

Abstract
53BP1 participates early in the DNA damage response and is involved in cell cycle checkpoint control. Moreover, the phenotype of mice and cells deficient in 53BP1 suggests a defect in DNA repair (Ward et al., 2003b). Therefore, we asked whether or not 53BP1 would be required for the efficient repair of DNA double strand breaks. Our data indicate that homologous recombination by gene conversion does not depend on 53BP1. Moreover, 53BP1-deficient mice support normal V(D)J recombination, indicating that 53BP1 is not required for “classic” nonhomologous end joining. However, class switch recombination is severely impaired in the absence of 53BP1, suggesting that 53BP1 facilitates DNA end joining in a way that is not required or redundant for the efficient closing of RAG-induced strand breaks. These findings are similar to those observed in mice or cells deficient in the tumor suppressors ATM and H2AX, further suggesting that the functions of ATM, H2AX, and 53BP1 are closely linked.
Keywords: NHEJ; ATM; H2AX; V(D)J recombination; DNA repair
Introduction
Eukaryotic cells are constantly exposed to DNA-damaging agents. DNA double strand breaks (DSBs) are considered the most genotoxic form of DNA damage. They can arise endogenously from reactive oxygen intermediates or through exogenous exposure of cells to ionizing radiation (IR). DSBs can also be generated when DNA replication forks encounter DNA lesions, such as DNA single strand breaks or DNA cross-links (Khanna and Jackson, 2001). In addition, DNA DSBs occur as part of the normal development of the immune repertoire in B and T lymphocytes. During the process of V(D)J recombination, the recombination-activating gene proteins RAG1 and RAG2 introduce DSBs between observed recombination signal sequences and flanking V, D, or J coding segments of the antigen-combining sites of immunoglobulin and T cell receptor (TCR) genes (Gellert, 2002). A second type of DNA recombination, class switch recombination (CSR), also involves the generation of DNA DSBs. Stimulated mature B cells replace the heavy chain constant region of the initially expressed IgM antibodies with a different constant region. This isotype switching allows antibodies to change their effector functions while maintaining their antigen specificity (Honjo et al., 2002).
Improper processing of DSBs gives rise to chromosomal instability that can result in carcinogenesis. To maintain genomic integrity, eukaryotic cells have evolved different pathways for the repair of DNA DSBs. In the yeast Saccharomyces cerevisiae, DSBs seem to be repaired almost exclusively through high fidelity homologous recombination (HR), a process that uses the undamaged sister chromatid or homologous chromosome as a DNA template (Lin et al., 1999; Khanna and Jackson, 2001). In mammalian cells, nonhomologous end joining (NHEJ), which is the error-prone joining of DNA ends without the requirement for sequence homology, plays an important role in DSB repair, especially during the G1 phase of the cell cycle when no sister chromatid is available (Hendrickson, 1997; Khanna and Jackson, 2001).
53BP1 participates early in the DNA damage response. It rapidly localizes to sites of DNA strand breaks in response to IR and interacts with phosphorylated histone H2AX (γ-H2AX; Schultz et al., 2000; Xia et al., 2000; Anderson et al., 2001; Rappold et al., 2001; Abraham, 2002). Studies using siRNA directed against 53BP1 implicate a role of 53BP1 in checkpoint control (DiTullio et al., 2002; Fernandez-Capetillo et al., 2002; Wang et al., 2002). Notably, 53BP1-deficient mice are hypersensitive to IR and exhibit an increased predisposition for T cell lymphomas. Moreover, lack of 53BP1 protein is accompanied by immunodeficiency and increased chromosomal instability (Morales et al., 2003; Ward et al., 2003b). This phenotype is reminiscent of the phenotype observed in mice with a defect in the NHEJ pathway.
Therefore, we examined if 53BP1 plays a role in DNA DSB repair. Our results indicate that HR by gene conversion does not require 53BP1. Moreover, NHEJ-dependent V(D)J recombination is not affected in the absence of 53BP1, but CSR is severely impaired in 53BP1−/− mice. These results suggest that 53BP1 facilitates DNA end joining in a way that is not essential for V(D)J recombination.
Results and discussion
Role of 53BP1 in DNA repair
We have shown earlier that 53BP1−/− mice are hypersensitive to radiation and die within 2 wk after exposure to 8 Gy of IR (Ward et al., 2003b). Similarly, thymocytes isolated from irradiated 53BP1-deficient mice show a 2.11 ± 0.79-fold higher rate of apoptosis when compared with thymocytes isolated from irradiated wild-type littermates (Fig. 1 A). No significant difference in apoptosis was found in thymi of un-irradiated animals (unpublished data). Moreover, primary mouse embryonic fibroblasts derived from 53BP1−/− embryos exhibit a delayed exit from the G2 phase of the cell cycle after radiation compared with wild-type mouse embryonic fibroblasts, presumably extending the time available for repair before entry into mitosis (Ward et al., 2003b). This radiation-induced G2 delay is also observed in 53BP1−/− embryonic cells derived from 53BP1−/− blastocysts (unpublished data). This evidence suggests a possible repair defect in 53BP1−/− cells.
Figure 1.
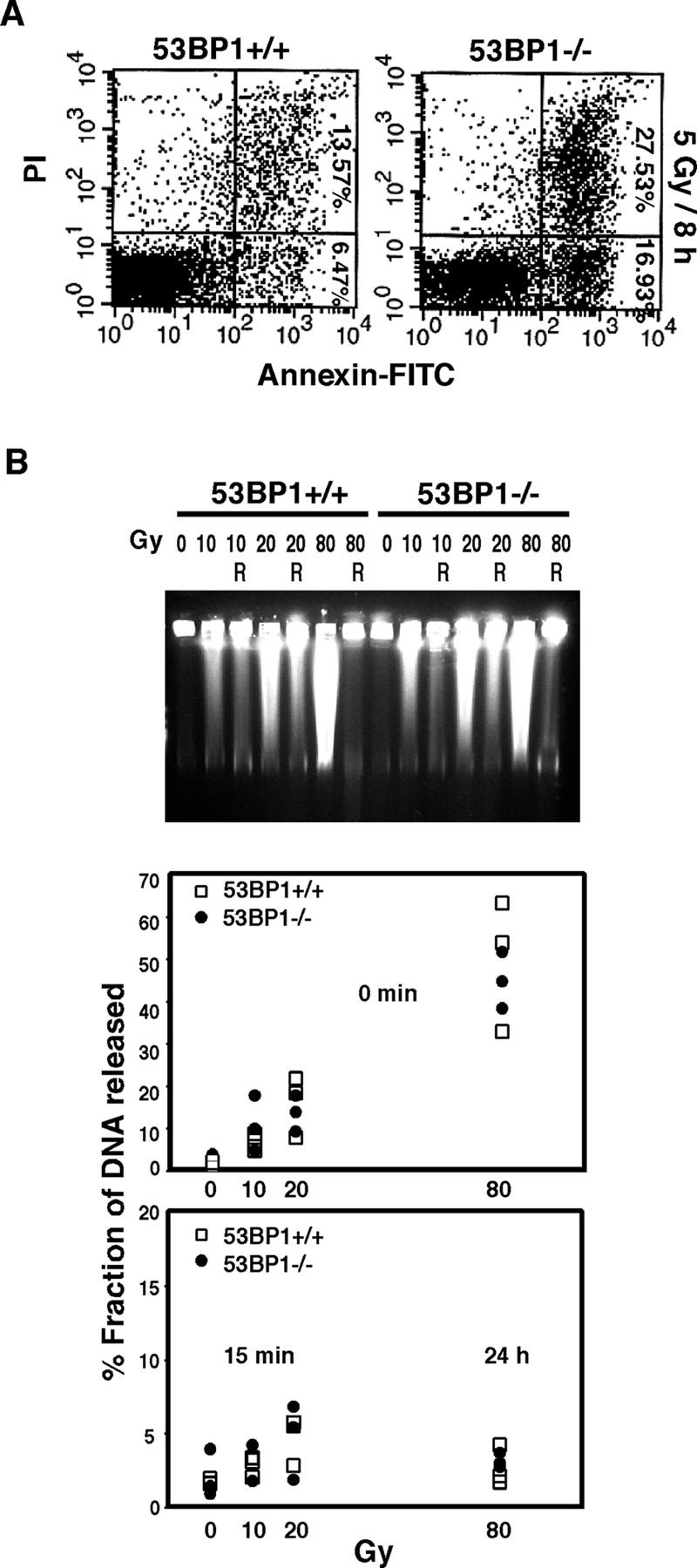
53BP1 and DNA damage repair. (A) Increased apoptosis in thymocytes derived from irradiated 53BP1−/− mice. Thymocytes were isolated from wild-type and 53BP1-deficient mice 8 h after irradiation with 5 Gy of IR and stained with annexin–FITC and PI. (B) 53BP1−/− embryonic cells show normal levels of DNA DSB rejoining. Wild-type and 53BP1-deficient cells were irradiated with different doses of IR and either allowed to recover (R) for 15 min or 24 h, as indicated, or immediately processed for PFGE analysis.
To examine whether or not the rejoining of DNA DSBs depends on 53BP1, we used the embryonic 53BP1+/+ and 53BP1−/− cell lines to perform pulse field gel electrophoresis (PFGE) assays. As shown in Fig. 1 B, neither DNA DSB induction nor DSB rejoining appeared to be defective in 53BP1-deficient cells. The majority of DNA DSBs was rejoined within 15 min after exposure of cells to 10 or 20 Gy of IR, or within 24 h after exposure to 80 Gy of IR. Because the sensitivity of the PFGE assay may not be sufficient to detect subtle defects in DNA DSB repair, we performed additional assays to assess whether or not 53BP1 plays a role in HR or NHEJ, the two major DNA DSB repair pathways.
53BP1 is not required for HR
To test if 53BP1 is involved in HR, we used the recombination repair substrate DR-GFP designed to model HR-directed repair by using a tandem GFP repeat. The first GFP gene is inactivated by the introduction of an I-SceI recognition site, while the adjacent GFP gene is differentially mutated. After the introduction of a DSB at the I-SceI site, the GFP gene can be reconstituted by HR using the downstream inactivated GFP gene as a template.
We transfected 53BP1+/+ or 53BP1−/− cells with DR-GFP and an I-SceI expression plasmid (pCBASce) and analyzed GFP expression 48 h later by flow cytometry. As a control, cells were transfected with DR-GFP alone or with a plasmid expressing an intact GFP gene. Although GFP-positive cells were very rare in both the wild-type (0.03–0.06%) and 53BP1-deficient cells (0.02–0.08%) transfected with DR-GFP alone (Fig. 2), an average of 4.80 ± 0.22% of 53BP1+/+ cells and 4.21 ± 1.42% of 53BP1−/− cells expressed GFP 48 h after cotransfection with the I-SceI expression vector (Fig. 2). Similar numbers of GFP-positive cells were also observed after transfection of 53BP1+/+ and 53BP1−/− cells with an intact control GFP expression vector, indicating that the transfection efficiency did not differ between 53BP1+/+ and 53BP1−/− cells (Fig. 2). Thus, our results suggest that DSB repair by gene conversion is not impaired in 53BP1-deficient cells.
Figure 2.

53BP1 is not required for homology-directed repair. Wild-type or 53BP1-deficient embryonic cells were cotransfected with an I-SceI repair substrate (DR-GFP) composed of two differentially mutated GFP and an I-SceI expression plasmid (pCBASce). Reconstitution of the GFP reporter gene by HR was assessed 46 h later by flow cytometry. As a control, cells were transfected with either DR-GFP alone or with a functional GFP expression plasmid (pIRES GFPpuro).
53BP1 is not required for “classic” NHEJ and V(D)J recombination
Cells with a defect in the classic NHEJ pathway tend to use microhomologies near the DNA ends to promote end-joining reactions. To determine the relative efficiency of classic versus microhomology-directed NHEJ, we transiently transfected cells with a linearized plasmid substrate designed to create a BstX1 restriction site when joined by microhomology (Verkaik et al., 2002). Whereas plasmids recovered from DNA-PKcs–deficient cells indicated high levels of microhomology-directed joining, as previously described (Verkaik et al., 2002), direct end joining dominated in 53BP1-deficient cells as well as in wild-type or DNA-PKcs–reconstituted cells (Fig. 3), suggesting that 53BP1−/− cells have no major defect in NHEJ.
Figure 3.

53BP1 is not required for NHEJ. Wild-type or 53BP1-deficient embryonic cells as well as DNA-PKcs–deficient and –reconstituted cells were transfected with plasmid pDVG94 linearized in such a way that joining on a particular microhomology creates a novel BstXI restriction site. 48 h later, the plasmid was recovered from the cells and the joining region was amplified by PCR. An aliquot of the PCR reaction was digested with BstXI, and uncut (180 bp) or BstXI-cut (120 bp) fragments were separated by gel electrophoresis.
Another good indicator of NHEJ proficiency is V(D)J recombination. RAG-initiated site-specific V(D)J recombination is impaired in NHEJ-deficient cells and mice (Bassing et al., 2002). Notably, 53BP1−/− mice have up to a 50% reduction in the number of thymocytes and mature T cells compared with wild-type littermates (Ward et al., 2003b), a phenotype that could arise from impaired TCR V(D)J rearrangement. Therefore, we analyzed the rearrangement of different TCR loci in 53BP1−/− and 53BP1+/+ mice. Overall, the level of V(D)J coding formation and the levels of TCRα excised signal joints appeared very similar in wild-type and knockout animals (Fig. 4). Quantitative analysis of the data confirmed that the level of the various TCR gene rearrangements tested did not differ between 53BP1+/+ and 53BP1−/− mice (unpublished data). Together, our data indicate that 53BP1 is not essential for V(D)J recombination.
Figure 4.

Normal TCR gene rearrangements in the absence of 53BP1. Serial dilutions of thymus DNA from 1-mo-old 53BP1-deficient mice and their wild-type littermates were PCR-amplified with primers specific for various TCRα, β, γ, or δ rearrangements or TCRα signal joints. The PCR products were Southern blotted and hybridized with the different gene-specific reverse probes as described in Materials and methods. PCR of the nonrearranging gene RAG-2 served as control for DNA quantity and integrity.
53BP1 is required for efficient CSR
Unlike V(D)J recombination, the initiating factors and the mechanisms leading to the joining of DNA ends in CSR are only partially understood. Recent evidence suggests that CSR is triggered by AID (activation-induced deaminase)-induced deamination of cytosine residues in single strand DNA exposed by transcription of the immunoglobulin switch region. Excision of the resulting dU/dG mismatches on complementary DNA strands is thought to introduce DSBs (Muramatsu et al., 2000; Honjo et al., 2002; Petersen-Mahrt et al., 2002; Chaudhuri et al., 2003; Dickerson et al., 2003; Ramiro et al., 2003). Comparison of switch junction sequences indicates that switch recombination does not depend on either sequence-specific or HR mechanisms (Dunnick et al., 1993). Some evidence points to the NHEJ pathway because Ku-deficient B-cells show almost no detectable CSR (Casellas et al., 1998; Manis et al., 1998; Reina-San-Martin et al., 2003), and CSR to most constant region genes is severely impaired in DNA-PK–deficient B cells (Manis et al., 2002). To determine whether or not CSR is impaired in the absence of 53BP1, we stimulated wild-type and 53BP1−/− B cells to undergo switching to IgG1 in vitro with LPS and IL-4. Cells were also labeled with CFSE to track cell divisions by flow cytometry. CFSE dye dilution histograms were similar between 53BP1−/− and wild-type B cells, and no proliferation defects or increased mortality were observed (Fig. 5 A). Despite equivalent proliferation, the percentage of IgG1 positive cells after 96 h of incubation was 30% in wild-type cells but only 2% in 53BP1−/− B cells (Fig. 5 B), suggesting severely impaired CSR in the absence of 53BP1.
Figure 5.
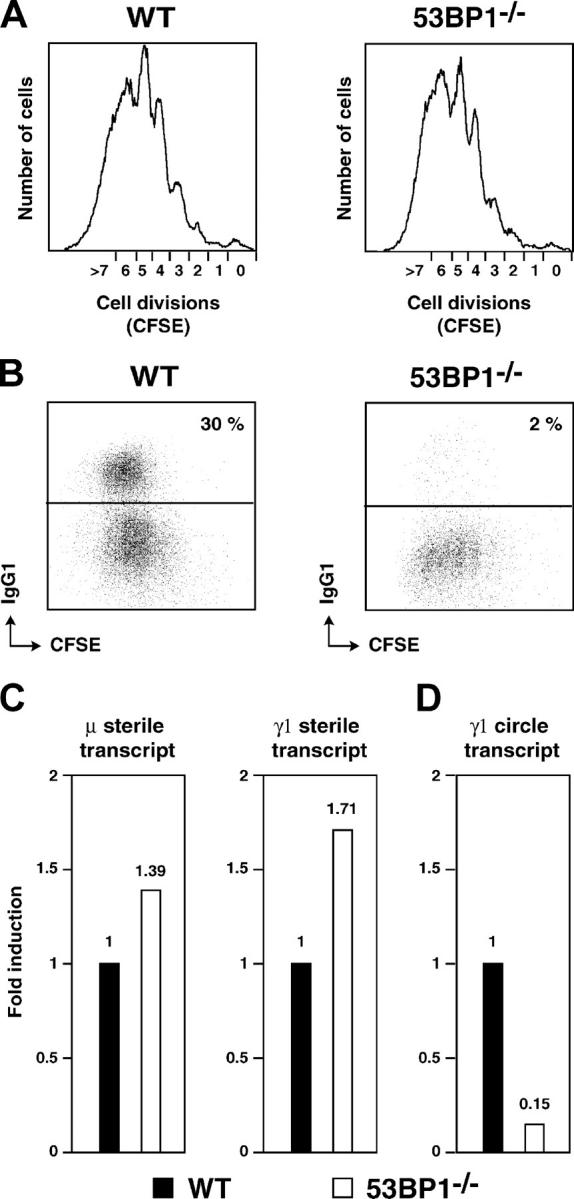
CSR is dependent on 53BP1. Cell division (A) and surface IgG1 expression (B) as measured by flow cytometry on live CFSE-labeled wild type (WT) and 53BP1−/− B cells stimulated with LPS plus IL-4 for 4 d. Results are representative of four experiments. Real-time RT-PCR for μ and γ1 sterile transcripts (C) and γ1 circle transcript (D) in wild-type (solid bars) and 53BP1−/− (open bars) B cells stimulated with LPS and IL-4 for 3 d. Results from four independent cultures are expressed as fold induction relative to wild-type B cells.
CSR is dependent on the rate of switch region transcription, and recombination is targeted to individual switch regions by transcription from intronic (I) promoters located upstream of each switch region. To determine if sterile transcription is normal in 53BP1−/− B cells, we measured the relative amounts of the μ and γ1 preswitch sterile transcripts (Reina-San-Martin et al., 2003) by quantitative real-time RT-PCR. As shown in Fig. 5 C, μ and γ1 sterile transcripts were expressed at comparable levels in wild-type and 53BP1−/− B cells, suggesting that sterile transcription of the IgM and IgG1 switch regions is not altered in the absence of 53BP1.
CSR is a deletional recombination reaction that results in the looping out and deletion of intervening DNA sequences as a circular episome (Iwasato et al., 1990; Matsuoka et al., 1990; von Schwedler et al., 1990). The looped-out circular DNA contains segments of Sμ and the target S region, including its I promoter. This promoter is still active in the looped-out circle and drives the synthesis of the circle transcript, a hybrid containing the I and Cμ exons (Kinoshita et al., 2001). The circle transcript appears only after productive CSR, and its level is proportional to the frequency of successful joining events (Kinoshita et al., 2001). To determine if 53BP1 deficiency has an impact on the frequency of joining during CSR, we used real-time RT-PCR to quantitate γ1 circle transcripts in B cells stimulated with LPS and IL-4. The level of γ1 circle transcript in 53BP1−/− B cells was 6.6-fold reduced when compared with wild-type (Fig. 5 D) and is consistent with decreased CSR in the absence of 53BP1 (Fig. 5 B). Together, these data indicate that 53BP1 is required for CSR at the DNA level and that impaired CSR in 53BP1−/− B cells is not due to abnormal B cell proliferation.
The impairment in CSR observed in 53BP1-deficient cells is less severe than the CSR defects in B cells deficient for components of the NHEJ pathway (Casellas et al., 1998; Manis et al., 1998, 2002). However, it appears to be more severe than the defect described in H2AX-deficient mice (Celeste et al., 2002; Reina-San-Martin et al., 2003). Moreover, the fact that 53BP1-null mice, like H2AX-deficient mice, support normal V(D)J recombination implies that the joining of class switch junctions differs from the rejoining of RAG-induced strand breaks. The repair pathways could be different for V(D)J recombination and CSR. Alternatively, 53BP1 and H2AX could be indirectly involved in the repair process by facilitating chromosomal accessibility or influencing chromatin organization and the loading of repair proteins. Because RAG proteins bind directly to DNA ends during V(D)J recombination, the involvement of RAG proteins may alleviate the requirement of a structural role of H2AX and 53BP1 in V(D)J recombination. Of course, it is also possible that the role of 53BP1 or H2AX in chromatin alterations is specific to CSR. Further analysis of H2AX and 53BP1 in the regulation of chromatin structure will be necessary to test these hypotheses.
Both 53BP1 and H2AX become rapidly phosphorylated by ATM (ataxia telangiectasia mutated) after IR and interact with each other at sites of DNA breaks (Burma et al., 2001; Rappold et al., 2001; Ward et al., 2003a). Similar to H2AX- or 53BP1-deficient cells, ATM-deficient cells also display a defect in CSR but support normal V(D)J recombination (Pan-Hammarstrom et al., 2003), suggesting that the functions of these proteins are closely linked. Although the exact role of 53BP1, ATM, and H2AX in DNA DSB repair remains to be determined, all three proteins appear to function in facilitating certain aspects of DNA end joining. Unraveling their mode of action in DNA repair will be a step further in our understanding of the process of carcinogenesis.
Materials and methods
Cell lines
53BP1-deficient and 53BP1 wild-type embryonic cell lines were established from day 3 blastocysts by a standard procedure. DNA-PKcs–deficient cells (V3.3) and DNA-PKcs reconstituted cells (V3.155) were a gift of D.C. Chen, Lawrence Berkeley National Laboratory, Berkeley, CA.
Apoptosis assay
To assess apoptosis in irradiated thymocytes, 6-wk-old 53BP1−/− mice and wild-type littermates were exposed to 5 Gy of IR. 8 h later, the animals were killed and the thymocytes were isolated and stained with annexin-FITC (Molecular Probes) and PI before analysis on a FACScan.
PFGE assay
For determination of DSB rejoining, equal numbers of exponentially growing cells labeled with [14C]thymidine were exposed to IR at the indicated doses. After various recovery times, the cells were embedded in agarose plugs and lysed for 16 h in 1% sarcosyl, 0.5 M EDTA, and 1mg/ml proteinase K. For determination of break induction, cells were embedded in plugs before irradiation and lysed immediately. The plugs were washed in TE buffer and electrophoresis was performed in a CHEF DRII system (Bio-Rad Laboratories) for 65 h in 0.8% agarose in 0.5× TBE at 14°C with a field strength of 1.5 V/cm and pulse times increasing from 50 to 5,000 s. For quantification, single lanes were cut into ∼1-cm-thick slices, melted, and analyzed on a scintillation counter. The level of DNA breakage was estimated by the fraction of activity released from the plug into the gel.
HR repair assay
The efficiency of HR was assessed using an I-SceI repair substrate (DR-GFP) composed of two differentially mutated GFP and an I-SceI expression plasmid (pCBASce; both gifts from M. Jasin, Memorial Sloan-Kettering Cancer Center and Cornell University Graduate School of Medical Sciences, New York, NY). 53BP1+/+ and 53BP1−/− embryonic cells were transfected by electroporation with either 5 μg DR-GFP plus 5 μg pCBASce or 5 μg DR-GFP alone. In addition, an aliquot of the cells was transfected with a functional GFP expression plasmid (pIRES-GFP Puro) to monitor transfection efficiency. 46 h later, cells were harvested and GFP expression was assessed by flow cytometry.
NHEJ repair and V(D)J assays
The assay for microhomology-directed end joining was performed as described previously (Verkaik et al., 2002). 5 μg of blunt-ended linear pDVH94 plasmid (a gift from D.C. van Gent, Erasmus Medical Center, Rotterdam, Netherlands) was electroporated into 4 × 106 cells, and extrachromosomal DNA was isolated 48 h later. DNA end-joining regions were amplified by PCR, and microhomology-directed end joining was assessed by BstXI restriction digestion.
V(D)J recombination in vivo was tested by a semiquantitative PCR method as described previously (Livak et al., 1995, 1999; Livak and Schatz, 1996).
Primer sequences not published before are as follows: Vβ4, 5′ GAAGCCTCTAGAGTTCATGTTTTC 3′; Jβ2.5, 5′ GAGCCGAGTGCCTGGCCCAAAGTA 3′; Vα2, 5′ GCCGGATCCAGGAGAAACGTGACCAGCAG 3′; Vα10, 5′ AGCGAATTCCCGCGTCCTTGGTTCTGCA 3′; Vα2 signal joint, 5′ CTCTGGATCCGAATTCATYTAAACTAGTTAA 3′, where Y = C or T; Vα10 signal joint, 5′ CCTGGATCCAGAATTCTACCAATACARGAAAG 3′, where R = A or G; and Jα26/27, 5′ CCTGGATCCTTACTGTCATATATCGAA 3′.
Lymphocyte cultures and flow cytometry
Resting B lymphocytes were isolated from the spleen using CD43 microbeads (Miltenyi Biotec), labeled with CFDA-SE for 10 min at 37°C (5 μM; Molecular Probes), and cultured (106 cells/ml) with LPS (25 ng/ml; Sigma-Aldrich) and IL-4 (5 ng/ml; Sigma-Aldrich) for 4 d. Percentage of switching to IgG1 was determined by flow cytometry by using Biotin-anti-IgG1 (BD Biosciences) and Streptavidin-PE-Cy7 (Caltag). Dead cells were excluded from the analysis by staining with Topro-3 (Molecular Probes).
Quantitative real-time RT-PCR
Total RNA was extracted with TRIzol (Invitrogen) and reverse transcribed with random hexamers and superscript II reverse transcriptase (Invitrogen). First strand cDNA was used for SYBR green fluorogenic dye real-time PCR (Applied Biosystems). Primers used and PCR conditions are described in Reina-San-Martin et al. (2003).
Acknowledgments
We thank David C. Chen, Maria Jasin, and Dik C. van Gent for valuable reagents. We are grateful to Larry Karnitz, Scott Kaufmann, and members of the Chen laboratory for helpful discussions.
This work was supported by a grant from the National Institutes of Health to J. Chen (CA100109). J. Chen is a recipient of a Department of Defense (DOD) breast cancer career development award (DAMD17-02-1-0472). I. Ward is supported by a postdoctoral fellowship from the DOD Breast Cancer Research Program (DAMD17-01-1-0317).
Abbreviations used in this paper: CSR, class switch recombination; DSB, double strand break; HR, homologous recombination; I, intronic; IR, ionizing radiation; NHEJ, nonhomologous end joining; PFGE, pulse field gel electrophoresis; TCR, T cell receptor.
References
- Abraham, R.T. 2002. Checkpoint signalling: focusing on 53BP1. Nat. Cell Biol. 4:E277–E279. [DOI] [PubMed] [Google Scholar]
- Anderson, L., C. Henderson, and Y. Adachi. 2001. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol. Cell. Biol. 21:1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing, C.H., W. Swat, and F.W. Alt. 2002. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 109:S45–S55. [DOI] [PubMed] [Google Scholar]
- Burma, S., B.P. Chen, M. Murphy, A. Kurimasa, and D.J. Chen. 2001. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276:42462–42467. [DOI] [PubMed] [Google Scholar]
- Casellas, R., A. Nussenzweig, R. Wuerffel, R. Pelanda, A. Reichlin, H. Suh, X.F. Qin, E. Besmer, A. Kenter, K. Rajewsky, and M.C. Nussenzweig. 1998. Ku80 is required for immunoglobulin isotype switching. EMBO J. 17:2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste, A., S. Petersen, P.J. Romanienko, O. Fernandez-Capetillo, H.T. Chen, O.A. Sedelnikova, B. Reina-San-Martin, V. Coppola, E. Meffre, M.J. Difilippantonio, et al. 2002. Genomic instability in mice lacking histone H2AX. Science. 296:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri, J., M. Tian, C. Khuong, K. Chua, E. Pinaud, and F.W. Alt. 2003. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 422:726–730. [DOI] [PubMed] [Google Scholar]
- Dickerson, S.K., E. Market, E. Besmer, and F.N. Papavasiliou. 2003. AID mediates hypermutation by deaminating single stranded DNA. J. Exp. Med. 197:1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiTullio, R.A., T.A. Mochan, M. Venere, J. Bartkova, M. Sehested, J. Bartek, and T.D. Halazonetis. 2002. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol. 4:998–1002. [DOI] [PubMed] [Google Scholar]
- Dunnick, W., G.Z. Hertz, L. Scappino, and C. Gritzmacher. 1993. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 21:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo, O., H.T. Chen, A. Celeste, I. Ward, P.J. Romanienko, J.C. Morales, K. Naka, Z. Xia, R.D. Camerini-Otero, N. Motoyama, et al. 2002. DNA damage-induced G(2)-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 4:993–997. [DOI] [PubMed] [Google Scholar]
- Gellert, M. 2002. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem. 71:101–132. [DOI] [PubMed] [Google Scholar]
- Hendrickson, E.A. 1997. Cell-cycle regulation of mammalian DNA double-strand-break repair. Am. J. Hum. Genet. 61:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo, T., K. Kinoshita, and M. Muramatsu. 2002. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu. Rev. Immunol. 20:165–196. [DOI] [PubMed] [Google Scholar]
- Iwasato, T., A. Shimizu, T. Honjo, and H. Yamagishi. 1990. Circular DNA is excised by immunoglobulin class switch recombination. Cell. 62:143–149. [DOI] [PubMed] [Google Scholar]
- Khanna, K.K., and S.P. Jackson. 2001. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27:247–254. [DOI] [PubMed] [Google Scholar]
- Kinoshita, K., M. Harigai, S. Fagarasan, M. Muramatsu, and T. Honjo. 2001. A hallmark of active class switch recombination: transcripts directed by I promoters on looped-out circular DNAs. Proc. Natl. Acad. Sci. USA. 98:12620–12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y., T. Lukacsovich, and A.S. Waldman. 1999. Multiple pathways for repair of DNA double-strand breaks in mammalian chromosomes. Mol. Cell. Biol. 19:8353–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, F., and D.G. Schatz. 1996. T-cell receptor alpha locus V(D)J recombination by-products are abundant in thymocytes and mature T cells. Mol. Cell. Biol. 16:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, F., H.T. Petrie, I.N. Crispe, and D.G. Schatz. 1995. In-frame TCR delta gene rearrangements play a critical role in the alpha beta/gamma delta T cell lineage decision. Immunity. 2:617–627. [DOI] [PubMed] [Google Scholar]
- Livak, F., M. Tourigny, D.G. Schatz, and H.T. Petrie. 1999. Characterization of TCR gene rearrangements during adult murine T cell development. J. Immunol. 162:2575–2580. [PubMed] [Google Scholar]
- Manis, J.P., Y. Gu, R. Lansford, E. Sonoda, R. Ferrini, L. Davidson, K. Rajewsky, and F.W. Alt. 1998. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med. 187:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis, J.P., D. Dudley, L. Kaylor, and F.W. Alt. 2002. IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity. 16:607–617. [DOI] [PubMed] [Google Scholar]
- Matsuoka, M., K. Yoshida, T. Maeda, S. Usuda, and H. Sakano. 1990. Switch circular DNA formed in cytokine-treated mouse splenocytes: evidence for intramolecular DNA deletion in immunoglobulin class switching. Cell. 62:135–142. [DOI] [PubMed] [Google Scholar]
- Morales, J.C., Z. Xia, T. Lu, M.B. Aldrich, B. Wang, C. Rosales, R.E. Kellems, W.N. Hittelman, S.J. Elledge, and P.B. Carpenter. 2003. Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J. Biol. Chem. 278:14971–14977. [DOI] [PubMed] [Google Scholar]
- Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- Pan-Hammarstrom, Q., S. Dai, Y. Zhao, I.F. van Dijk-Hard, R.A. Gatti, A.L. Borresen-Dale, and L. Hammarstrom. 2003. ATM is not required in somatic hypermutation of VH, but is involved in the introduction of mutations in the switch mu region. J. Immunol. 170:3707–3716. [DOI] [PubMed] [Google Scholar]
- Petersen-Mahrt, S.K., R.S. Harris, and M.S. Neuberger. 2002. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 418:99–103. [DOI] [PubMed] [Google Scholar]
- Ramiro, A.R., P. Stavropoulos, M. Jankovic, and M.C. Nussenzweig. 2003. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 4:452–456. [DOI] [PubMed] [Google Scholar]
- Rappold, I., K. Iwabuchi, T. Date, and J. Chen. 2001. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage–signaling pathways. J. Cell Biol. 153:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin, B., S. Difilippantonio, L. Hanitsch, R.F. Masilamani, A. Nussenzweig, and M.C. Nussenzweig. 2003. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J. Exp. Med. 197:1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, L.B., N.H. Chehab, A. Malikzay, and T.D. Halazonetis. 2000. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 151:1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkaik, N.S., R.E. Esveldt-van Lange, D. van Heemst, H.T. Bruggenwirth, J.H. Hoeijmakers, M.Z. Zdzienicka, and D.C. van Gent. 2002. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur. J. Immunol. 32:701–709. [DOI] [PubMed] [Google Scholar]
- von Schwedler, U., H.M. Jack, and M. Wabl. 1990. Circular DNA is a product of the immunoglobulin class switch rearrangement. Nature. 345:452–456. [DOI] [PubMed] [Google Scholar]
- Wang, B., S. Matsuoka, P.B. Carpenter, and S.J. Elledge. 2002. 53BP1, a mediator of the DNA damage checkpoint. Science. 298:1435–1438. [DOI] [PubMed] [Google Scholar]
- Ward, I.M., K. Minn, K.G. Jorda, and J. Chen. 2003. a. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J. Biol. Chem. 278:19579–19582. [DOI] [PubMed] [Google Scholar]
- Ward, I.M., K. Minn, J. Van Deursen, and J. Chen. 2003. b. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 23:2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, Z., J.C. Morales, W.G. Dunphy, and P.B. Carpenter. 2000. Negative cell cycle regulation and DNA damage-inducible phosphorylation of the BRCT protein 53BP1. J. Biol. Chem. 276:2708–2718. [DOI] [PubMed] [Google Scholar]