR-α/β−/− mice, respectively, as shown in Fig. 4. All mice (10 per group) were infected by aerosol with 102 H37Rv.
IFN-γ and NOS2 mRNA Synthesis in Response to Infection.
According to the foregoing results, growth of Mtb in the lungs was maintained at a stationary level from day 20 on. It could be argued, therefore, that immunity would need to be continuously mediated and expressed in the lungs from day 20 on, as would be evidenced by the continuous synthesis of mRNA for IFN-γ and NOS2. This was investigated in WT and targeted mutant mice by multiprobe ribonuclease protection assay (RPA) analysis of RNA from lungs harvested at progressive stages of infection (Fig. 6 and Fig. 7). The results in Fig. 6 show that in the case of WT mice, NOS2 mRNA was constitutively synthesized in the lungs at low levels before infection was initiated, and that its synthesis was substantially upregulated from day 20 of infection on. IFN-γ mRNA was also synthesized in larger quantity in the lungs of WT mice from day 20 on, as was IL-4 mRNA, although in quantities too small to be visible in the figure. The presence of IL-4 mRNA was clearly visible in the original radioautograph. IL-10, IL-13, and IL-2 mRNAs were not detected in measurable quantities under the conditions of the assay. It will be noted (Fig. 6 and Fig. 7) that IL-15 mRNA was constitutively present in the lungs, and that its synthesis appeared not to be substantially altered in response to infection.
Figure 6.
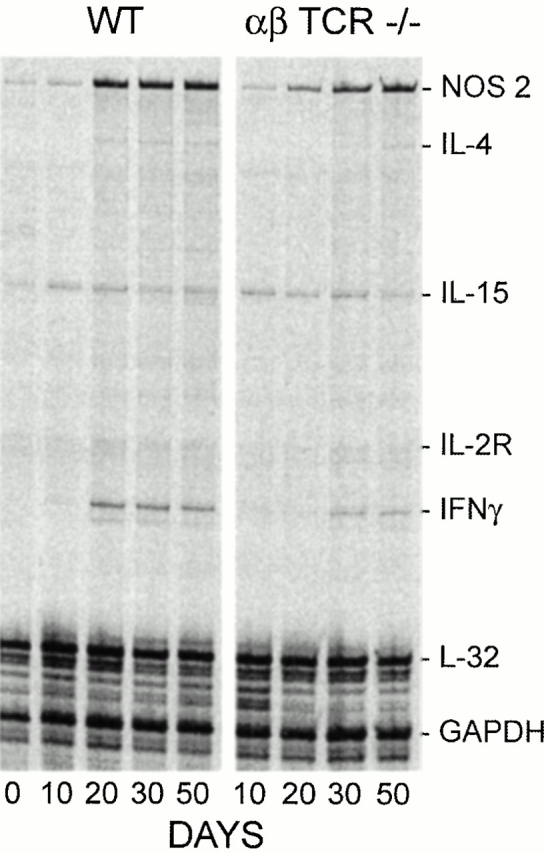
Radioautograph of multiprobe RPA run using 35S-labeled probes to detect mRNA for IFN-γ, NOS2, IL-4, IL-13, IL-15, and IL-2Rα in the lungs of WT and TCR-α/β−/− mice on days 0, 10, 20, 30, and 50 of infection. The results clearly show upregulation of mRNA for NOS2 and IFN-γ from day 20 of infection in WT mice. Much less IFN-γ mRNA was present in the lungs of TCR-α/β−/− mice from day 30 on, and the production of NOS2 mRNA was delayed. IL-15 mRNA was constitutively produced in the lungs of both groups of mice, and its synthesis appeared to be little altered by infection. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Figure 7.
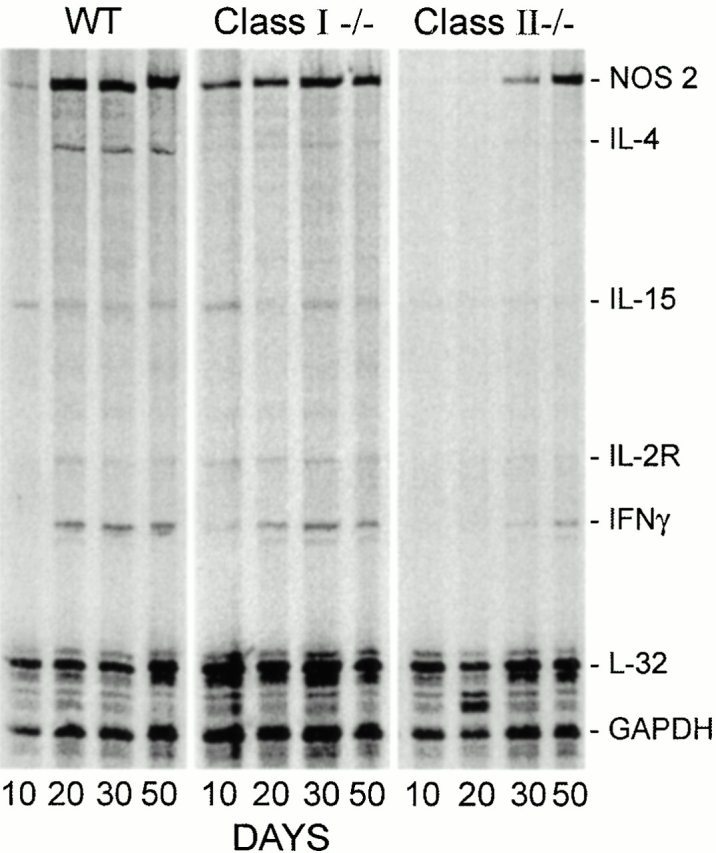
Changes in the quantity of mRNA for IFN-γ and NOS2 against time of infection in the lungs of WT, class I−/−, and class II−/− mice as determined by RPA. IFN-γ and NOS2 mRNA was upregulated from day 20 of infection on in WT mice. Likewise, IFN-γ and NOS2 mRNA was present in the lungs of class I−/− mice from days 20 on. In the lungs of class II−/− mice, in contrast, upregulation of IFN-γ mRNA was not evident until day 30, and relatively low levels were produced thereafter. Appreciable amounts of NOS2 mRNA were made in the lungs of these mice, but not until after day 30. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
In TCR-α/β−/− mice (Fig. 6), lung IFN-γ synthesis was not detected until day 30 and remained at a lower level than in WT mice through day 50. The synthesis of NOS2 mRNA was upregulated from day 20 through day 50 in the lungs of these mice, although apparently at a lower level than in WT mice. In the lungs of class I−/− mice (Fig. 7), increased synthesis of IFN-γ and NOS2 mRNA was upregulated from day 20 of infection to levels similar to those reached in WT mice. In class II−/− mice, in contrast, the level of IFN-γ mRNA synthesis in the lungs was almost undetectable at day 20, but increased to relatively low levels on days 30 and 50. Likewise, NOS2 mRNA in class II−/− mice was not synthesized in lungs of these mice in appreciable amounts before day 30.
It was considered improper to attempt to quantify relative changes in the levels of IFN-γ and NOS2 mRNA against time of infection by normalizing the density of their autoradiographic bands to those of the two housekeeping genes used in the assay. This was because it was obvious from the RPA results that synthesis of mRNAs for the housekeeping genes changed appreciably in response to infection. Problems associated with the use of housekeeping gene mRNAs as internal standards for determining changes in the quantities of other mRNA species in tissues under different experimental conditions has been discussed by others 26 27. Real-time PCR is currently being used in this laboratory to quantify changes in IFN-γ and NOS2 mRNA on the basis of whole lung mRNA, according to published methodology 28.
Lung Histopathology and Immunocytochemical Localization of NOS2.
Histology was performed on lungs harvested on day 50 and day 80 of infection when infection in WT mice was being held in stationary phase by acquired immunity. However, only day 50 histology is shown, because certain mutant mice were dead by day 80. In mice that survived, day 80 histology was essentially the same as that seen at day 50, except that the lesions were somewhat larger. Low power microscopy of day 50 lung sections (Fig. 8) serve to show the size of individual lesions and the extent to which infection-induced pathology occupied the lungs. For example, it can be seen in Fig. 8 that infection-induced pathology was confined to discrete sites that were similar in appearance, number, and size in the lungs of WT and class I−/− mice. In both types of mice, each lesion was composed of lightly stained and heavily stained areas known from previous studies 20 to represent collections of macrophages and lymphocytes, respectively. The lightly and densely stained areas are also evident from low power micrographs of sections stained for NOS2 by immunocytochemistry. This is shown in Fig. 9, where it can be seen that cells that stained positively for NOS2 in the lung lesions of WT and class I−/− mice were confined to the lightly stained macrophage areas of lesions. At higher power (Fig. 9), these cells were seen to be epithelioid macrophages with a foamy cytoplasm. Acid-fast staining showed that NOS2-positive macrophages contained Mtb bacilli, and that more bacilli were present in macrophages of class I−/− mice than in macrophages of WT mice. This is in keeping with higher level of lung infection in the former mice, in agreement with growth curves shown in a preceding section. Thus, in the lungs both of WT and class I−/− mice the histological response at each site of infection consisted of a localized alveolitis dominated by NOS2-positive macrophages and by lymphocytes. Examination of numerous sections indicated that at day 80 of infection, the lung lesions in class I−/− mice were somewhat larger than in WT mice. This was also evident from macroscopic examination of whole lungs at the time of sacrifice.
Figure 8.
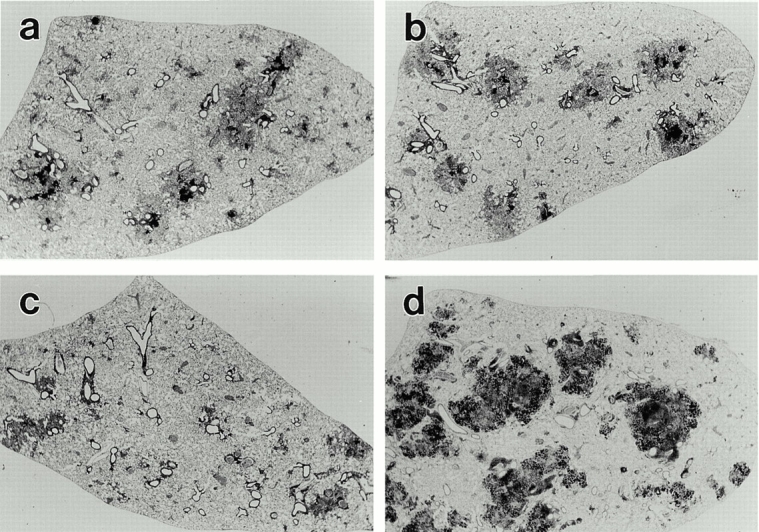
Low power (×6.5) of 15-μm thick sections of the left lobe of lungs of a WT (a) class I−/− (b), class II−/− (c), and TCR-α/β−/− (d) mouse on day 50 of infection initiated with 102 H37Rv by aerosol. The individual lesions in WT and class I−/− mice are very similar in size and appearance, being made up of darkly stained (lymphoid) and lightly stained (macrophage) areas. The lung lesions in class II−/− mice are smaller and less well defined, whereas the lung lesions in TCR-α/β−/− mice are large, darkly stained, and granular in appearance.
Figure 9.
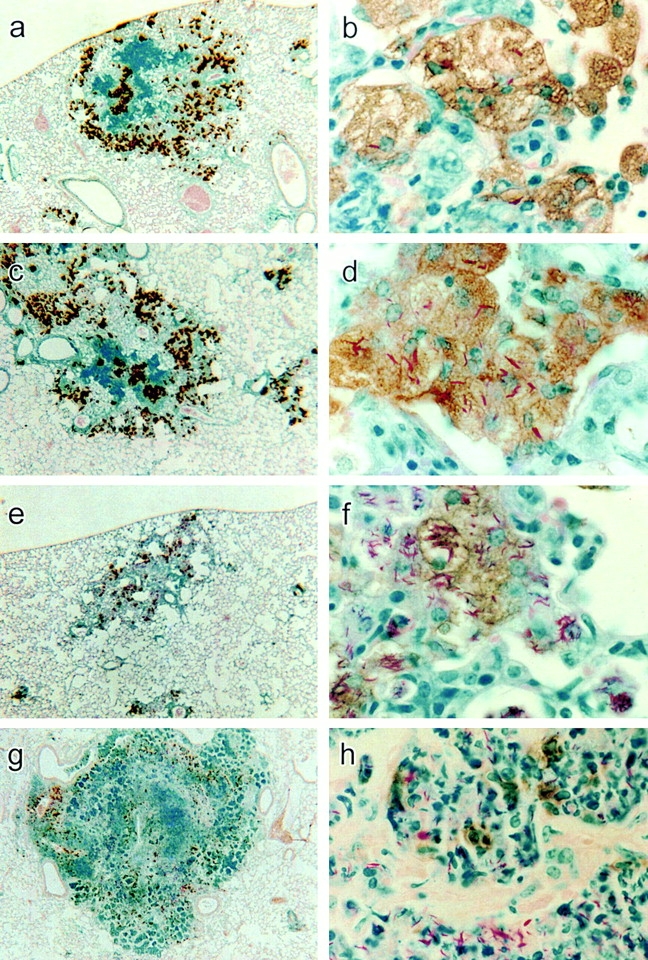
Low power (×25) and high power (×550) micrographs of lung sections of a WT (a and b), class I−/− (c and d), class II−/− (e and f), and TCR-α/β−/− mouse (g and h) stained for NOS2 by immunocytochemistry. At low power, the immunocytochemical reaction product is seen to be located in cells in the lightly stained macrophage areas of the lesions of WT and class I−/− mice. In class II−/− and TCR-α/β−/− mice, the reaction product is scattered in smaller quantity in cells throughout the lesions. At high power, the reaction product is seen confined to macrophages in lesions of WT and class I−/− mice, with some of the macrophages containing acid-fast bacilli. The NOS2-positive macrophages of the class I−/− mouse contain more acid-fast bacilli than NOS2-positive macrophages in WT lesion. In the lesion of the class II−/− mouse, NOS2-positive macrophages are heavily infected with Mtb, although some heavily infected macrophages have little or no reaction product. In the lesion of the TCR-α/β−/− mouse, most of the NOS2 reaction product appears to be concentrated in scattered mononuclear cells.
The lung lesions of class II−/− mice on day 50 (Fig. 8) were smaller, more numerous, less well organized, and occupied a smaller proportion of the lung volume than in the case of WT and class I−/− mice. The lesions were almost completely made up of lightly stained cells that at higher power (Fig. 9) were seen to be macrophages, many of which stained positively for NOS2. It is also evident that macrophages that were positive for the enzyme were heavily infected with acid-fast bacilli. However, some infected macrophages contained little or no NOS2 reaction product (not shown). Moreover, many of the infected macrophages did not display a foamy cytoplasm typical of that of epithelioid macrophages seen in the lungs of WT and class I−/− mice. Overall, the smaller number of cells showing NOS2 reaction product in class II−/− mice is in keeping with the low levels of lung NOS2 mRNA demonstrated by RPA.
The lung lesions of TCR-α/β−/− mice at low power (Fig. 8) were distinctly different in appearance from those in other types of mice. At day 50 they were larger and more numerous and collectively appeared to occupy a substantial proportion (∼50%) of the lung volume. Their histology showed no resemblance to that of lesions in WT, class I, or class II mutant mice. It was made up (Fig. 9) of an exudative, necrotic pneumonitis dominated by neutrophils. Moreover, the lungs as a whole were obviously edematous and individual air sacs were swollen to the extent that their outpocketings (alveoli) were abolished. The cells within these air sacs were heavily infected with acid-fast bacteria, in keeping with the very large numbers of Mtb CFUs in the lungs, as shown in a preceding section. Immunocytochemistry showed (Fig. 9) that NOS2 staining was concentrated in mononuclear cells scattered throughout the lesions. However, the majority of cells in lesions, including those that were infected, did not stain for NOS2.
Discussion
It is known that tuberculosis in mice 20 29, like tuberculosis in >85% of immunocompetent humans 30, is a progressive chronic disease confined to the lungs. It is only in this organ that infection-induced pathology is progressive and extensive enough to compromise organ function and cause death. However, in order for the disease in mice to resemble that in humans, it is necessary to infect mice with a small enough number of bacilli to ensure long-term survival, otherwise chronic lung pathology does not have time to develop. This was the rationale for infecting mice with 1–2 × 102 CFU of H37Rv via the respiratory route in the study reported here. It is shown that implantation of this number of Mtb in the lungs of WT mice resulted in progressive bacterial multiplication for ∼20 d, after which multiplication was controlled and infection caused to become approximately stationary under the influence of acquired immunity. That lung infection with Mtb in mice enters a stationary phase under the influence of host immunity was convincingly demonstrated many years ago by McCune and Tompsett 31, and more recently by Schell et al. 32. The same has been shown to be true for Mtb infection in guinea pigs 33 and rabbits 34. However, because progressive lung disease (pathology) develops in all three-host species, stationary infection does not represent a state of tuberculosis latency. It needs to be mentioned, moreover, that conversion of lung infection from progressive to stationary phase is not achieved by mice belonging to strains that are genetically more susceptible than the C57BL/6 strain 35.
In keeping with previous studies 20 35, each lung lesion in WT mice at day 50 of infection is shown here to consist of a discrete area of alveolitis dominated by epithelioid macrophages, some of which contain Mtb bacilli. Large accumulations of lymphoid cells at the periphery and within the lesions obviously represented a major cellular component of the response, although it is not known whether all of the lymphocytes contributed to protective immunity. According to previous studies 20 36, stationary infection in the lungs of WT mice causes lesions to increase progressively in size and the interstitium within lesions to expand and become fibrotic. Eventually, lesions coalesce and lung consolidation becomes extensive enough to kill mice with a MST of >250 d. Therefore, these results, like those published previously 20, show that immunity to primary infection neither causes infection to resolve nor prevents disease from developing.
It was expected, as shown here, that the onset of control of Mtb growth at infectious foci in the lungs of WT mice from day 20 of infection on would be temporally associated with increased synthesis of mRNA for IFN-γ and for NOS2. It was also expected that stationary infection would be associated with the synthesis of NOS2 in macrophages at infectious foci, as revealed by immunohistochemistry. Thus, stationary lung infection is shown here to be associated with the continuous synthesis of mRNA for a key Th1 cytokine involved in mediation of immunity 34 25 and with the synthesis by macrophages of an inducible enzyme necessary for the rapid production of NO and reactive nitrogen intermediates essential for the expression of immunity 22. We have also shown (to be published) that NOS2 and IFN-γ mRNA are continuously synthesized in the lungs for at least 150 d of infection. Relevant to these findings is a published finding 22 showing that inhibition of NOS2-dependent NO production in mice with stationary infection by treatment with N6-(iminoethyl) lysine resulted in resumption of Mtb growth. A similar resumption of Mtb growth was shown to occur in mice treated with the less specific inhibitor of NOS2-dependent NO production, aminoguanidine 37, and in mice selectively depleted of CD4 T cells 38.
As for the response to Mtb infection in class I−/− mice, there is no doubt from this study that the pathology seen in the lungs of these mice was qualitatively similar to that seen in WT mice. This was to be expected, given that class I−/− mice, like WT mice, were able to eventually stabilize infection and cause it to become approximately stationary, albeit at a 1 log higher level. As this higher level of infection was caused by about a 30-d delay before infection became stationary, it follows that CD8 T cells, in addition to CD4 T cells, contributed significantly to the control bacterial growth during this period. Whether CD8 cells help to maintain infection at a stationary level at later times of infection is not known. However, the fact that stabilization of infection is achieved by class I−/− mice means that CD8 T cells are not needed to eventually stop infection from progressing, even after it reaches a 1 log higher level. A recently published study of mice infected with much higher numbers of Mtb via the intravenous route 39 also shows that lung infection is controlled and becomes stationary in the absence of CD8 T cell–mediated immunity. The presence of a higher level of stationary infection in class I−/− mice undoubtedly was the reason why lung lesions in these mice eventually increase in size at a faster rate than those in WT mice. This is not surprising, given that a 1 log higher level of stationary infection represents 10 times more bacilli per lesion and a larger bacterial load per macrophage. This presumably would represent a larger antigen load per macrophage and a larger intracellular stimulus for the production of chemokines and proinflammatory cytokines by macrophages. It is not surprising, therefore, that the survival time of class I−/− mice was shorter, although not much shorter (25%) than that of WT mice. Presumably, both types of mice died of Th1-mediated chronic lung disease.
The survival time of class II−/− mice, in contrast, was substantially shorter (86%) than that of WT mice. Moreover, because infection was progressive in all organs in class II−/− mice, it seems likely that death was caused by pathology in multiple organs induced by overwhelming systemic infection. According to the histology of the lungs of class II−/− mice, there was not enough disease in these organs between days 50 and 80 to cause death. On the other hand, there was extensive pathology in the liver and spleen (unpublished results). Regardless, there can be little doubt from this study that class II−/− mice were greatly more susceptible to Mtb infection than class I−/−. Indeed, if successful immunity in the mouse model of tuberculosis is defined as the ability to cause infection to become stationary rather than to resolve, then class I−/− mice, but not class II−/− mice, were capable of expressing successful immunity. The delayed synthesis of lower levels of IFN-γ mRNA in the lungs of class II−/− mice is in keeping with their failure to mediate protective immunity. The inability of these mice to stabilize lung infection when detectable amounts of IFN-γ and NOS2 mRNA were eventually synthesized might indicate that activated macrophages are capable of exerting only a limited inhibitory effect on intracellular Mtb growth after the load of Mtb per macrophage increases above a certain threshold. Threshold numbers were reached because of the delay in the expression of immunity, as evidenced by the delay in synthesis of IFN-γ and NOS2.
It seems likely that most of the IFN-γ mRNA that was present in the lungs of class II−/− mice was synthesized by CD8 T cells and possibly NK cells. Even so, the exact way that CD8 T cells mediate immunity in WT and class II−/− mice is not known. According to one study 40, the ability of these cells to synthesize IFN-γ is essential for their ability to express immunity. Whether they also function to lyse infected host cells in vivo remains to be shown, although evidence obtained with gene knockout mice indicates that neither perforin-dependent nor Fas-dependent lysis is likely to be involved 41 42. It is possible, on the other hand, that CD8 T cells are protective by virtue of their ability to make granulysin, a molecule known to be directly toxic for Mtb in vitro according to studies with human CD8 T cells 43. However, according to the study presented here none of these functions of CD8 T cells is essential for stabilization of Mtb infection in mice. Presumably, the absence of CD8 T cells is compensated for by an expansion in the number of CD4 T cells in lesions, or by upregulation of cytokine and chemokine production by individual CD4 T cells in response to a higher level of stationary infection at infectious foci.
Perhaps the most compelling evidence presented here that CD8 T cells contribute to immunity is the demonstration that their presence in class II−/− mice prevented the massive exudative, inflammatory response seen in the lungs of mice devoid of both subsets of αβ T cells. The strikingly greater resistance of class II−/− mice compared with αβ T cell−/− mice presumably was due to the ability of CD8 T cells to create conditions that favor the extravasation and accumulation of macrophages, rather than neutrophils, at sites of Mtb growth. Indeed, the results show that lung lesions in class II−/− mice, although small and disorganized, were dominated by macrophages, at least up to day 80 of infection. However, unlike macrophages in lesions of WT and class I−/− mice, many infected macrophages in lesions of class II−/− mice appeared morphologically less mature and contained much larger numbers of acid-fast bacilli. Moreover, although many of these heavily infected macrophages stained for NOS2, many did not. It was also apparent that Mtb had multiplied just as much in NOS2-positive as in NOS2-negative macrophages, presumably because Mtb numbers had become too high for macrophages to control by the time NOS2 synthesis was upregulated.
Because survival results obtained with class II−/− and class I−/− mice were reproduced with mice depleted of CD4 and CD8 T cells by mAb treatment, it is unlikely that cells in addition to CD8 T cells contribute significantly to resistance in class II−/− mice. It was also found as part of this study (results not presented) that CD8 and CD4 gene–deleted mice were somewhat less susceptible to tuberculosis than class I−/− and class II−/− mice respectively. This presumably was because of the ability of T cells to respond to a limited extent to Ags in the absence CD4- and CD8-mediated costimulation. Taken as a whole, the results presented show that the absence of CD8 T cells in mice infected with a small number of Mtb via the respiratory route was of much less consequence to survival than was shown by others 7 15 39 for mice infected with a larger number via the intravenous route. A possible explanation for this disagreement is that inoculation of 105 or 106 CFU of Mtb via the intravenous route results in a much larger total bacterial burden with which CD4 T cells need to deal. According to the bacterial growth curves in the publications cited, giving over 105 Mtb via the intravenous route resulted in over 3 logs of bacteria implanting in the lungs at the start of infection. Assuming that each CFU that implants gives rise to a separate site of infection at which pathology can develop, it is not surprising that mice so infected died much earlier than mice with 10-fold fewer lung lesions, as shown in the study reported here. Unfortunately, it is not known whether mice devoid of CD4 T cell–mediated immunity would have died earlier or later from a 105 or 106 intravenous inoculum than mice devoid of CD8 T cell–mediated immunity, as class II−/− mice were not included in the studies for comparison. Regardless, even in the case of large intravenous inocula, the absence of CD8 T cell–mediated immunity did not prevent mice from stabilizing infection at a higher level and causing it to become stationary 39.
Lastly, the finding that mice devoid of γδ T cells were not deficient in an ability to stabilize infection is in keeping with results published by others 11. The additional finding that these mice had the same survival time as WT mice suggests that if γδ T cells function to regulate infection-induced inflammation in the lung, as has been proposed 11, it is unlikely that this regulatory function is relevant to either the expression of protective immunity or to infection-induced pathology that eventually causes death.
Acknowledgments
This work was supported by National Institutes of Health grants AI37844 and HL64565.
Footnotes
Abbreviations used in this paper: MST, median survival time; Mtb, Mycobacterium tuberculosis; NOS2, inducible nitric oxide synthase; RPA, ribonuclease protection assay; WT, wild-type.
References
- Boom W.H. The role of T cell subsets in Mycobacterium tuberculosis infection. Infect. Agents Dis. 1996;5:73–81. [PubMed] [Google Scholar]
- Murray P.J. Defining the requirements for immunological control of mycobacterial infections. Trends Micribiol. 1999;7:366–372. doi: 10.1016/s0966-842x(99)01567-x. [DOI] [PubMed] [Google Scholar]
- Stenger S., Modlin R.L. T cell-mediated immunity to Mycobacterium tuberculosis . Curr. Opin. Microbiol. 1999;2:89–93. doi: 10.1016/s1369-5274(99)80015-0. [DOI] [PubMed] [Google Scholar]
- Cooper A.M., Saunders B.M., D'Souza C.D., Frank A.A., Orme I.M. Mycobacterium tuberculosis-driven processes in gene-disrupted mice. Bull. Inst. Pasteur. 1997;95:85–95. [Google Scholar]
- Orme I.M., Collins F.M. Adoptive protection of the Mycobacterium tuberculosis-infected lung. Cell. Immunol. 1984;84:113–120. doi: 10.1016/0008-8749(84)90082-0. [DOI] [PubMed] [Google Scholar]
- Caruso A.M., Serbina N., Klein E., Triebold K., Bloom B.R., Flynn J. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-γ, yet succumb to tuberculosis. J. Immunol. 1999;162:5407–5416. [PubMed] [Google Scholar]
- Flynn J., Goldstein M.M., Triebold K.J., Koller B., Bloom B. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA. 1992;89:12013–12017. doi: 10.1073/pnas.89.24.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveton C., Barnass S., Champion B., Lucas S., de Souza B., Nicol M., Banerjee D., Rook G. T-cell mediated protection of mice against virulent Mycobacterium tuberculosis . Infect. Immun. 1989;57:390–395. doi: 10.1128/iai.57.2.390-395.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boom W.H. Gamma delta T cells and Mycobacterium tuberculosis . Microbes Infect. 1999;1:187–195. doi: 10.1016/s1286-4579(99)80033-1. [DOI] [PubMed] [Google Scholar]
- Ladel C.H., Blum C., Dreher A., Reifenberg K., Kaufmann S.H.E. Protective role of γδ T cells and αβ T cells in tuberculosis. Eur. J. Immunol. 1995;25:2877–2881. doi: 10.1002/eji.1830251025. [DOI] [PubMed] [Google Scholar]
- D'Souza C.D., Cooper A.M., Frank A.A., Mazzaccaro R.J., Bloom B.R., Orme I.M. An anti-inflammatory role for γδ T lymphocytes in acquired immunity to Mycobacterium tuberculosis . J. Immunol. 1997;158:1217–1221. [PubMed] [Google Scholar]
- Johnson C.M., Cooper A.M., Frank A.A., Bonorino C.B., Wysoki L.J., Orme I.M. Mycobacterium tuberculosis aerogenic rechallenge infections in B cell-deficient mice. Tubercle Lung Dis. 1997;78:257–261. doi: 10.1016/s0962-8479(97)90006-x. [DOI] [PubMed] [Google Scholar]
- Porcelli S., Morita C.T., Brenner M.B. CD1b restricts the response of human CD4−CD8− T lymphocytes to a microbial antigen. Nature. 1992;360:593–597. doi: 10.1038/360593a0. [DOI] [PubMed] [Google Scholar]
- Thomssen H., Ivanyi J., Espitia C., Arya A., Londei M. Human CD4−CD8− αβ+ T cell receptor T cells recognize different mycobacteria strains in the context of CD1b. Immunology. 1995;85:33–40. [PMC free article] [PubMed] [Google Scholar]
- Behar S.M., Dascher C.C., Grusby M.J., Wang C.R., Brenner M.B. Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis . J. Exp. Med. 1999;189:1973–1980. doi: 10.1084/jem.189.12.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrie D.B., Silva C.L., Tascon R.E. DNA vaccines against tuberculosis. Immunol. Cell Biol. 1997;75:591–594. doi: 10.1038/icb.1997.93. [DOI] [PubMed] [Google Scholar]
- Denis O., Tanghe A., Palfliet K., Jurion F., T-P. van den Berg A., Vanonckelen J., Ooms E., Saman J.B., Ulmer J., Content, Huygen K. Vaccination with plasmid DNA encoding mycobacterial antigen 85A stimulates a CD4 and CD8 T cell epitopic repertoire broader than that stimulated by Mycobacterium tuberculosis H37Rv infection. Infect. Immun. 1998;66:1527–1533. doi: 10.1128/iai.66.4.1527-1533.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess J., Kaufmann S.H.E. Live antigen carriers as tools for improved anti-tuberculosis vaccines. FEMS Immunol. Med. Microbiol. 1999;23:165–173. doi: 10.1111/j.1574-695X.1999.tb01236.x. [DOI] [PubMed] [Google Scholar]
- Silva C.L., Bonato V.L., Lima V.M., Faccioli L.H., Leao S.C. Characterization of the memory/activated T cells that mediate the long-lived host response against tuberculosis after bacillus Calmette-Guerin or DNA vaccination. Immunology. 1999;97:573–581. doi: 10.1046/j.1365-2567.1999.00840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn P.L., North R.J. Virulence ranking of some Mycobacterium tuberculosis and Mycobacterium bovis strains according to their ability to multiply in the lungs, induce lung pathology, and cause mortality in mice. Infect. Immun. 1995;63:3428–3437. doi: 10.1128/iai.63.9.3428-3437.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R.C., Zabrowarny L.A. A safer staining method for acid fast bacilli. J. Clin. Pathol. 1993;46:559–560. doi: 10.1136/jcp.46.6.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking J.D., North R.J., LaCourse R., Mudgett J.S., Shah S.K., Nathan C.F. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA. 1997;94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson S., Bonecini-Almeida M., Silva J.R.L., Nathan C., Xie Q.W., Mumford R., Weidner J.R., Calaycay J., Geng J., Boechat N. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J. Exp. Med. 1996;183:2293–2302. doi: 10.1084/jem.183.5.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A.M., Dalton D.K., Stewart T.A., Griffen J.P., Russell D.G., Orme I.M. Disseminated tuberculosis in IFN-γ gene disrupted mice. J. Exp. Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J.L., Chan J., Triebold K.J., Dalton D.K., Stewart T.A., Bloom B.R. An essential role for interferon-γ in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanakis E. Problems related to the interpretation of autoradiographic data on gene expression using common constitutive transcripts as controls. Nucleic Acids Res. 1993;16:3809–3819. doi: 10.1093/nar/21.16.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thellin O., Zorzi W., Lakaye B., De Borman B., Coumans B., Hennen G., Grisar T., Igout A., Heinen E. Housekeeping genes as internal standardsuse and limits. J. Biotechnol. 1999;75:291–295. doi: 10.1016/s0168-1656(99)00163-7. [DOI] [PubMed] [Google Scholar]
- Serazin-Leroy V., Denis-Henriot D., Morot M., de Mazancourt P., Giudicelli Y. Semi-quantitative RT-PCR for comparison of mRNA in cells with different amounts of housekeeping gene transcripts. Mol. Cell. Probes. 1998;12:283–291. doi: 10.1006/mcpr.1998.0182. [DOI] [PubMed] [Google Scholar]
- Pierce C.H., Dubos R.J., Scheafer W.B. Multiplication and survival of tubercle bacilli in the organs of mice. J. Exp. Med. 1953;97:189–205. doi: 10.1084/jem.97.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farer L.S., Lowell A.M., Meador M.P. Extrapulmonary tuberculosis in the United States. Am. J. Epidemiol. 1979;109:205–217. doi: 10.1093/oxfordjournals.aje.a112675. [DOI] [PubMed] [Google Scholar]
- McCune R.M., Tompsett R. Fate of mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. J. Exp. Med. 1956;104:737–762. doi: 10.1084/jem.104.5.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell R.F., Ealey W.F., Harding G.E., Smith D.W. The influence of vaccination on the course of experimental airborne tuberculosis in mice. J. Reticuloendothel. 1974;Soc. 16:131–138. [PubMed] [Google Scholar]
- Balasubramanian V., Wiegeshaus E.H., Smith D.W. Growth characteristics of recent sputum isolates of Mycobacterium tuberculosis in guinea pigs infected by the respiratory route. Infect. Immun. 1992;60:4762–4767. doi: 10.1128/iai.60.11.4762-4767.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurie M.B., Zappasodi P., Tickner C. On the nature of genetic resistance to tuberculosis in the light of the host-parasite relationships in natively resistant and susceptible rabbits. Am. Rev. Tuberc. Pulm. Dis. 1955;72:297–329. doi: 10.1164/artpd.1955.72.3.297. [DOI] [PubMed] [Google Scholar]
- Medina E., North R.J. Evidence inconsistent with a role for the Bcg gene (Nramp1) in resistance of mice to infection with Mycobacterium tuberculosis . J. Exp. Med. 1996;183:1045–1051. doi: 10.1084/jem.183.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes E.R., Frank A.A., Orme I.M. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis . Tubercle Lung Dis. 1997;78:57–66. doi: 10.1016/s0962-8479(97)90016-2. [DOI] [PubMed] [Google Scholar]
- Flynn J.L., Scanga C.A., Tanaka K.E., Chan J. Effects of aminoguanidine on latent murine tuberculosis. J. Immunol. 1998;160:1796–1803. [PubMed] [Google Scholar]
- Scanga C.A., Mohan V.P., Yu K., Joseph H., Tanaka K., Chan J., Flynn J.L. Depletion of CD4+ T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon γ and nitric oxide synthase 2. J. Exp. Med. 2000;192:347–358. doi: 10.1084/jem.192.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa A.O., Mazzaccaro R.C., Russell R.G., Lee F.K., Turner O.C., Hong S., Van Kaer L., Bloom B.R. Relative contributions of distinct MHC class I-dependent cell populations in protection to tuberculosis infection in mice. Proc. Natl. Acad. Sci. 2000;USA. 97:4204–4208. doi: 10.1073/pnas.97.8.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tascon R.E., Stavropoulos E., Lukacs K.V., Colston M.J. Protection against Mycobacterium tuberculosis infection by CD8+ T cells requires the production of gamma interferon. Infect. Immun. 1998;66:830–834. doi: 10.1128/iai.66.2.830-834.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A.M., D'Souza C., Frank A.A., Orme I.M. The course of mycobacterium tuberculosis infection in the lungs of mice lacking expression of either perforin- or granzyme-mediated cytolytic mechanisms. Infect. Immun. 1997;65:1317–1320. doi: 10.1128/iai.65.4.1317-1320.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laochumroonvorapong P., Wang J., CC. Liu W., Ye A.L., Moreira K.B., Elkon V.H., Freedman, Kaplan G. Perforin, a cytotoxic molecule which mediates cell necrosis, is not required for the early control of mycobacterial infection in mice. Infect. Immun. 1997;65:127–132. doi: 10.1128/iai.65.1.127-132.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenger S., Hanson D.A., Teitelbaum R., Dewan P., Niazi K.R., Froelich C.J., Ganz T., Thoma-Uszynski S., Melian A., Bogdan C. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. 1998;282:121–125. doi: 10.1126/science.282.5386.121. [DOI] [PubMed] [Google Scholar]