

Abstract
Infection with murine cytomegalovirus (MCMV) has contributed to understanding many aspects of human infection and, additionally, has provided important insight to understanding complex cellular responses. Dendritic cells (DCs) are a major target for MCMV infection. Here, we analyze the effects of MCMV infection on DC viability, and show that infected DCs become resistant to apoptosis induced by growth factor deprivation. The precise contribution of changes in the expression of Bcl-2 family proteins has been assessed and a new checkpoint in the apoptotic pathway identified. Despite their resistance to apoptosis, MCMV-infected DCs showed Bax to be tightly associated with mitochondria and, together with Bak, forming high molecular weight oligomers, changes normally associated with apoptotic cell death. Exposure of a constitutively occluded Bax NH2-terminal epitope was blocked after infection. These results suggest that MCMV has evolved a novel strategy for inhibiting apoptosis and provide evidence that apoptosis can be regulated after translocation, integration, and oligomerization of Bax at the mitochondrial membrane.
Keywords: dendritic cell; cytomegalovirus; apoptosis; Bcl-2; Bax
Introduction
Apoptosis is a highly regulated control process involving the removal of damaged or unwanted cells from multicellular organisms. Elimination of infected cells through a process of rigorously controlled cell death provides a powerful defense mechanism against intracellular pathogens, such as viruses. Not surprisingly, viruses have evolved strategies to modulate apoptosis, and thereby circumvent host immune defense (Roulston et al., 1999). The relevance of apoptosis in the control of viral replication is demonstrated by the known antiviral effects of agents that trigger apoptosis, such as TNF and Fas ligand, and by the identification of viral proteins with anti-apoptotic activity (Xu et al., 2001). In particular, large DNA viruses, such as baculovirus, adenovirus, pox viruses, and herpes viruses, encode a number of proteins that interfere at various stages of the apoptotic pathway (Roulston et al., 1999; Tortorella et al., 2000).
Cytomegalovirus (CMV), a member of the β herpes virus family, is a prime candidate for possessing anti-apoptotic functions, because its replication cycle is relatively slow and the virus persists lifelong within its host (Mocarski, 1996). Previously, infection of human umbilical-vein endothelial or mouse neuronal cells with human CMV (HCMV) and murine CMV (MCMV), respectively, was found to render infected cells resistant to apoptosis (Kovacs et al., 1996; Kosugi et al., 1998; Wang et al., 2000). Unlike other herpes viruses, CMV does not encode homologues of Bcl-2 or FLIP (Roulston et al., 1999). Anti-apoptotic properties have been ascribed to the M45 gene of MCMV, and the UL36 and UL37 genes of HCMV (Goldmacher et al., 1999; Brune et al., 2001; Skaletskaya et al., 2001). Although the mechanism of action for M45 is unknown, UL36 appears to act by inhibiting caspase-8 activation, and UL37 prevents mitochondrial permeabilization (Goldmacher et al., 1999; Skaletskaya et al., 2001). Despite having ascribed anti-apoptotic activities to several CMV proteins, the precise checkpoints targeted by the virus in apoptotic pathways have not been defined.
Central to apoptotic pathways is the action of caspases, a group of aspartate-specific cysteine proteases whose activation is regulated by the Bcl-2 family of proteins (Cory and Adams, 2002; Shi, 2002). The Bcl-2 family is composed of both pro- and anti-apoptotic proteins that share homology in the Bcl-2 homology (BH) regions (Marsden and Strasser, 2003). The current paradigm suggests that pro-survival Bcl-2 family proteins prevent caspase activation by maintaining mitochondrial integrity (Gross et al., 1999). The activity of pro-survival Bcl-2 proteins is counteracted by two groups of pro-apoptotic proteins, namely, Bax/Bak and the BH3-only group of proteins. BH3-only proteins are regulated by a variety of mechanisms, such as sequestration to the cytoskeleton, which favorably positions these molecules to act as sensors for apoptotic stimuli (Puthalakath and Strasser, 2002). In response to apoptotic stimuli, BH3-only proteins translocate to the mitochondria where it appears they antagonize the function of pro-survival Bcl-2 proteins (Puthalakath and Strasser, 2002).
The BH3-only proteins require the presence of the Bax/Bak proteins for killing. Bax, normally a monomer located in the cytoplasm, undergoes a conformational change, translocates to the mitochondria, and oligomerizes once activated (Hsu et al., 1997; Wolter et al., 1997). Similarly, apoptotic stress results in conformational changes and oligomerization of Bak, although Bak is constitutively localized to mitochondria (Griffiths et al., 1999; Nechushtan et al., 2001). Activation of Bax/Bak at mitochondria is thought to induce permeabilization of the outer mitochondrial membrane, thereby allowing the release of pro-apoptotic proteins, such as cytochrome c and Smac/DIABLO, and the subsequent activation of caspases (Gross et al., 1999). Retention of Bax in the cytoplasm most likely acts as a safe guard against inappropriate activation of the cell death machinery. Consistent with this theory is the observation that enforced dimerization of Bax induces apoptosis, and Bax mutants that are constitutively localized to mitochondria are more effective killers (Gross et al., 1998; Suzuki et al., 2000). However, cells exposed to the apoptosis inducing drug taxol were shown to survive despite the fact that Bax dimerized and translocated to the mitochondria (Makin et al., 2001). Furthermore, detachment of epithelial cells from ECM resulted in reversible changes to Bax conformation and localization (Gilmore et al., 2000; Valentijn et al., 2003). These results suggest that activation of Bax is not sufficient to commit to apoptosis, and that an additional checkpoint may exist.
The ability of CMV to successfully escape immune responses and to establish persistent infection of its hosts led us to hypothesize that this virus would be capable of interfering with apoptosis in cells that are key immune effectors. DCs are specialized antigen presenting cells critical for the initiation and regulation of both innate and adaptive immune responses (Banchereau et al., 2000). Our recent studies have identified DCs as a major target for MCMV infection, both in vivo and in vitro (Andrews et al., 2001). Here, we have demonstrated that MCMV-infected DCs become resistant to apoptosis. Furthermore, we have assessed the precise contribution of changes in the expression of Bcl-2 family proteins and have defined the specific point at which MCMV interferes in the apoptotic pathway. Our studies provide evidence that translocation of Bax to mitochondria, and oligomerization of Bax and Bak are not sufficient for the initiation of apoptotic cell death.
Results
MCMV prevents apoptosis in DCs
Recently, we have demonstrated that DCs are permissive to MCMV infection, and that infection results in functional impairment of these cells (Andrews et al., 2001). To successfully replicate several viruses have evolved mechanisms to prevent apoptosis of host cells (Tortorella et al., 2000). We sought to determine whether MCMV affected the ability of DCs to undergo apoptosis. The well-characterized D1 DC culture system was initially used for these experiments because, unlike freshly isolated DCs which undergo spontaneous maturation and cell death, D1 cells maintain an immature DC phenotype and proliferate when grown in the presence of growth factor enriched conditioned medium (CM; Winzler et al., 1997). Removal of CM causes D1 cells to undergo apoptosis (Rescigno et al., 1998). Hence, D1 DC cultures provide a unique system to study the effects of MCMV infection on immature DCs, a cell type we have shown to be a major target of MCMV infection in vivo (Andrews et al., 2001).
Initially, we determined whether MCMV could prevent apoptosis of D1 cells after growth factor withdrawal. D1 DCs were infected with MCMV at a multiplicity of infection (MOI) >3 and cultured for 4 d, by which time 100% of the cells are MCMV infected (Andrews et al., 2001). MCMV-infected D1, and uninfected control D1 cells, were washed and replated in either normal growth medium or in medium lacking CM. After 16 h the number of viable cells was determined by flow cytometry using annexin V-FITC staining. Consistent with previous reports, a significant decrease in the percentage of viable cells was observed when CM was removed from DC cultures (Fig. 1 a; Rescigno et al., 1998). Conversely, withdrawal of CM from DCs infected with MCMV did not affect cell viability (Fig. 1 a).
Figure 1.

MCMV inhibits apoptosis in DCs. (a) The viability of D1 DCs infected with MCMV (MOI >3) for 4 d was measured after culture in the presence or absence of growth factor CM for 16 h. Data represent mean ± SD; n = 6. (b) The viability of uninfected, MCMV-infected or LPS-stimulated D1 DCs cultured in the presence or absence of CM for the indicated times was measured. Data represent mean ± SD; n = 6. (c) Caspase activity in uninfected, LPS-stimulated or MCMV-infected D1 DCs grown in the presence or absence of CM for 12 h was determined by cleavage of the caspase-3 substrate Ac-DEVD-pA. Data represent mean ± SD; n = 3. (d) Viability of Flt3L expanded MCMV-infected or control DCs cultured in medium containing 10% FCS and Flt3L (cells grown in CM), or 1% FCS (cells grown in the absence of CM). Data represent mean ± SD; n = 3. Shaded bars represent cells grown in CM. White bars represent cells grown in the absence of CM.
Stimulation of D1 DCs with LPS induces functional maturation, arrests DC proliferation, and promotes survival after growth factor deprivation (Winzler et al., 1997; Rescigno et al., 1998). The effect of MCMV infection on DC viability after growth factor withdrawal was compared with the effect of LPS-induced maturation. D1 cells were either infected with MCMV (MOI >3 for 4 d), activated with LPS (10 μg/ml for 2 d), or left untreated. Cells were then replated in the presence or absence of CM and the number of viable cells determined (Fig. 1 b). LPS-stimulated D1 cells were cell cycle arrested and became resistant to apoptosis induced by growth factor withdrawal (Rescigno et al., 1998). Infection of D1 cells with MCMV also resulted in cell cycle arrest, and was as effective as LPS stimulation at protecting against apoptosis (Fig. 1 b).
Although activation of caspases is considered a crucial effector of apoptosis, some evidence exists for a caspase-independent apoptotic pathway (Marsden and Strasser, 2003). We examined whether caspases are activated during apoptosis of DCs. Uninfected, LPS-stimulated, and MCMV-infected D1 DCs were grown in the presence or absence of growth factor for 12 h and total cell lysates prepared. Caspase activity in the lysates was measured using a colorimetric substrate (Ac-DEVD-pA) for caspase-3 and related caspases. After growth factor withdrawal from uninfected immature D1 cells, a significant increase in caspase activity was observed (Fig. 1 c). No change in caspase activity was observed after growth factor removal from LPS-stimulated or MCMV-infected cells (Fig. 1 c).
We then tested the effect of MCMV infection on the viability of in vitro expanded DCs. These experiments were performed using DCs generated from the bone marrow by Flt3L expansion (Brasel et al., 2000). This method generates cells with phenotypic and functional characteristics of DCs found in lymphoid tissues (Brasel et al., 2000). After in vitro expansion, DCs were infected with MCMV (MOI >3) and cultured in the presence of Flt3L for a further 4 d. MCMV-infected DCs and uninfected control DCs were then washed and replated in growth medium containing 10% FCS and Flt3L or in medium containing 1% FCS only. The number of viable cells was determined 16 h later by annexin V-FITC staining. Removal of FCS and Flt3L resulted in a significant reduction in the percentage of viable cells in the control cultures (Fig. 1 d). No change in the viability of MCMV-infected DCs was observed after withdrawal of FCS and Flt3L (Fig. 1 d). These studies, using bone marrow–derived DCs, confirmed that MCMV inhibits DC cell death.
MCMV-infected DCs are resistant to apoptosis induced by inhibition of PI3 kinase
Next we sought to determine which cellular signal transduction pathways are involved in protecting MCMV-infected DCs from apoptosis. Activation of extracellular signal regulated kinase (ERK) is required to maintain the viability of LPS-stimulated DCs after growth factor withdrawal (Rescigno et al., 1998). However, inhibition of ERK does not affect the viability of immature or LPS-stimulated DCs grown in the presence of growth factors (Rescigno et al., 1998). Similarly, inhibition of ERK kinases does not affect the viability of freshly isolated splenic DCs or human monocyte-derived DCs (Ardeshna et al., 2000; Park et al., 2002). The protection from apoptosis provided by stimulation with LPS or CpG DNA in these systems can be blocked after inhibition of the phosphatidylinositide-3-OH kinase (PI3 kinase) pathway (Ardeshna et al., 2000; Park et al., 2002).
The role of PI3 kinase activity in the context of DC survival was tested using LY294002, a specific inhibitor of PI3 kinase. MCMV-infected D1, or uninfected control D1 cells, were plated into fresh medium containing growth factor in the absence or presence of 50 μM LY294002, and the percentage of viable cells determined by annexin V-FITC staining after 16 h. Incubation of D1 DCs with LY294002 lead to reduced viability, suggesting that PI3 kinase activity is critical for maintaining DC viability (Fig. 2 a). MCMV-infected DCs were found to be resistant to the effects of LY294002, indicating that the virus can bypass the requirement for a signal from PI3 kinase in maintaining cell viability (Fig. 2 a). The contribution of the ERK MAP kinase pathway to viability was also assessed using the MEK1/2 inhibitor U0126. The viability of control or MCMV-infected DCs after inhibition of the ERK pathway was not altered (Fig. 2 a).
Figure 2.
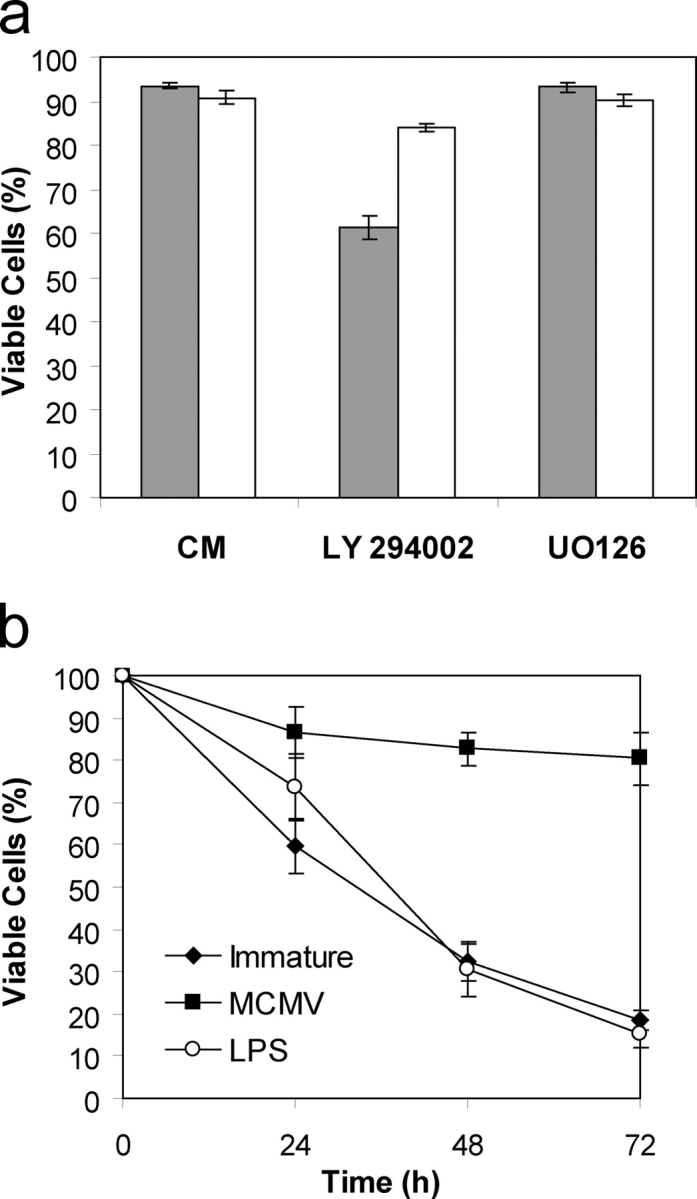
Inhibition of PI3 kinase does not affect the viability of MCMV-infected DCs. (a) Percentage viability of immature or MCMV-infected D1 DCs cultured in normal growth medium, medium containing 50 μM LY 294002, or medium containing 10 μM UO126 for 16 h. (b) Viability of MCMV-infected, LPS-stimulated or control D1 DCs grown in medium containing 25 μM LY 294002. Data presented in a and b represent mean ± SD; n = 6. Shaded bars represent uninfected immature DCs. White bars represent MCMV-infected DCs.
Inhibition of PI3 kinase induced apoptosis in LPS-stimulated DCs (Fig. 2 b). The viability of LPS-stimulated DCs grown in the presence of 25 μM LY294402 decreased at a rate similar to that observed for immature DCs. Thus, PI3 kinase activity is required for the survival of both immature and LPS-stimulated DCs. Conversely, inhibition of PI3 kinase did not affect the viability of MCMV-infected DCs (Fig. 2 b).
Expression of Bim increases after growth factor withdrawal
We next sought to characterize the molecular events leading to apoptosis of DCs after growth factor withdrawal. Given the role of Bcl-2 proteins in regulating apoptosis induced by growth factor deprivation, we monitored the expression of Bcl-2 family members in DCs during apoptosis induced by withdrawal of growth factor. No change in the protein level of Bcl-2, Bcl-xL, or Bax was observed after growth factor withdrawal at any of the time points tested (Fig. 3 a). Similarly, no change in the expression of pro-survival Bcl-w or Bfl-1/A1 protein was detected, whereas expression of Mcl-1 was not detected in these cells (not depicted). An increase in the expression of Bim, a BH3 only pro-apoptotic protein was observed (Fig. 3 a). Alternative splicing of Bim gives rise to three variants (BimEL, BimL, and BimS) each of which contains the BH3 domain and is capable of functioning as a death inducer (O'Connor et al., 1998). Bim activity is regulated at a posttranslational level and, in healthy cells most Bim molecules are sequestered to the microtubular dynein motor complex (Puthalakath et al., 1999). Certain apoptotic stimuli result in translocation of Bim to the mitochondria where the protein is able to antagonize the function of pro-survival Bcl-2 molecules (Puthalakath et al., 1999). Therefore, the proportion of Bim protein localizing to mitochondria in DCs grown in the presence or absence of growth factor was examined by subcellular fractionation. As expected, very little Bim protein could be detected in the mitochondrial fraction of cells grown in the presence of growth factor, whereas a significant amount was observed when DCs were grown in the absence of growth factor for 12 h (Fig. 3 b). These results are consistent with observations made in lymphocyte and neuronal cultures, where Bim expression has been found to be critical for the initiation of apoptosis after growth factor withdrawal (Bouillet et al., 1999; Villunger et al., 2000; Putcha et al., 2001; Shinjyo et al., 2001).
Figure 3.
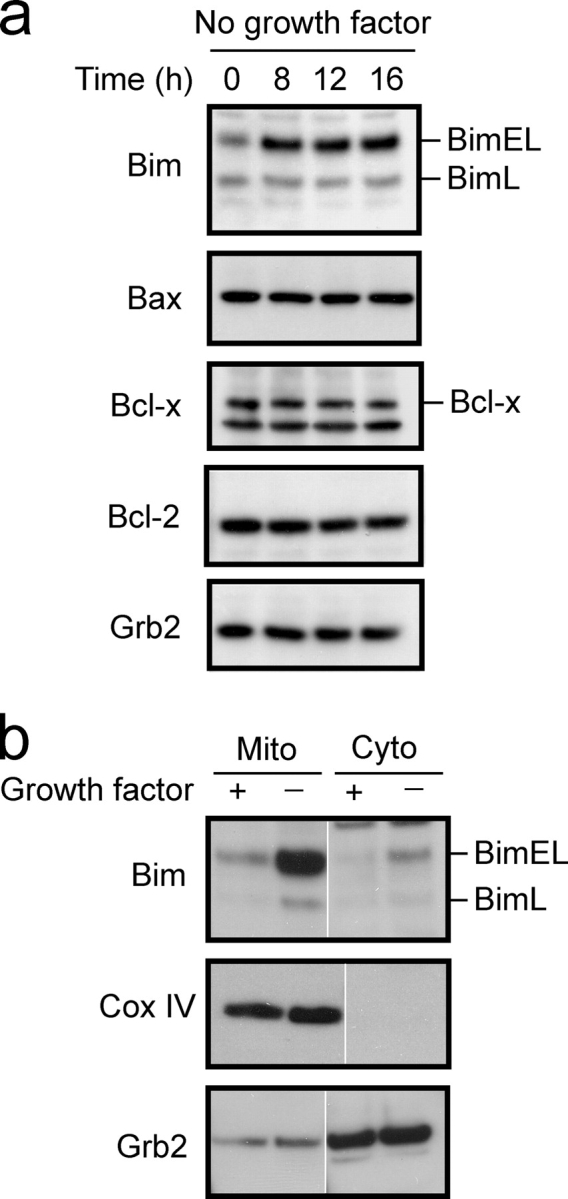
Withdrawal of growth factor from DCs leads to increased Bim expression. (a) Total cell lysates were prepared from uninfected immature D1 DCs grown in the presence or absence of growth factor for the indicated times. Cell lysates were resolved by SDS-PAGE, transferred to PVDF membrane, and immunoblotted with antibodies against the indicated proteins. (b) D1 DCs were grown in the presence or absence of growth factor for 8 h. Cells were subjected to isotonic lysis and mitochondrial (Mito) and cytoplasmic (Cyto) fractions isolated. Proteins were resolved by SDS-PAGE, transferred to PVDF membrane, and expression of the indicated proteins determined by immunoblot. White lines indicate that intervening lanes have been spliced out.
Protection from apoptosis correlates with increased expression of pro-survival Bcl-2 proteins
A number of reports have demonstrated that DC survival is enhanced after activation induced by inflammatory signals, or by T cell–derived factors, such as CD40L or TNF-related activation-induced cytokine (Bjork et al., 1997; Wong and Choi, 1997; Pirtskhalaishvili et al., 2000; Park et al., 2002). Increased expression of Bcl-2 and/or Bcl-xL in these cells might explain these results (Bjork et al., 1997; Wong and Choi, 1997; Pirtskhalaishvili et al., 2000; Park et al., 2002). Therefore, we compared the expression of Bcl-2 family proteins in immature, LPS-stimulated, and MCMV-infected DCs by immunoblot analysis to determine whether alterations in the expression of these proteins might explain the observed inhibition of apoptosis. Stimulation of DCs with LPS resulted in increased expression of the pro-survival Bcl-2 family members Bcl-xL and Bfl-1/A1, whereas expression of Bcl-2 and Bcl-w was not affected (Fig. 4). An increase in the expression of Bim was also observed after LPS stimulation (Fig. 4). The increased expression of pro-survival Bcl-2 family proteins may allow these cells to tolerate the increased expression of Bim (O'Connor et al., 1998). Although MCMV infection of DCs had no effect on the expression of Bcl-xL, an increase in Bfl-1/A1 and Bim protein expression was observed (Fig. 4).
Figure 4.
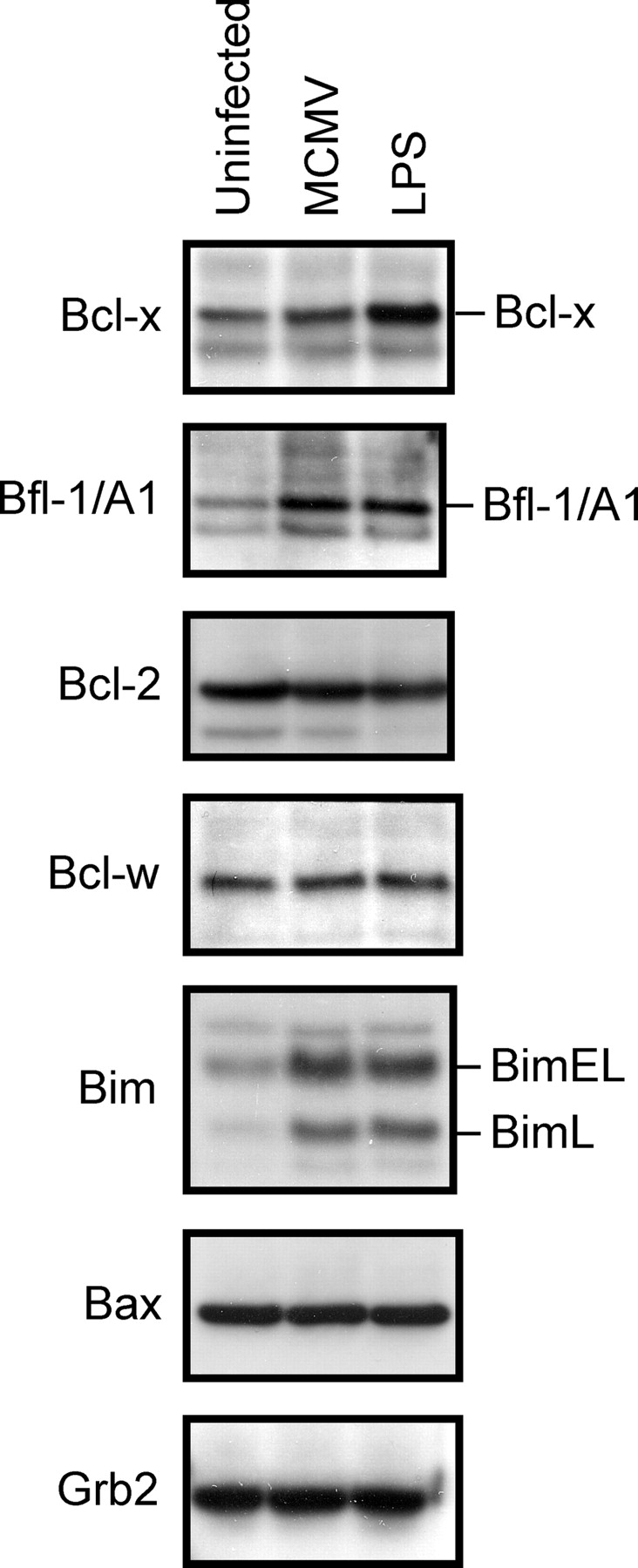
MCMV infection or LPS stimulation of DCs affects the expression of Bcl-2 family members. Total cell lysates were prepared from uninfected, LPS-stimulated, or MCMV-infected D1 DCs, resolved by SDS-PAGE and transferred to PVDF membrane. Membranes were immunoblotted with the indicated antibodies.
Bax is targeted to mitochondria in MCMV-infected DCs
MCMV infection might inhibit apoptosis by preventing the mitochondrial dysfunction normally associated with the apoptotic process. A key event in mitochondrial dysfunction is the translocation of Bax from the cytosol to the mitochondria, where it facilitates the release of pro-apoptotic molecules (Wang, 2001). Inhibition of cell death by Bcl-xL has been associated with its ability to inhibit the formation of Bax clusters at the mitochondrial membrane (Nechushtan et al., 2001). Given the observed increase in the expression of Bcl-xL and/or Bfl-1/A1 in DCs that had become resistant to apoptosis, we investigated the possibility that Bax translocation might be inhibited in these cells. Cellular fractionation and immunoblot analysis were used to monitor the subcellular distribution of Bax in uninfected, LPS-stimulated, or MCMV-infected DCs. As expected the majority of Bax protein localized to the cytoplasmic fraction of immature and LPS-stimulated DCs grown in the presence of growth factor (Fig. 5 a). Removal of the growth factor from immature DCs resulted in translocation of Bax from cytoplasmic to mitochondrial fractions, along with a concomitant increase in the amount of cytochrome c that could be detected in the cytoplasmic fraction (Fig. 5 a). Conversely, growth factor withdrawal from LPS-stimulated DCs did not alter the subcellular localization of Bax nor did it increase cytoplasmic cytochrome c levels. Surprisingly, Bax was constitutively localized to the mitochondrial fraction in MCMV-infected DCs, both in the presence and absence of growth factor. Despite the translocation of Bax to the mitochondrial fraction, cytochrome c levels did not increase in the cytoplasm, suggesting that mitochondrial integrity was not disrupted. Bax was also constitutively associated with mitochondria in Flt3L expanded DCs after MCMV infection (Fig. 5 b). Together, these results indicate that in DCs MCMV inhibits apoptosis after Bax translocation to the mitochondria.
Figure 5.
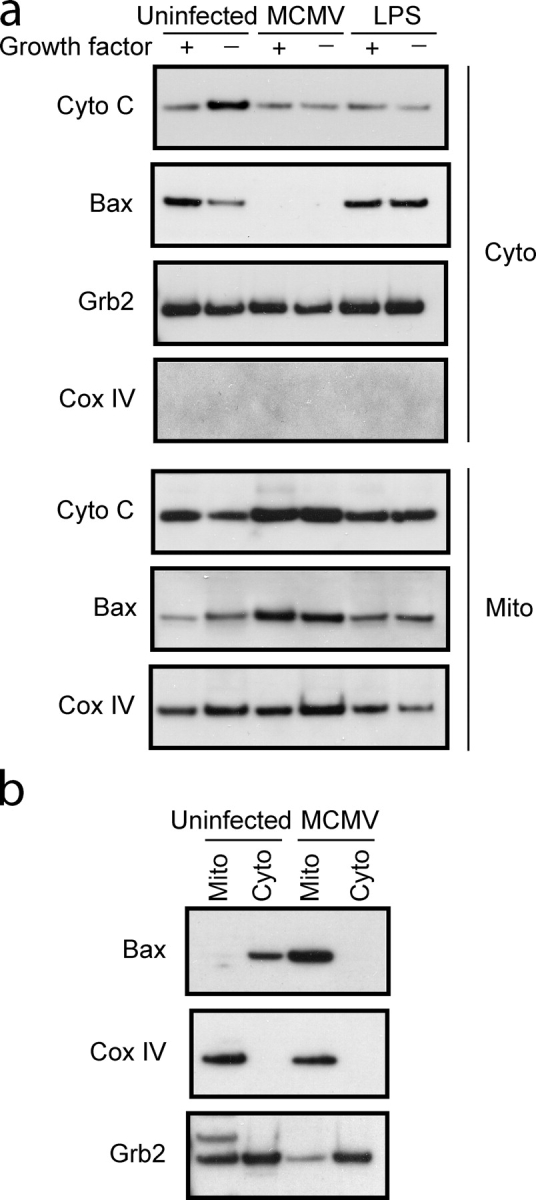
Bax localizes to the mitochondria in MCMV-infected DCs. Mitochondrial (Mito) and cytoplasmic (Cyto) fractions were isolated from control or MCMV-infected (a) D1 DCs or (b) Flt3L expanded DCs. Lysates were separated by SDS-PAGE, transferred to PVDF membrane, and immunoblotted with the indicated antibodies.
Mitochondrial membrane potential (ΔΨm) is maintained in MCMV-infected DCs
The translocation of Bax from the cytosol to the mitochondria occurs at an early stage of the apoptotic pathway (Hsu et al., 1997; Wolter et al., 1997). However, Bax has also been reported to be in peripheral association with the mitochondria in healthy cells (Goping et al., 1998; Desagher et al., 1999). Regardless of its subcellular localization in healthy cells, after an apoptotic stimulus, Bax undergoes a conformational change and integrates into the mitochondrial membrane. Thus, we tested whether the Bax protein localized to the mitochondria of MCMV-infected DCs was merely peripherally associated with mitochondrial membranes. Surprisingly, the Bax protein localized to the mitochondria of MCMV-infected DCs was resistant to alkaline extraction, indicating that in infected DCs Bax integrates into the mitochondrial membrane (Fig. 6 a). Integration of Bax into the mitochondrial membrane is normally associated with the loss of ΔΨm. ΔΨm was examined in control and MCMV-infected DCs using MitoTracker red CMXRos, a dye whose accumulation in mitochondria depends on mitochondrial transmembrane potential (Marzo et al., 1998). Uninfected and MCMV-infected DCs accumulated the MitoTracker dye to a similar extent (Fig. 6 b). The specificity of the MitoTracker staining was confirmed by incubating DCs with the mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP). Exposure of control and MCMV-infected DCs to CCCP resulted in a significant reduction in MitoTracker signal. Together, these results indicate that the mitochondria of MCMV-infected DCs maintain normal membrane potential despite the tight association of Bax with mitochondrial membranes.
Figure 6.


ΔΨm is not altered despite Bax integration to mitochondrial membranes. (a) Mitochondrial fractions prepared from uninfected or MCMV- infected D1 DCs were incubated in 0.1 M Na2CO3, pH 11.5, and centrifuged at 100,000 g for 45 min to yield supernatant (S) and pellet (P) fractions containing alkali-sensitive and -resistant proteins, respectively. Samples were resolved by SDS-PAGE, transferred to PVDF membrane, and immunoblotted with the indicated antibodies. (b) ΔΨm was analyzed in uninfected or MCMV- infected DCs labeled with MitoTracker red CMXRos in the presence or absence of the mitochondrial uncoupler CCCP, as indicated. Dual staining with the nuclear dye Hoechst 33258 allows visualization of all the cells in the field.
Bax oligomerization occurs in the absence of NH2-terminal epitope exposure
In addition to changes in the subcellular localization of Bax, exposure of an occluded NH2-terminal epitope and oligomerization have been correlated with apoptosis. The NH2-terminal conformation of Bax was assessed using the conformation-specific 6A7 antibody. Uninfected or MCMV-infected DCs were grown in the presence or absence of growth factor for 8 h, and cell lysates prepared in the presence of CHAPS detergent. As expected, the 6A7 antibody could not efficiently detect Bax from control DCs (uninfected, plus growth factor), whereas Bax in the “active” conformation was readily detected in DCs undergoing apoptosis (uninfected, no growth factor; Fig. 7 a). In contrast, in MCMV-infected DCs the 6A7 antibody failed to detect Bax, even in the absence of growth factor (Fig. 7 a).
Figure 7.

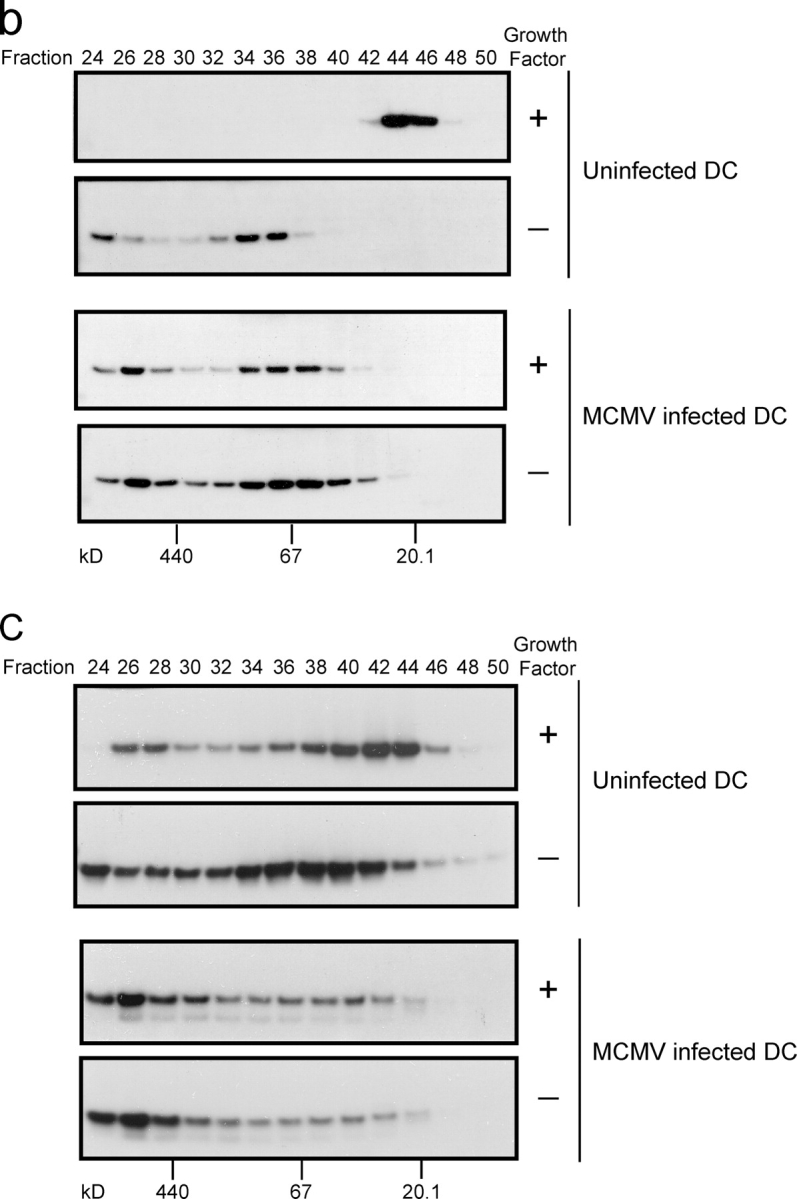
Bax and Bak elute as high molecular weight oligomers in MCMV-infected DCs, but exposure of a Bax NH2-terminal epitope is inhibited. (a) MCMV-infected or uninfected control D1 DCs were grown in the presence or absence of growth factor for 8 h and total cell lysates prepared using 2% CHAPS lysis buffer. Bax protein was immunoprecipitated using the 6A7 conformation-specific antibody. Precipitated proteins were resolved by SDS-PAGE and detected by immunoblot. (b and c) Uninfected or MCMV-infected DCs were grown in the presence or absence of growth factor for 12 h. Mitochondria were isolated by differential centrifugation, treated with 2% CHAPS lysis buffer, and solubilized proteins were separated by gel filtration. The indicated fractions were resolved by SDS-PAGE and immunoblotted with antibodies against (b) Bax and (c) Bak.
Size exclusion chromatography was then used to characterize the oligomerization state of Bax. Mitochondria from control or MCMV-infected DCs, grown in the presence or absence of growth factor, were isolated by differential centrifugation. Mitochondrial proteins were solubilized using 2% CHAPS and the extracts analyzed by gel filtration. In healthy DCs Bax eluted as a monomer, with an apparent molecular mass of ∼20 kD (Fig. 7 b). Removal of growth factor from uninfected DCs resulted in Bax forming complexes with molecular masses >67 kD. Surprisingly, the majority of Bax in MCMV-infected cells was contained in high molecular weight complexes, even before growth factor withdrawal (Fig. 7 b). These results demonstrated that MCMV infection of DCs leads to the oligomerization of Bax at the mitochondria; however, exposure of the NH2-terminal epitope, normally observed during the induction of apoptosis, is inhibited as a result of MCMV infection.
Knockout studies have demonstrated a redundant role for Bax and Bak, with either protein sufficient to induce apoptosis in a variety of cell types (Lindsten et al., 2000). Like Bax, induction of apoptosis results in a conformational change to the NH2 terminus of Bak and its oligomerization. Exposure of the Bak NH2-terminal epitope after MCMV infection could not be assessed because appropriate mouse reagents do not exist. Gel filtration analysis was used to determine the oligomerization status of Bak. In healthy DCs Bak eluted over a broad range (Fig. 7 c), implying that monomeric and oligomeric forms of Bak are present in normal cells, a result consistent with previous reports (Antonsson et al., 2001). After induction of apoptosis in DCs a small change in the elution profile of Bak was observed with a larger proportion of high molecular weight Bak complexes being detected. In MCMV-infected DCs Bak was predominantly contained within high molecular weight complexes even before growth factor withdrawal (Fig. 7 c). Thus, MCMV infection of DCs results in the oligomerization of both Bax and Bak.
MCMV M36, M37, and M45 do not interfere with Bax function
Three MCMV proteins (M36, M37, and M45) have been ascribed possible anti-apoptotic functions (Goldmacher et al., 1999; Brune et al., 2001; Skaletskaya et al., 2001; McCormick et al., 2003), and thus represent potential candidates for the inhibition of Bax function we observed after MCMV infection. To test the relevance of these proteins, we determined their ability to interact with Bax and to prevent apoptosis induced via the mitochondrial pathway. The viral ORFs were transfected into COS-7 cells and interaction with Bax determined by coimmunoprecipitation. Bax did not interact with either M36, M37 or M45 (Fig. 8 a). Furthermore, overexpression of these MCMV proteins did not protect from staurosporine-induced apoptosis (Fig. 8 b). The inability of the viral proteins to interact with Bax and prevent apoptotic cell death was confirmed by transfection studies in murine fibroblasts (not depicted).
Figure 8.
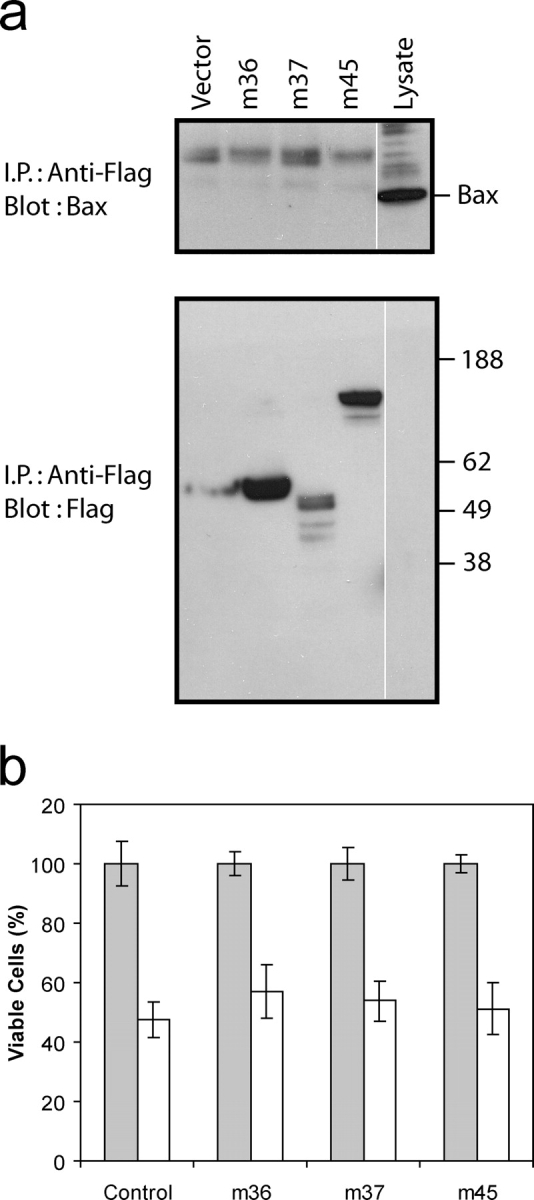
Candidate MCMV anti-apoptotic proteins do not bind Bax or inhibit apoptosis. Flag-tagged M36, M37, and M45 MCMV viral ORFs were transiently transfected into COS cells. (a) Cells were lysed in the presence of 2% CHAPS and Flag-tagged proteins immunoprecipitated. Immunoprecipitated proteins were resolved by SDS-PAGE and immunoblotted with anti-Bax specific antibody. The expression of the Flag-tagged constructs was confirmed by immunoblotting of total cell lysates. White lines indicate that intervening lanes have been spliced out. (b) Cells were grown in the presence (shaded bars) or absence (white bars) of staurosporine (250 nM) for 18 h and the number of viable cells counted. Data represent the mean ± SD, n = 4.
Discussion
Interference with DC function is a strategy used by a number of viruses to disarm the host immune response. Here, we have investigated the effects of MCMV on the viability of DCs, a primary target of infection in vivo (Andrews et al., 2001). MCMV infection protects DCs from apoptosis induced by growth factor withdrawal. Protection from apoptosis is also observed in DCs after bacterial encounter. Although the PI3 kinase pathway is essential for the protection induced by bacterial encounter, it is not required for the protection induced by MCMV infection suggesting that signal transduction pathways other than those affected during bacterial infection are targeted by the virus. Characterization of the molecular events associated with DC apoptosis induced by growth factor withdrawal indicated that Bim expression is increased, prompting us to analyze the expression of Bcl-2 family proteins in LPS-stimulated and MCMV-infected DCs. Our finding that expression of Bcl-xL and Bfl-1/A1 was increased after LPS stimulation provides a mechanism to explain the anti-apoptotic effects of LPS stimulation. Typically, increased expression of pro-survival Bcl-2 proteins inhibits the translocation of Bax to the mitochondria, thus, preventing mitochondrial dysfunction (Marsden and Strasser, 2003). Indeed, no significant translocation of Bax to mitochondria was observed in LPS-stimulated DCs after growth factor withdrawal. Similar observations have recently been reported using GM-CSF expanded DCs, where alterations in Bcl-2 protein expression were proposed to act as a molecular timer regulating DC life span (Hou and Van Parijs, 2004). In contrast, despite the fact that Bfl-1/A1 expression is increased in MCMV-infected DCs, in these cells Bax predominantly localized to mitochondria. These findings provide compelling evidence that different molecular mechanisms protect DCs from apoptosis after LPS stimulation and MCMV infection.
The significance of Bax and Bak to the initiation of apoptosis is underscored by the finding that mice deficient for both proteins display multiple developmental defects (Lindsten et al., 2000). The key event in apoptosis mediated through mitochondrial dysfunction is a conformational change in Bax and Bak and the formation of high molecular weight complexes (Hsu et al., 1997; Nechushtan et al., 2001; Sundararajan and White, 2001). After MCMV infection, Bax was found in tight association with the mitochondrial membrane, and both Bax and Bak were in an oligomerized form. Despite both Bax and Bak displaying characteristics typically observed during apoptosis, ΔΨm was not affected, nor was cytochrome c released into the cytoplasm. Importantly, a conformational change which exposes a constitutively occluded epitope at the NH2 terminus of Bax, and which normally occurs during apoptosis, was not observed in MCMV-infected DCs. Translocation of Bax to mitochondria and formation of Bax complexes without a loss of mitochondrial integrity were reported in a taxol-resistant cell line (Makin et al., 2001). The percentage of taxol-resistant cells which had oligomerized Bax, and how much Bax protein was in this conformation was difficult to determine, and indeed the majority of Bax in these cells was monomeric (Makin et al., 2001). In our MCMV-infected DC cultures all the Bax protein was oligomerized, despite the complete resistance to apoptosis. The exposure of the constitutively occluded NH2-terminal epitope of Bax observed in taxol-resistant cells (Makin et al., 2001), did not occur in MCMV-infected DCs. Our findings raise the interesting possibility that MCMV sequesters Bax and Bak in oligomeric complexes which are structurally distinct from those required to induce apoptosis. Alternatively, additional proteins may be required to cooperate with Bax/Bak oligomers to induce mitochondrial permeabilization.
Thus, MCMV infection reveals the existence of a checkpoint in the Bcl-2–regulated apoptotic pathway after translocation/oligomerization of Bax and Bak. This checkpoint is specifically targeted by MCMV to inhibit apoptosis in DCs.
Given the importance of Bax/Bak in the apoptotic process it is not surprising that a number of viruses have evolved strategies to interfere with the function of these proteins. Several viruses encode Bcl-2 homologues that appear to interfere with apoptosis by directly binding to Bax/Bak and antagonizing their function (Cuconati et al., 2002). The best characterized of these is the E1B 19K protein of adenovirus (Perez and White, 2000). CMVs do not encode sequence homologues of Bcl-2, suggesting that this family of viruses has evolved alternative strategies to interfere with Bax/Bak functions. The product of the HCMV gene UL37 encodes a mitochondria-localized death suppressor termed vMIA (Goldmacher et al., 1999; Goldmacher, 2002). Recent reports have demonstrated that vMIA functions by sequestering Bax at the mitochondrial membrane in an inactive complex (Arnoult et al., 2004; Poncet et al., 2004). Although M37, the MCMV homologue of UL37, would seem an ideal candidate for mediating the observed inhibition of Bax function in MCMV-infected DCs, it should be noted that the homology between M37 and UL37 is restricted to the region encoding UL37 exon 3. Both anti-apoptotic and mitochondrial localizing functions of UL37 have been mapped to exon 1, a region lacking obvious sequence homology in the MCMV genome (Rawlinson et al., 1996; Goldmacher, 2002). Indeed, we were unable to demonstrate an association between M37 and Bax, and overexpression of M37 in COS-7 or NIH-3T3 cells did not inhibit apoptosis induced by staurosporine treatment. Similarly, coimmunoprecipitation experiments showed that Bax does not associate with either M36 or M45, the other known anti-apoptotic proteins encoded by MCMV.
Because MCMV infection of DCs results in changes to Bax similar to those induced by vMIA, we hypothesize that a functional homologue of the HCMV vMIA is encoded by MCMV. Identification of this protein is the focus of our ongoing studies. Unlike vMIA, which does not bind Bak or inhibit its function (Arnoult et al., 2004), infection of DCs with MCMV resulted in the oligomerization of both Bax and Bak. A possible explanation for this observation is that the putative MCMV vMIA homologue can interact with both Bax and Bak; alternatively, MCMV might encode an additional proteins which specifically interferes with Bak function. Finally, because activation of Bak in some cells has been shown to require Bax activation (Arnoult et al., 2004), an alternative possibility is that interference with Bax function is sufficient to prevent apoptosis in DCs.
In conclusion, we have shown that CMV interferes with apoptosis in DCs, cells that are major targets of infection and viral replication both in vitro and in vivo. The ability of MCMV to increase the life span of DCs confers the virus a significant survival advantage, because in these cells MCMV replicates to high titres for long periods of time (Andrews et al., 2001). A further exciting outcome of our studies is the clear definition of a checkpoint in the Bcl-2–regulated apoptotic pathway after Bax/Bak oligomerization, but before mitochondrial disruption. Our results provide unequivocal evidence that translocation, integration, and oligomerization of Bax at the mitochondrial membrane, generally thought to represent a terminal commitment to cell death, are in fact not sufficient to force cells to die, because in MCMV-infected DCs these events occur, but the cells remain viable. Analysis of viral infections has provided many novel and important clues about complex cellular processes and pathways. Further elucidation of the mechanism used by MCMV to interfere with mitochondrial functions will undoubtedly contribute important insights into the regulation of apoptosis.
Materials and methods
Cell lines and reagents
Mouse embryonic fibroblasts were cultured in minimal essential medium (GIBCO BRL) supplemented with neonatal calf serum (GIBCO BRL). D1 DCs were derived from murine splenic DCs and maintained in vitro as growth factor–dependent immature DCs (Winzler et al., 1997). D1 DCs were cultured in complete Iscove's modified Dulbecco's medium (IMDM; Sigma-Aldrich) containing 10% heat inactivated FBS (GIBCO BRL); 100 IU/ml penicillin and 100 μg/ml streptomycin (Amersham Biosciences and Upjohn); 2 mM l-glutamine and 50 μM 2-mercaptoethanol and supplemented with 30% CM containing 10–20 ng/ml GM-CSF (Winzler et al., 1997). Flt3L expanded DCs were generated from the bone marrow of C57BL/6 mice as described previously (Brasel et al., 2000). COS-7 cells were cultured in DME (GIBCO BRL), 10% FBS, and transfected by the standard DEAE-dextran method with 15 μg of each relevant construct. The MCMV viral ORFs m36, m37, and m45 were cloned into pCDNA3 in frame with a COOH terminus FLAG.
Preparation and purification of MCMV stocks
Mouse embryonic fibroblasts were infected with MCMV-K181-Perth salivary gland passaged stocks with an MOI of 0.05 for 1 h at 37°C. Cells were monitored daily for evidence of cytopathic effect and the supernatant removed when 100% cpe was observed. Supernatants were filtered through 0.45-μm filters and centrifuged at 35,000 g for 2 h at 4°C. The supernatant was removed, and the viral pellet resuspended in ice-cold medium, aliquoted, and stored at −180°C. Viral titers were determined on embryonic fibroblasts as described previously (Farrell et al., 1997).
Induction and detection of apoptosis
Apoptosis was induced in D1 or Flt3L expanded DCs by washing cells twice in serum-free medium. D1 DCs were reseeded in complete IMDM without R1 CM, whereas Flt3L DCs were reseeded in IMDM/1% FCS. PI3 kinase and MEK1/2 activity was inhibited with LY294002 (Calbiochem) and U0126 (Cell Signaling Technology), respectively. Apoptosis was assessed by staining cells with annexin V-FLUOS (Roche) according to the manufacturer's recommendation and cells analyzed on a FACSCalibur (Becton Dickinson). Caspase activity was determined using the caspase-3 substrate Ac-DEVD-pNA (Calbiochem) according to the manufacturer's instructions. Apoptosis was induced in transfected COS-7 cells by staurosporine treatment (250 nm, 18 h). Viability was assessed by trypan blue exclusion.
Antibodies
The following antibodies were used for this work: anti-Grb2 (clone C-23) obtained from Santa Cruz Biotechnology; anti–Bfl-1/A1 (clone 78616.11) obtained from R&D Systems; anti–Cox IV (clone 20E8) obtained from Molecular Probes; anti–cytochrome c (7H8.2C12), anti–Bcl-x (clone 44), and anti-Bax (6A7) obtained from BD Biosciences; anti-Bax (clone 5B7) obtained from Sigma-Aldrich; and anti-Bax and anti-Bak obtained from Upstate Biotechnology. The anti–Bcl-2 (clone 3F11), anti-Bim (clone 14A8), and anti–Bcl-w antibodies were gifts from D. Wang and L. O'Reilly (Walter and Eliza Hall Institute, Melbourne, Australia). Anti–mouse and anti–rabbit peroxidase conjugates were purchased from Amersham Biosciences; the anti–rat and anti–Armenian hamster peroxidase conjugates were obtained from Jackson ImmunoResearch Laboratories.
Preparation of cell lysates and subcellular fractionation
Total cell lysates were prepared by resuspending cells in Triton X-100 lysis buffer (150 mM NaCl, 10 mM Tris, pH 7.4, 1% Triton X-100, 1× complete protease inhibitors [Roche]) and incubating on ice for 30 min. Nuclei and cell debris was removed by centrifugation (1,000 g for 5 min) and protein concentration determined using a Micro BCA assay kit (Pierce Chemical Co.). Cytoplasmic and mitochondrial fractions were prepared by resuspending 107 cells in 500 μl isotonic lysis buffer (210 mM mannitol, 70 mM sucrose, 20 mM Hepes, pH 7.2, 10 mM KCl, 1.5 mM MgCl2, 1 mM EGTA, 1× complete protease inhibitors) and passing the sample through a G27 needle 30 times. The resulting sample was centrifuged at 1,500 g for 5 min to remove nuclei and unbroken cells. The supernatant was subsequently centrifuged at 14,000 g for 10 min. The pellet, containing mitochondria was resuspended in Triton X-100 lysis buffer, while the supernatant was centrifuged at 100,000 g for 45 min at 4°C with the resulting supernatant used as the cytoplasmic fraction.
Alkali extraction of mitochondria
Mitochondria from healthy or apoptotic cells were isolated as described above and resuspended in freshly prepared 0.1 M NaCO3, pH 11.5, and incubated for 20 min on ice. Mitochondrial membranes were pelleted by centrifugation (100,000 g for 2 h at 4°C). The resulting pellet (alkali–resistant fraction) was resuspended in Triton X-100 lysis buffer, while the supernatant contained alkali–sensitive proteins.
Immunoblotting
Protein samples were resolved on 4–12% Bis-Tris gradient gels (Novex) and transferred to PVDF plus membrane (Osmonics). Immunodetection was performed using the appropriate peroxidase-conjugated secondary antibody and ECL detection reagent (Amersham Biosciences). Equal loading of lanes was confirmed by Grb2 or Cox IV immunoblot.
Confocal microscopy
Analysis of ΔΨm was performed by resuspending cells (3 x105 cells/ml) in medium containing 300 nM MitoTracker red CMXRos in the presence or absence of 50 μM CCCP for 20 min at 37°C. Cells were then washed in medium twice and plated onto silane-(ICN Biomedicals)-coated coverslips and incubated at 37°C for 20 min. Cells were fixed in 3.7% PFA (37°C for 15 min), and washed twice in PBS. Cells were permeabilized with PBS/0.2% Triton X-100 and stained with 2 μM Hoechst 33258 to visualize nuclei and mounted. All analysis was performed on an MRC 1000/1024 UV laser scanning confocal microscope (Bio-Rad Laboratories) with a 40× objective (NA 1.3; Nikon) and images saved as Biorad.PIC files. Subsequent analysis was performed using Adobe Photoshop v. 7.0.
Gel filtration
Gel filtration was performed at 4°C using Toyopearl HW-65F resin (Tosoh Bioscience) equilibrated in column buffer (25 mM Hepes-NaOH, pH 7.5, 300 mM NaCl, 200 μM DTT, 2% CHAPS). The column was calibrated using ferritin (440 kD), BSA (67 kD), and trypsin inhibitor (20.1 kD). The void volume of the column was determined using blue dextran. Mitochondria were isolated by differential centrifugation as outlined above and solubilized using CHAPS lysis buffer (150 mM NaCl, 25 mM Hepes, pH 7.5, 2% CHAPS, 1× complete protease inhibitors). The sample was then centrifuged at 21,000 g for 20 min to remove insoluble material. Samples of 500 μl were loaded onto the column and the eluate was monitored at 280 nm. Fractions of 25 drops (680 μl) were collected and aliquots analyzed by immunoblot.
Acknowledgments
The authors are grateful to Dr. M. Wikstrom, and Drs. K. Heel and P. Rigby from the Biomedical Imaging and Analysis Facility. The authors are grateful to Dr. A. Strasser, Dr. D. Huang, and Dr. L. O'Reilly for providing reagents and helpful insights, and Dr. G. Stewart for technical assistance with the gel filtration analysis.
This project was supported by grants from the National Health and Medical Research Council of Australia and the Wellcome Trust. M.A. Degli-Esposti is supported by a Wellcome Trust Overseas Senior Research Fellowship in Biomedical Science.
Abbreviations used in this paper: BH, Bcl-2 homology; CCCP, carbonyl cyanide m-chlorophenylhydrazone; CM, conditioned medium; CMV, cytomegalovirus; DC, dendritic cell; ΔΨm, mitochondrial membrane potential; ERK, extracellular signal regulated kinase; IMDM, Iscove's modified Dulbecco's medium; MCMV, murine CMV; MOI, multiplicity of infection; PI3, phosphatidylinositide-3-OH kinase.
References
- Andrews, D.M., C.E. Andoniou, F. Granucci, P. Ricciardi-Castagnoli, and M.A. Degli-Esposti. 2001. Infection of dendritic cells by murine cytomegalovirus induces functional paralysis. Nat. Immunol. 2:1077–1084. [DOI] [PubMed] [Google Scholar]
- Antonsson, B., S. Montessuit, B. Sanchez, and J.C. Martinou. 2001. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J. Biol. Chem. 276:11615–11623. [DOI] [PubMed] [Google Scholar]
- Ardeshna, K.M., A.R. Pizzey, S. Devereaux, and A. Khwaja. 2000. The PI3 kinase, p38 SAP kinase, and NF-kappa B signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood. 96:1039–1046. [PubMed] [Google Scholar]
- Arnoult, D., L.M. Bartle, A. Skaletskaya, D. Poncet, N. Zamzami, P.U. Park, J. Sharpe, R.J. Youle, and V.S. Goldmacher. 2004. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. USA. 101:7988–7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebecque, Y.T. Liu, B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811. [DOI] [PubMed] [Google Scholar]
- Bjork, P., J. Banchereau, and L. Flores-Romo. 1997. CD40 ligation counteracts Fas-induced apoptosis of human dendritic cells. Int. Immunol. 9:365–372. [DOI] [PubMed] [Google Scholar]
- Bouillet, P., D. Metcalf, D.C. Huang, D.M. Tarlinton, T.W. Kay, F. Kontgen, J.M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 286:1735–1738. [DOI] [PubMed] [Google Scholar]
- Brasel, K., T. De Smedt, J.L. Smith, and C.R. Maliszewski. 2000. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood. 96:3029–3039. [PubMed] [Google Scholar]
- Brune, W., C. Menard, J. Heesemann, and U.H. Koszinowski. 2001. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science. 291:303–305. [DOI] [PubMed] [Google Scholar]
- Cory, S., and J.M. Adams. 2002. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer. 2:647–656. [DOI] [PubMed] [Google Scholar]
- Cuconati, A., K. Degenhardt, R. Sundararajan, A. Anschel, and E. White. 2002. Bak and Bax function to limit adenovirus replication through apoptosis induction. J. Virol. 76:4547–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desagher, S., A. Osen-Sand, A. Nichols, R. Eskes, S. Montessuit, S. Lauper, K. Maundrell, B. Antonsson, and J.C. Martinou. 1999. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144:891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell, H.E., H. Vally, D.M. Lynch, P. Fleming, G.R. Shellam, A.A. Scalzo, and N.J. Davis-Poynter. 1997. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature. 386:510–514. [DOI] [PubMed] [Google Scholar]
- Gilmore, A.P., A.D. Metcalfe, L.H. Romer, and C.H. Streuli. 2000. Integrin-mediated survival signals regulate the apoptotic function of Bax through its conformation and subcellular localization. J. Cell Biol. 149:431–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmacher, V.S. 2002. vMIA, a viral inhibitor of apoptosis targeting mitochondria. Biochimie. 84:177–185. [DOI] [PubMed] [Google Scholar]
- Goldmacher, V.S., L.M. Bartle, A. Skaletskaya, C.A. Dionne, N.L. Kedersha, C.A. Vater, J. Han, R.J. Lutz, S. Watanabe, E.D. McFarland, et al. 1999. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA. 96:12536–12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goping, I.S., A. Gross, J.N. Lavoie, M. Nguyen, R. Jemmerson, K. Roth, S.J. Korsmeyer, and G.C. Shore. 1998. Regulated targeting of bax to mitochondria. J. Cell Biol. 143:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, G.J., L. Dubrez, C.P. Morgan, N.A. Jones, J. Whitehouse, B.M. Corfe, C. Dive, and J.A. Hickman. 1999. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144:903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, A., J. Jockel, M.C. Wei, and S.J. Korsmeyer. 1998. Enforced dimerization of bax results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 17:3878–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, A., X.M. Yin, K. Wang, M.C. Wei, J. Jockel, C. Millman, H. Erdjument-Bromage, P. Tempst, and S.J. Korsmeyer. 1999. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-X-L prevents this release but not tumor necrosis factor-R1/Fas death. J. Biol. Chem. 274:1156–1163. [DOI] [PubMed] [Google Scholar]
- Hou, W.S., and L. Van Parijs. 2004. A Bcl-2-dependent molecular timer regulates the lifespan and immunogenicity of dendritic cells. Nat. Immunol. 5:583–589. [DOI] [PubMed] [Google Scholar]
- Hsu, Y.T., K.G. Wolter, and R.J. Youle. 1997. Cytosol-to-membrane redistribution of bax and bcl-x-l during apoptosis. Proc. Natl. Acad. Sci. USA. 94:3668–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi, I., Y. Shinmura, R.Y. Li, S. Aibamasago, S. Baba, K. Miura, and Y. Tsutsui. 1998. Murine cytomegalovirus induces apoptosis in non-infected cells of the developing mouse brain and blocks apoptosis in primary neuronal culture. Acta Neuropathol. (Berl.). 96:239–247. [DOI] [PubMed] [Google Scholar]
- Kovacs, A., M.L. Weber, L.J. Burns, H.S. Jacob, and G.M. Vercellotti. 1996. Cytoplasmic sequestration of p53 in cytomegalovirus-infected human endothelial cells. Am. J. Pathol. 149:1531–1539. [PMC free article] [PubMed] [Google Scholar]
- Lindsten, T., A.J. Ross, A. King, W.X. Zong, J.C. Rathmell, H.A. Shiels, E. Ulrich, K.G. Waymire, P. Mahar, K. Frauwirth, et al. 2000. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol. Cell. 6:1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makin, G.W.J., B.M. Corfe, G.J. Griffiths, A. Thistlethwaite, J.A. Hickman, and C. Dive. 2001. Damage-induced Bax N-terminal change, translocation to mitochondria and formation of Bax dimers/complexes occur regardless of cell fate. EMBO J. 20:6306–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden, V.S., and A. Strasser. 2003. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu. Rev. Immunol. 21:71–105. [DOI] [PubMed] [Google Scholar]
- Marzo, I., C. Brenner, N. Zamzami, J.M. Jurgensmeier, S.A. Susin, H.L. Vieira, M.C. Prevost, Z. Xie, S. Matsuyama, J.C. Reed, and G. Kroemer. 1998. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 281:2027–2031. [DOI] [PubMed] [Google Scholar]
- McCormick, A.L., A. Skaletskaya, P.A. Barry, E.S. Mocarski, and V.S. Goldmacher. 2003. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology. 316:221–233. [DOI] [PubMed] [Google Scholar]
- Mocarski, E.S. 1996. Cytomegaloviruses and their replication. Fields Virology. Vol. 2. B.N. Fields, D.M. Knipe, and P.M. Howley, editors. Lippincott-Raven, Philadelphia. 2447–2492.
- Nechushtan, A., C.L. Smith, I. Lamensdorf, S.H. Yoon, and R.J. Youle. 2001. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 153:1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, L., A. Strasser, L.A. O'Reilly, G. Hausmann, J.M. Adams, S. Cory, and D.C. Huang. 1998. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17:384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, Y., S.W. Lee, and Y.C. Sung. 2002. Cutting Edge: CpG DNA inhibits dendritic cell apoptosis by up-regulating cellular inhibitor of apoptosis proteins through the phosphatidylinositide-3′-OH kinase pathway. J. Immunol. 168:5–8. [DOI] [PubMed] [Google Scholar]
- Perez, D., and E. White. 2000. TNF-alpha signals apoptosis through a bid-dependent conformational change in Bax that is inhibited by E1B 19K. Mol. Cell. 6:53–63. [PubMed] [Google Scholar]
- Pirtskhalaishvili, G., G.V. Shurin, C. Esche, Q. Cai, R.R. Salup, S.N. Bykovskaia, M.T. Lotze, and M.R. Shurin. 2000. Cytokine-mediated protection of human dendritic cells from prostate cancer-induced apoptosis is regulated by the Bcl-2 family of proteins. Br. J. Cancer. 83:506–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poncet, D., N. Larochette, A.L. Pauleau, P. Boya, A.A. Jalil, P.F. Cartron, F. Vallette, C. Schnebelen, L.M. Bartle, A. Skaletskaya, et al. 2004. An anti-apoptotic viral protein that recruits Bax to mitochondria. J. Biol. Chem. 279:22605–22614. [DOI] [PubMed] [Google Scholar]
- Putcha, G.V., K.L. Moulder, J.P. Golden, P. Bouillet, J.A. Adams, A. Strasser, and E.M. Johnson. 2001. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 29:615–628. [DOI] [PubMed] [Google Scholar]
- Puthalakath, H., and A. Strasser. 2002. Keeping killers on a tight leash: transcriptional and posttranslational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 9:505–512. [DOI] [PubMed] [Google Scholar]
- Puthalakath, H., D.C.S. Huang, L.A. O'Reilly, S.M. King, and A. Strasser. 1999. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell. 3:287–296. [DOI] [PubMed] [Google Scholar]
- Rawlinson, W.D., H.E. Farrell, and B.G. Barrell. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 70:8833–8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescigno, M., M. Martino, C.L. Sutherland, M.R. Gold, and P. Ricciardi-Castagnoli. 1998. Dendritic cell survival and maturation are regulated by different signaling pathways. J. Exp. Med. 188:2175–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulston, A., R.C. Marcellus, and P.E. Branton. 1999. Viruses and apoptosis. Annu. Rev. Microbiol. 53:577–628. [DOI] [PubMed] [Google Scholar]
- Shi, Y. 2002. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell. 9:459–470. [DOI] [PubMed] [Google Scholar]
- Shinjyo, T., R. Kuribara, T. Inukai, H. Hosoi, T. Kinoshita, A. Miyajima, P.J. Houghton, A.T. Look, K. Ozawa, and T. Inaba. 2001. Downregulation of Bim, a proapoptotic relative of Bcl-2, is a pivotal step in cytokine-initiated survival signaling in murine hematopoietic progenitors. Mol. Cell. Biol. 21:854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletskaya, A., L.M. Bartle, T. Chittenden, A.L. McCormick, E.S. Mocarski, and V.S. Goldmacher. 2001. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA. 98:7829–7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan, R., and E. White. 2001. E1B 19K blocks Bax oligomerization and tumor necrosis factor alpha-mediated apoptosis. J. Virol. 75:7506–7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M., R.J. Youle, and N. Tjandra. 2000. Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell. 103:645–654. [DOI] [PubMed] [Google Scholar]
- Tortorella, D., B.E. Gewurz, M.H. Furman, D.J. Schust, and H.L. Ploegh. 2000. Viral subversion of the immune system. Annu. Rev. Immunol. 18:861–926. [DOI] [PubMed] [Google Scholar]
- Valentijn, A.J., A.D. Metcalfe, J. Kott, C.H. Streuli, and A.P. Gilmore. 2003. Spatial and temporal changes in Bax subcellular localization during anoikis. J. Cell Biol. 162:599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villunger, A., L.A. O'Reilly, N. Holler, J. Adams, and A. Strasser. 2000. Fas ligand, Bcl-2, granulocyte colony-stimulating factor, and p38 mitogen-activated protein kinase: regulators of distinct cell death and survival pathways in granulocytes. J. Exp. Med. 192:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., P.H. Marker, J.D. Belcher, D.E.L. Wilcken, L.J. Burns, G.M. Vercellotti, and X.L. Wang. 2000. Human cytomegalovirus immediate early proteins upregulate endothelial p53 function. FEBS Lett. 474:213–216. [DOI] [PubMed] [Google Scholar]
- Wang, X. 2001. The expanding role of mitochondria in apoptosis. Genes Dev. 15:2922–2933. [PubMed] [Google Scholar]
- Winzler, C., P. Rovere, M. Rescigno, F. Granucci, G. Penna, L. Adorini, V.S. Zimmermann, J. Davoust, and P. Ricciardi-Castagnoli. 1997. Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J. Exp. Med. 185:317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter, K.G., Y.T. Hsu, C.L. Smith, A. Nechushtan, X.G. Xi, and R.J. Youle. 1997. Movement of bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 139:1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, B., and Y. Choi. 1997. Pathways leading to cell death in T cells. Curr. Opin. Immunol. 9:358–364. [DOI] [PubMed] [Google Scholar]
- Xu, X.N., G.R. Screaton, and A.J. McMichael. 2001. Virus infections: escape, resistance, and counterattack. Immunity. 15:867–870. [DOI] [PubMed] [Google Scholar]