

Abstract
Induction of molecular chaperones is the characteristic protective response to environmental stress, and is regulated by a transcriptional program that depends on heat shock factor 1 (HSF1), which is normally under negative regulatory control by molecular chaperones Hsp70 and Hsp90. In metazoan species, the chaperone system also provides protection against apoptosis. We demonstrate that the dual function co-chaperone/ubiquitin ligase CHIP (C-terminus of Hsp70-interacting protein) regulates activation of the stress-chaperone response through induced trimerization and transcriptional activation of HSF1, and is required for protection against stress-induced apoptosis in murine fibroblasts. The consequences of this function are demonstrated by the phenotype of mice lacking CHIP, which develop normally but are temperature-sensitive and develop apoptosis in multiple organs after environmental challenge. CHIP exerts a central and unique role in tuning the response to stress at multiple levels by regulation of protein quality control and transcriptional activation of stress response signaling.
Keywords: apoptosis/chaperone/proteasome/stress response/ubiquitin
Introduction
Protection against environmental challenge is a universal necessity for free-living organisms, and conserved networks exist to detect and respond to stress. Induction of the cytosolic molecular chaperones (including heat shock proteins Hsp70 and Hsp90) is among the most evident response to environmental challenge, and these proteins can account for several percent of all proteins within a cell in times of stress. Although originally appreciated as a mechanism for protection against thermal stress, the so-called heat shock response is activated by a range of other stressors (including osmotic changes, ischemia and aging) all of which have the accumulation of misfolded proteins as a central feature. The cytosolic chaperones buffer misfolded proteins and aid their refolding. In addition, functions distinct from their chaperone activity have emerged. Both Hsp70 and Hsp90 assist in the delivery of fatally damaged proteins to the ubiquitin–proteasome protein degradation machinery and modulate the apoptotic response (Schneider et al., 1996; Bercovich et al., 1997; Gabai et al., 1997; Mosser et al., 1997; Beere and Green, 2001), so that the ultimate effect is to confer a protected environment within the cell until non-native proteins can be folded or cleared.
Both the activation and suppression of this pathway occur at the transcriptional level through regulation of heat shock factor 1 (HSF1), which binds to heat shock elements (HSEs) located in the promoters of genes in this network (Morimoto, 1998). Activation of HSF1 requires trimerization, phosphorylation and, in some cases, subcellular relocalization. Several molecular chaperones, including Hsp70, Hsp90 and HDJ1 (Hsp40), play a role in the attenuation phase of HSF1 activation (Rabindran et al., 1994; Shi et al., 1998; Zou et al., 1998). In particular, Hsp70 forms a stable complex with the transactivation domain of HSF1, and thus may attenuate HSF1 activity, in part by preventing recruitment of the general transcriptional machinery and also by stabilizing the monomeric conformation (Shi et al., 1998). Attenuation of HSF1 activity by stress-inducible chaperones provides a means for autoregulation of this pathway, which is under tight control at multiple levels (Zuo et al., 1995).
The activity of Hsp70 is determined by its interactions with accessory proteins called co-chaperones [reviewed in (Luders et al., 1998)]. Together, these co-chaperones regulate a reaction that requires iterative cycles of ATP binding, hydrolysis and nucleotide exchange, which in turn control substrate affinity and folding activity. One such co-chaperone, CHIP (C-terminus of Hsp70-interacting protein), interacts with Hsp70 (and also Hsp90) via three tandem tetratricopeptide repeat (TPR) motifs. CHIP, which is highly conserved in metazoan species, regulates the Hsp70 folding cycle by attenuating Hsp70 ATPase activity and substrate affinity and therefore reducing the rate of refolding measured in vitro (Ballinger et al., 1999). In addition to its N-terminus TPR motifs, CHIP also contains a U-box at its C-terminus (Aravind and Koonin, 2000); this RING finger-like domain has ubiquitin ligase activity, and CHIP facilitates the ubiquitination and subsequent proteasome-dependent degradation of several chaperone substrates (Connell et al., 2001; Jiang et al., 2001; Meacham et al., 2001). Thus, CHIP provides a direct link between the chaperone and proteasome systems, and is postulated to assist in regulating the cellular balance between folding and degradation (McClellan and Frydman, 2001).
Although the biochemical effects of co-chaperones are well characterized, there are surprisingly few data on which to predict the ultimate cellular and physiologic consequences of their activities in multicellular organisms. Overexpression of Hsp70 itself leads to protection against stress in a variety of circumstances (Hutter et al., 1996). However, the functional redundancy of different Hsp70 isoforms has made loss-of-function studies less revealing (Dix et al., 1996; Huang et al., 2001). Similarly, with the exception of BAG-1 [which has antiapoptotic effects that are not clearly linked to its effect on chaperone function (Takayama et al., 1995)] we know little about the cellular effects of co-chaperones (and nothing about their physiologic roles) in mammalian systems.
Results
CHIP increases Hsp70 expression by activation of transcription factor HSF1
We examined the cellular functions of one such co-chaperone, CHIP, utilizing an adenoviral approach to overexpress CHIP in COS7 cells, which express CHIP endogenously at low but detectable levels. When protein extracts were analyzed by Coomassie Blue staining, we noted that overexpression of CHIP elevated the levels of a highly inducible 70 kDa protein (Figure 1A). Based on the size and abundance of this protein, we entertained the possibility that CHIP could regulate the expression of stress-responsive chaperones. Indeed, western blot analysis demonstrated that several chaperones were regulated by CHIP, with the effects on Hsp70 being most marked (Figure 1B). CHIP’s induction of Hsp70 was comparable to and not additive with the effects of heat shock (Figure 1C), making it likely that CHIP and heat shock activate Hsp70 through the same pathway. This unique observation of co-chaperone-dependent Hsp70 expression led us to consider its mechanisms and functional consequences in more detail.

Fig. 1. Activation of HSF1 by CHIP. (A) Coomassie Blue gel of lysates from COS7 cells infected with an adenovirus expressing CHIP demonstrates increased expression of a 70-kDa protein compared with cells infected with a control adenovirus. (B) Western blot analysis indicates that expression of the chaperones Hsp27, Hsp90α and especially Hsp70 is increased by CHIP. (C) COS7 cells infected for 24 h with an adenovirus expressing CHIP or a control adenovirus were subjected to heat shock at 42°C for the indicated times. Hsp70 induction by CHIP was comparable to, and not additive with, that induced by heat shock. (D) Hsp70 promoter:reporter constructs containing (+HSE) or lacking (–HSE) the HSF1 response element were transiently co-transfected with CHIP expression vectors and reporter activity was determined 48 h after transfection, with or without heat shock (42°C). CHIP transactivates the Hsp70 promoter in an HSE-dependent fashion, as does heat shock. (E) HSF1 DNA binding activity to the HSE was assessed by gel shift assay. Expression of CHIP enhances binding of two specific bands (SB), which are competed away by unlabeled HSE but not by an NFκB consensus sequence. The binding activities can be supershifted (SS) with an HSF1, but not an HSF2, antibody. NS, nonspecific. (F) Upregulation of Hsp70 in response to CHIP 48 h after transfection was determined in wild-type or HSF1-null murine fibroblasts by western blot analysis.
A trivial explanation is that overexpression of CHIP increases the concentration of misfolded proteins, which in turn induce the stress response; however, this was not observed in metabolic labeling studies (Meacham et al., 2001), neither has general inhibition of proteasome function been detected (Jiang et al., 2001). To address the mechanism of Hsp70 regulation by CHIP, we performed reporter gene assays using the Hsp70 promoter, as Hsp70 is predominantly regulated at the transcriptional level by nuclear proteins such as HSF1 and STAT family members (Stephanou et al., 1999; Pirkkala et al., 2001). CHIP expression potently activated Hsp70 promoter activity (Figure 1D). The activation by CHIP was comparable to that induced by heat shock and was abolished in a promoter fragment that lacked the HSE, indicating that this effect is dependent on HSE-binding nuclear protein(s). We addressed the role of the Hsp70 HSE in mediating the transcriptional effects of CHIP more directly by gel mobility shift analysis. Specific binding of nuclear proteins to the radiolabeled HSE was enhanced by CHIP overexpression (Figure 1E). This binding could be competed by an excess of unlabeled HSE but not by a non-specific sequence. The identity of this specific binding activity as HSF1 was verified by mobility supershift with an antibody recognizing HSF1. Finally, we confirmed that HSF1 was necessary for the induction of Hsp70 by CHIP in fibroblasts from mice deficient in HSF1 (McMillan et al., 1998; Xiao et al., 1999). CHIP upregulated Hsp70 appropriately in wild-type fibroblasts, but not in cells lacking HSF1 (Figure 1F). In fact, modest downregulation of basal Hsp70 levels by CHIP was observed in HSF1-null cells, which may imply that CHIP has effects on more than one signaling pathway that regulates Hsp70 expression, although HSF1-dependent regulation is clearly the dominant pathway and probably the only positive regulatory pathway affected by CHIP. This may reflect an activity of CHIP on other signaling pathways that also affect Hsp70 expression but that are only obvious when the effects of HSF1 are removed; such an interpretation is consistent with the results of Figure 1D as well.
Evidence from a variety of systems indicates that both Hsp70 and Hsp90 can repress the activation of HSF1, as can other co-chaperones, making the activation of HSF1 by CHIP an exceptional observation. The induction of Hsp70 by CHIP is specific, as BiP levels (which are inducible by activators of the endoplasmic reticulum stress response such as tunicamycin) were unaffected by CHIP (Figure 2A). Importantly, the inducibility of Hsp70 was completely abolished by a mutant form of CHIP (CHIP K30A). This mutant is expressed at similar levels to wild-type protein and differs only in a single amino acid residue in the TPR domain that abolishes interactions with Hsp70 (Scheufler et al., 2000); thus, non-specific activation of the stress response by protein overexpression can be excluded. To further test the specificity of this effect, we examined a panel of co-chaperones for their ability to increase Hsp70 expression and promoter activity. In comparison with other co-chaperones (including protein phosphatase 5, FKBP59 and HOP, each of which contains TPR repeats), only CHIP increased Hsp70 expression (Figure 2B), and no co-chaperone other than CHIP significantly activated the Hsp70 promoter (Figure 2C). The specificity of this effect raised the question of which features of CHIP contribute to HSF1 activation. To address this, we tested the ability of CHIP mutants to increase Hsp70 expression and to activate the Hsp70 promoter in transient transfection assays. CHIP K30A is ineffective in increasing Hsp70 expression or promoter activity, whereas the activity of CHIP D253N/R254G (which contains mutations in the U-box that abolish ubiquitin ligase activity (Xu et al., 2002)) was equivalent to that of wild-type CHIP (Figure 2D). Thus, the activation of HSF1 by CHIP is dependent on CHIP’s ability to interact with Hsp70 and/or Hsp90, whereas the ubiquitin ligase activity may not be as important, at least under these conditions.
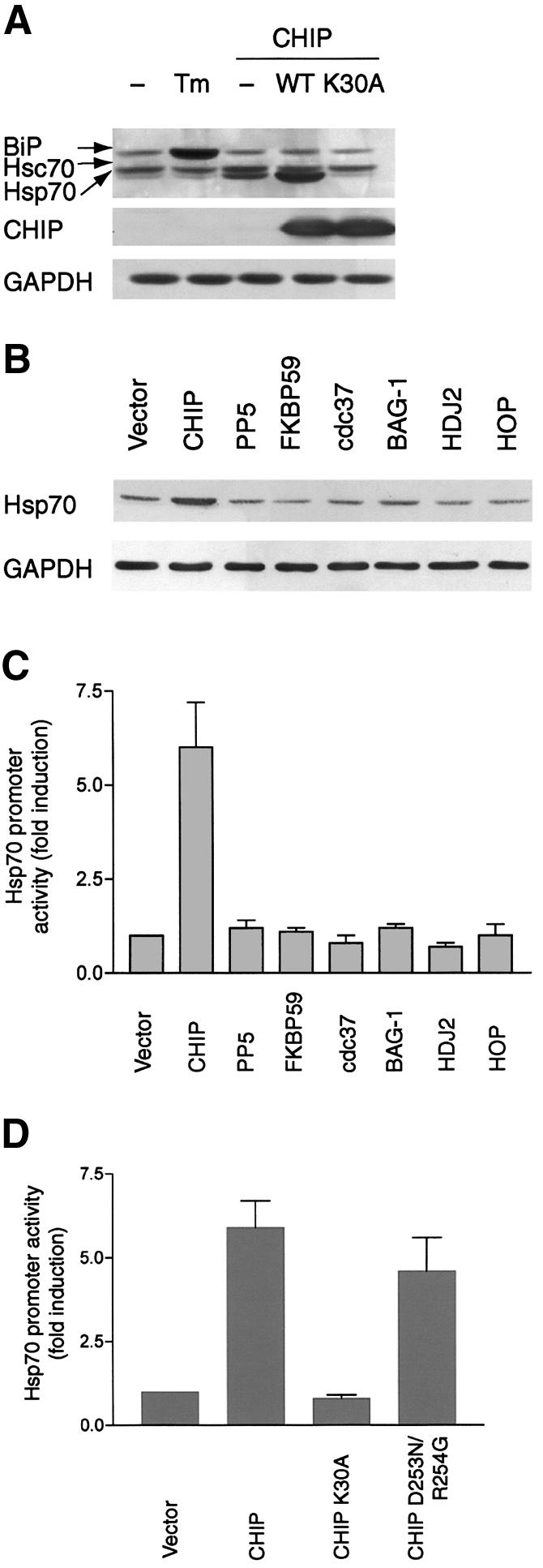
Fig. 2. Activation of HSF1 is specific to CHIP and dependent on chaperone interactions. (A) Western blot analysis using an antibody that recognizes BiP, Hsc70 and Hsp70 demonstrates that only wild-type CHIP, but not CHIP K30A, increases Hsp70 expression after adenoviral infection, whereas BiP levels are increased by tunicamycin (Tm) but not by CHIP. (B and C) Only overexpression of CHIP, but not other co-chaperones [protein phosphatase 5 (PP5), FKBP59, cdc37, BAG-1, HDJ2 or HOP], increases Hsp70 protein expression (B) and promoter activity (C) after transient transfection. (D) Hsp70 promoter activity is increased by wild-type CHIP and by a mutant lacking ubiquitin ligase activity (CHIP D253N/R254G), but not by a mutant that does not interact with Hsp70/Hsp90 (CHIP K30A).
Assembly of a CHIP-dependent activated HSF1 complex
One of the hallmarks of HSF1 activation is retarded mobility due to trimerization. Using a non-denaturing gel separation technique that allows discrimination of monomeric, dimeric, Hsp70-bound and trimeric HSF1 complexes (Zou et al., 1998; Liu and Thiele, 1999; Guo et al., 2001), we found that increasing levels of CHIP caused rapid and nearly quantitative accumulation of active, trimeric HSF1 (Figure 3A). Interestingly, accumulation of the activated form of HSF1 coincided with the earliest detectable expression of CHIP after adenoviral infection and persisted for at least 48 h, even though other ongoing stimuli for HSF1 activation (such as heat shock) have a self-terminating effect (in our experiments, within 4 h), suggesting that CHIP-induced trimeric HSF1 complexes may be resistant to attenuation. CHIP had no effect on the stability of HSF1, nor did we detect ubiquitylation of HSF1 by CHIP, but labeling studies did indicate that HSF1 was phosphorylated coincident with CHIP-induced trimerization (data not shown), as is suggested by the slightly retarded mobility of HSF1 we consistently noted by western blotting after denaturing gel electrophoresis. Several studies have demonstrated that both Hsp70 and Hsp90 can repress transcriptional activation by HSF1 (Rabindran et al., 1994; Shi et al., 1998; Zou et al., 1998). Given the importance of CHIP’s Hsp70/Hsp90 interaction domain in the activation of HSF1, one explanation for CHIP-dependent activation of HSF1 is that CHIP disrupts Hsp/HSF1 complexes. To test this, we examined the effects of CHIP expression on these interactions. Either with or without crosslinkers, Hsp90/HSF1 interactions remained below the limits of our detection. However, in contrast to the expectation that CHIP would disrupt HSF1/Hsp70 complexes, we found that overexpression of CHIP led to stable Hsp70 interactions with the activated form of HSF1 (Figure 3B). These interactions were abolished by mutation of CHIP’s TPR domain, suggesting that this HSF1/Hsp70 complex required interactions between CHIP and Hsp70. In converse experiments, CHIP (as well as Hsp70) was also found in HSF1 immunoprecipitates (Figure 3C), and the stability of these complexes was similar to that of CHIP-dependent HSF1 trimerization (Figure 3A). We could also coimmunoprecipitate HSF1 with transiently transfected Myc-tagged CHIP (Figure 3D).

Fig. 3. CHIP-dependent HSF1:chaperone complexes. (A) HSF1 expression was examined by non-denaturing gel electrophoresis in COS7 cells after heat shock (42°C) or after infection with CHIP-expressing adenovirus (or a control adenovirus). Depletion of inactive complexes [which consist of dimers and Hsp70-bound monomers (Liu and Thiele, 1999; Guo et al., 2001)] and quantitative accumulation of the activated trimeric form of HSF1 occurs in CHIP-expressing cells. (B) COS7 cell lysates were prepared 24 h after infection with wild-type (WT) or CHIP or the K30A mutant (or a control adenovirus), and whole cell lysates (WCL) or immunoprecipitates (IP) were probed by western blotting for the presence of HSF1, Hsp70 and CHIP. (C) COS7 cells were infected with CHIP adenovirus as in (B) for the indicated times and whole cell lysates or HSF1 immunoprecipitates were probed for HSF1, Hsp70 or CHIP. Hsp70 is stably associated with activated HSF1 in CHIP-expressing cells and CHIP could be precipitated with Hsp70 in these HSF1 immunoprecipitates. (D) HSF1 co-immunoprecipitates with CHIP following transient transfection of Myc-tagged CHIP in COS7 cells.
We have not identified direct interactions between CHIP and HSF1, and their association depends on the integrity of CHIP’s TPR domain, so these results are likely to indicate that CHIP, Hsp70 and activated HSF1 exist in an Hsp70-dependent ternary complex that resists HSF1 inactivation. To consider the mechanism of CHIP-dependent HSF1 activation in more detail, we evaluated the subcellular distribution of CHIP in the setting of cell stress. The activation of HSF1 in HeLa cells has been carefully considered, and requires cytoplasmic-to-nuclear translocation as an early event (Zuo et al., 1995; Cotto et al., 1997). A direct role for CHIP in HSF1 activation would require that a fraction of CHIP is nuclear-localizing. To examine this in more detail, we analyzed the subcellular distribution of CHIP and HSF1 after heat shock. In the unstimulated state, the majority of HSF1 is confined to the cytoplasm, as is the majority of CHIP (Figure 4A). As previously noted, HSF1 rapidly translocated to the nucleus after heat shock. Interestingly, there was an inversion of the cytoplasmic/nuclear ratio of CHIP as well, indicating that CHIP also relocalizes to the nucleus after heat shock. Both HSF1 and CHIP reaccumulate in the cytoplasm during recovery from heat shock. This observation was confirmed by immunofluorescence (Figure 4B), which demonstrates that cytoplasmic CHIP is largely localized in the region of the endoplasmic reticulum in unstressed conditions as previously described (Meacham et al., 2001). After heat shock, the cytoplasmic pool collapses and increased accumulation of CHIP is observed in punctate loci within the nucleus. During recovery, CHIP reaccumulates in the cytoplasm in a pattern similar to that observed in unstressed conditions.

Fig. 4. Nuclear transport and DNA binding of HSF1 induced by CHIP. (A) The subcellular localization of HSF1 and CHIP before and after heat shock (42°C for 30 min) or heat shock plus recovery for 1 h was determined by western blot analysis of nuclear (Nu) and cytoplasmic (Cy) fractions. (B) Immunocytochemical analysis of endogenous CHIP expression after heat shock (42°C for 30 min). The CHIP antibody stains red. (C) Association of endogenous CHIP and HSF1 in HeLa cells before and after heat shock (HS). WCL, whole cell lysate; NS, non-specific antibody. (D) Kinetics of association of HSF1 with CHIP after heat shock (HS) or heat shock plus recovery (R) for the indicated times. (E) The subcellular redistribution of HSF1 and Hsp70 was determined by western blot analysis of nuclear and cytoplasmic fractions of HeLa cells that were untreated (Control) or infected with a GFP-expressing adenovirus (Ad-Track) or with a virus expressing CHIP for 24 h. (F) Gel shift analysis of DNA-binding-competent HSF1 complexes with or without CHIP overexpression (24 h after infection) using HSE as a probe. Specific binding (SB) of complexes was determined by competition (Comp) with HSE or an NFκB binding site. DNA-bound HSF1 complexes were probed with antibodies to HSF1, Hsp70, CHIP or preimmune serum (NS). (G) Gel shift analysis of HSF1 complexes before or after heat shock (HS) for 30 min.
The synchronous nucleocytoplasmic transport of HSF1 and CHIP led us to consider whether there is a direct association between endogenous CHIP and HSF1 in the setting of cellular stress. Indeed, consistent with temporal trends in localization, we could detect HSF1-containing CHIP immunocomplexes both before and after heat shock (Figure 4C). Notably, the HSF1–CHIP heterocomplex was detectable under conditions in which HSF1 is entirely nuclear localized. The kinetics of this association paralleled activation of HSF1 by heat shock, and during the recovery period HSF1 became largely dissociated from CHIP (Figure 4D). This suggested to us that CHIP may facilitate HSF1 translocation. To test this, we overexpressed CHIP and examined the subcellular localization of HSF1. Increasing levels of CHIP resulted in complete redistribution of HSF1 to the nucleus (Figure 4E), indicating that CHIP is indeed sufficient to induce HSF1 translocation. This observation provides a necessary mechanism for CHIP-dependent HSF1 activation.
Several arguments can be made to support the transcriptional competence of nuclear CHIP–HSF1 complexes: CHIP essentially induces quantitative HSF1 trimerization (Figure 3A), HSF1 DNA-binding activity is markedly increased in the presence of CHIP (Figure 1E) and CHIP-dependent upregulation of Hsp70 by HSF1 is potent and sustained (Figures 1B and 3C). To address this directly, we probed for the presence of CHIP in HSF1 complexes bound to the HSE. As previously observed (Figure 1E), overexpression of CHIP markedly enhanced HSF1 binding to an oligonucleotide containing its canonical sequence (Figure 4F). In addition to HSF1, we could detect both Hsp70 and CHIP in these complexes, indicating that the trimeric complex (Figures 3B and C) was competent for binding DNA. In addition, and consistent with the interaction of CHIP and activated HSF1 in the nucleus, endogenous CHIP was detected in DNA-bound HSF1-containing complexes that were transcriptionally activated by heat shock (Figure 4G). Interestingly, we detect less Hsp70 in DNA-bound HSF1 complexes after heat shock than when these complexes are directly induced by CHIP, presumably because Hsp70 is more likely to be sequestered by unfolded proteins under these circumstances and therefore is most likely to be dispensable for the DNA-binding activity of HSF1. The ability of CHIP to activate HSF1, and its interaction with HSF1 and participation in DNA-bound HSF1 complexes after heat shock, argues for a previously unanticipated role for CHIP in modulating the stress response that can best be tested by examining the cellular and physiologic consequences when CHIP is deficient.
Perinatal lethality in mice deficient in CHIP
If CHIP has such a central role in regulation of the general transcriptional stress response, as our studies suggest, then the cellular response to stress should be abated in the absence of CHIP. We generated mice deficient in CHIP to test this hypothesis. The first three exons of the CHIP gene were replaced with a PGK–Neo selection cassette by homologous recombination (Figure 5A). Germline transfer of the targeted allele was successful (Figure 5B) and deletion of CHIP expression was confirmed by western blotting (Figure 5C). Mice heterozygous at the CHIP locus were maintained on a mixed 129 Sv/Ev X C57BL/6 background. CHIP (–/–) mice were recovered in Mendelian frequencies from heterozygous crosses, indicating that CHIP is not required for essential developmental events in these mice (Figure 5D). However, 5% of mice were quickly cannibalized in the immediate peripartum period, most likely after perinatal death, and these mice could not be genotyped. We did not observe this rate of perinatal lethality in litters from wild-type crosses nor when wild-type males were bred with CHIP (+/–) females, excluding a maternal effect for the CHIP (+/–) genotype. We therefore presume the ungenotyped mice were predominantly CHIP (–/–). The remaining CHIP (–/–) offspring suckled normally; however, 20% of these mice also died in the peripartum period, which greatly exceeded the perinatal mortality of CHIP (+/+) or (+/–) mice (Figure 5E). This lethality was not rescued by transferring litters to wild-type foster mothers of mixed or FVB background. The dead CHIP (–/–) mice had no obvious morphologic abnormalities and had milk in their stomachs at necropsy. Normal surfactant production was confirmed by PAS staining in these mice.

Fig. 5. Viability of CHIP (–/–) mice generated by homologous recombination. (A) CHIP gene, targeting vector and disrupted allele The PGK–Neo cassette was inserted in place of a 0.8 kb BglII (BA)–HpaI (H) fragment, resulting in the deletion of the first three exons of CHIP. (B) Recombinant clones were initially identified by Southern blot analysis with 3′ external probes and F1 mice by Southern blot analysis with external probes or by PCR analysis for wild-type and mutant alleles. (C) Deletion of CHIP expression was confirmed by western blot analysis. (D) Tabular results of intercrosses of CHIP (+/–) parents. (E) Survival curves of live-born mice from CHIP (+/–) intercrosses indicate that a fraction of CHIP (–/–) mice die in the immediate peripartum period. (F) The thymus gland (arrows) overlying the heart is atrophied in organ preparations of deceased neonatal CHIP (–/–) mice, but not in wild-type mice (upper panels) or in surviving CHIP (–/–) mice (not shown). Histologic examination reveals hypocellularity and shrunken, pyknotic nuclei in atrophic thymuses from CHIP (–/–) mice, characteristic of thymocyte apoptosis (lower panels).
The most striking structural lesion noted in the dead CHIP (–/–) mice was marked thymic atrophy (Figure 5F, upper panel). Histologic analysis demonstrated that thymic remnants were hypocellular and contained dense, pyknotic nuclear clumps, the hallmarks of massive thymocyte apoptosis (Figure 5F, lower panels). Surviving CHIP (–/–) mice had no obvious thymocyte abnormalities and were morphologically normal. Measurement of development of CD4(+) and CD8(+) by flow cytometry indicated that these cells developed normally in thymuses of CHIP (–/–) mice and that their numbers were not different from wild-type (data not shown), making a primary thymocyte abnormality unlikely. Peripartum thymocyte apoptosis is often a non-specific response to abnormal stress tolerance, and is due in part to increased levels of circulating corticosteroids (Tybulewicz et al., 1991). Based on additional studies described below, we interpret these results as indicators that CHIP (–/–) mice may adapt poorly to the stress of parturition.
CHIP is required for maximal Hsp70 induction and protection against stress-induced apoptosis
To address the role of CHIP in regulation of physiologic stress response pathways more directly, we isolated fibroblasts from CHIP (–/–) mice or wild-type littermates. CHIP (–/–) fibroblasts had typical morphologic, growth and viability characteristics under standard culture conditions (37°C). However, in comparison with wild-type fibroblasts, CHIP (–/–) fibroblasts exhibited markedly diminished responses to stress. For example, the ability of Hsp70 to be induced in response to heat shock was reduced by 50–60% in CHIP (–/–) fibroblasts (Figure 6A). This biochemical indication of impaired response to stress is similar to that which occurs in HSF1-null cells (McMillan et al., 1998), as would be expected from our overexpression studies (Figure 1). We also found that viability of CHIP (–/–) fibroblasts after heat shock (45°C) was sharply reduced compared with wild-type cells (Figure 6B). The stress-induced deficit in viability of cells lacking CHIP was accompanied by increased indices of apoptosis (Figure 6C), which is consistent with previous studies indicating that heat stress can activate an apoptotic cascade (Beere and Green, 2001). The stress-induced defect in CHIP (–/–) fibroblasts was not limited to a failure of tolerance to heat shock; cells lacking endogenous CHIP were also more susceptible to the amino acid analog l-canavanine, which incorporates into proteins during synthesis and induces misfolding (Figure 6D). We further examined whether CHIP (–/–) cells develop thermotolerance after preconditioning with a sublethal heat challenge (42°C), which is the primary defect in HSF1 (–/–) fibroblasts (McMillan et al., 1998). The loss of viability of CHIP (–/–) cells after thermal challenge was only partially rescued by preconditioning (Figure 6E). This suggested to us that CHIP may also be required for development of thermotolerance. The effect of CHIP on thermotolerance was more evident after prolonged (24 h) recovery after heat shock, at which time cell death after heat shock in the absence of preconditioning was maximal regardless of genotype (Figure 6F). To analyze biochemical markers of apoptosis, we measured caspase 3 activity after thermal stress with or without preconditioning. In comparison to wild-type cells, CHIP (–/–) cells markedly increased caspase 3 activity following thermal challenge at 45°C, and this increase is only modestly reduced by preconditioning (Figure 6G). Similarly, cleaved (and therefore activated) caspase 3 is only detected in CHIP (–/–) cells after heat shock, with or without preconditioning (Figure 6H). These in vitro studies support our interpretation of CHIP overexpression studies, and indicate that CHIP has an unanticipated central role in regulating the cellular response to stress.

Fig. 6. Impaired stress response and increased apoptosis in CHIP-deficient fibroblasts. (A) Western blot analysis of CHIP (+/+) or CHIP (–/–) fibroblasts after heat shock (HS) at 42°C for the indicated times. Induction of Hsp70 by heat shock is attenuated in cells lacking CHIP. (B and C) The viability of CHIP (–/–) fibroblasts is markedly decreased in response to heat stress (45°C) for the indicated times (B). The decrease in viability is associated with increased accumulation of oligonucleosomes during recovery from heat shock (45°C for 45 min) for the indicated times as a marker of apoptosis (C). (D) CHIP (–/–) cells are also sensitive to challenge with l-canavanine, an amino acid analog that causes accumulation of misfolded proteins. (E and F) Viability in CHIP (–/–) and CHIP (+/+) cells was determined after 45 min of heat shock (45°C) with or without preconditioning at 42°C for 30 min and 6 h (E) or 24 h (F) of recovery at 37°C, as indicated. CHIP (–/–) fibroblasts suffer decrements in viability in the presence or absence of preconditioning. NS, without heat stress. (G and H) Caspase 3 activity (G) and cleaved caspase 3 levels (H) were measured using a colorimetric assay after thermal challenge (HS) with or without thermal preconditioning (PRE), as indicated.
Impaired responses to thermal challenge in CHIP (–/–) mice
We tested the physiologic significance of these in vitro studies in mice deficient in CHIP. Because we observed no obvious anatomic abnormalities in mature CHIP (–/–) mice, we considered the possibility that these mice might have a generally impaired response to physiologic stress, especially given the neonatal events that occurred in a subset of CHIP-deficient mice (Figure 5). Sensitivity to thermal challenge is a well-characterized physiologic stressor. Hsp70 levels were very low or undetectable in sham-treated mice, as is to be expected (Hotchkiss et al., 1993; Huang et al., 2001), regardless of their genotype. After sublethal elevation of core body temperatures, induction of Hsp70 was consistently lower in CHIP (–/–) mice than in wild-type mice (Figure 7A), which is similar to the responses of CHIP (–/–) cells in culture (Figure 6A). To test the physiologic response to hyperthermia, core body temperatures of mice were raised to 42°C for 15 min. All wild-type mice survived this challenge, and death during recovery was only 25%. (No deaths were noted after 24 h of recovery.) In marked contrast, 100% of CHIP (–/–) mice died rapidly, either during thermal challenge or immediately thereafter (Figure 7B). Necropsy revealed characteristic stress-responsive lesions. The primary abnormalities noted were edema, hyperemia and friability of the small intestine (Figure 7C), which are common lesions in humans suffering from heat stroke (Shibolet et al., 1967). In contrast to the normal villous architecture observed in heat-shocked wild-type mice (Figure 7D), marked blunting of villi was noted in CHIP (–/–) mice, as was thrombosis of subvillous arterioles (Figure 7E). Many cells within the damaged villi of CHIP (–/–) mice were shrunken with pyknotic nuclei (Figure 7F). TUNEL staining indicated that these cells were undergoing apoptosis (Figure 7G). It is most likely that the rapid death of CHIP (–/–) mice after thermal challenge is secondary to fluid shifts or endotoxemia from compromise of the gastrointestinal barrier. Apoptosis in heat-stressed CHIP (–/–) mice was not limited to the gastrointestinal tract. For example, we also observed sheets of dying splenocytes in CHIP-deficient mice after thermal challenge (compare Figures 7H and I). Most splenocytes of CHIP (–/–) mice were TUNEL-positive (Figure 7K) whereas this finding was rare in wild-type mice (Figure 7J).

Fig. 7. Impaired stress response and stress-induced apoptosis in CHIP (–/–) mice. (A) Induction of Hsp70 in organs from CHIP (+/+) or (–/–) mice before (C) or 24 h after transient heat shock (HS) to 42°C. (B) Survival curves of CHIP (–/–) and CHIP (+/+) mice (n = 16) subjected to sustained core hyperthermia of 42°C for 15 min. No CHIP (–/–) mice recovered from hyperthermia. (C) Pathologic analysis of CHIP (–/–) mice after hyperthermia demonstrated friability and hyperemia of the small intestine. (D–G). Histologic analysis of small intestinal mucosal of CHIP (+/+) (D) and CHIP (–/–) (E–G) mice after hyperthermic challenge. Normal villous structure is preserved in CHIP (+/+) mice (D), whereas atrophy of the terminal villi and thrombosis (arrows) are noted in CHIP (–/–) mice (E). Dense, shrunken nuclei are observed in villous tissues of CHIP (–/–) mice (F) that stain TUNEL-positive (G), indicating stress-induced apoptosis. (H–K) Histologic analysis of splenic tissues from CHIP (+/+) (H and J) and CHIP (–/–) (I and K) mice after hyperthermic challenge. Hypocellularity and pyknosis of splenocytes from CHIP (–/–) mice (I) stain strongly TUNEL-positive (K), whereas the architecture and viability of splenocytes from CHIP (+/+) mice is preserved after hyperthermia (H and J).
Discussion
The cellular response to stress encompasses not only the expression of cytoplasmic chaperones that buffer the cell against proteotoxic damage, but also induction of cell cycle arrest and inhibition of translation, the latter effects providing a cell with the opportunity to recover from stress-induced damage. Although essential in the short term, this response must be tightly regulated so that a cell can return to other functions appropriately. The prevailing model indicates that the stress response is attenuated by interactions between HSF1 and the chaperones Hsp70 and Hsp90, which repress the transactivation function of HSF1 and stabilize the inactive HSF1 monomer (Morimoto, 1998). However, this model is complicated by the fact that HSF1–Hsp70 interactions occur during both the activation and attenuation phases of the heat shock response [(Rabindran et al., 1994) and our own observations], and HSF1-bound Hsp70 has little effect on transcriptional activity during the activation phase. Thus, the transition to an attenuation state is either a very slow process or, more likely, additional remodeling of HSF1–chaperone complexes must occur to attenuate HSF1. The latter model is supported by observations that chaperone-dependent remodeling and transcriptional inhibition of steroid receptor transcriptional complexes results from recruitment of the co-chaperone p23 (Freeman and Yamamoto, 2002); perhaps similar co-chaperone-dependent remodeling occurs following HSF1 activation that results in transition to the attenuation phase.
The studies here demonstrate that an additional HSF1–chaperone complex can assemble in the presence of CHIP and that this complex maintains a stably activated form of HSF1 that resists attenuation. CHIP may remodel these complexes so that domains otherwise masked by Hsp70 become accessible due to CHIP-dependent conformational changes. It is equally plausible that the effects of CHIP on HSF1 activation are due to modulation of the catalytic activity of Hsp70—HSF1 repression by Hsp70 may be dependent on the folding or ATPase activity of Hsp70, both of which are modified by CHIP (Ballinger et al., 1999). In any event, the activation of HSF1 by CHIP represents another layer of regulation, in addition to that conferred by Hsp70 or Hsp90 alone, for controlling the effects of HSF1, and markedly contrasts with other co-chaperones tested in this context, which universally repress HSF1 activation (Rabindran et al., 1994; Nair et al., 1996; Shi et al., 1998; Zou et al., 1998; Bharadwaj et al., 1999; Marchler and Wu, 2001). This additional regulation may be necessary to fine-tune the stress response (perhaps through networking of chaperone–co-chaperone interactions) or to coordinate expression of cytoplasmic chaperones in circumstances not classically associated with protein misfolding (such as in transformed cells or during cell cycle progression (Jaattela, 1999; Helmbrecht et al., 2000). HSF1 activation by CHIP may be especially relevant when CHIP levels are regulated, for example in metabolically active tissues such as striated muscle (Ballinger et al., 1999) or after proteasome inhibition (Imai et al., 2002).
The cellular studies presented here provide a context for the known biochemical activities of CHIP. A model has emerged in which CHIP participates in cytoplasmic quality control decisions by diverting proteins from the chaperone-dependent folding pathway to the ubiquitin-proteasome system for degradation (Hohfeld et al., 2001; McClellan and Frydman, 2001). The ability to switch a peptide off the folding pathway provides a means for the cell to dispose of damaged or slowly folding proteins. Because the chaperone system itself participates in CHIP-dependent protein degradation, one can envision that the activity of CHIP would come at the expense of folding capacity, even for peptides that are otherwise competent for folding. A global diversion of chaperone substrates to the proteasome for degradation would undoubtedly be an enormous burden for the cell to bear, given the large number of proteins that require cytoplasmic chaperones to achieve an active conformation. By activating HSF1 coincidently with the acceleration of ubiquitination of chaperone substrates, CHIP increases the total buffering and folding capacity of the cell, providing a means for CHIP to regulate both folding and degradation capacity simultaneously. This model, which was not anticipated by previous studies examining the biochemical function of CHIP (Ballinger et al., 1999; Connell et al., 2001; Jiang et al., 2001), would account for the protective cellular effects of CHIP identified in this report, and suggests that protein folding and degradation must occur simultaneously and in coordination to buffer against proteotoxic damage.
The activation of HSF1 by CHIP reflects a common theme in co-chaperone interactions: that major and often diametrically opposed chaperone activities are regulated by recruitment of a single co-chaperone into the chaperone heterocomplex. Because the biochemical properties of the co-chaperones are so diverse, we can anticipate that their cellular and physiologic functions will also be distinct and unexpected. With respect to CHIP, the most important lesson of the present studies is that it activates a cytoprotective and antiapoptotic program in the setting of cellular stress. Thus, CHIP provides a means for the cell to coordinate folding, degradation and the global stress response, each of which probably contributes to its antiapoptotic effects. This activity is likely to have relevance pathophysiologically, since CHIP has been shown to assist parkin in inhibiting cell death caused by an unfolded form of the parkin substrate Pael-R in neuronal cells (Imai et al., 2002). A body of evidence is now available linking the cytoplasmic chaperones (especially Hsp70) with antiapoptotic events at multiple levels (Beere and Green, 2001), and at least one other co-chaperone, BAG-1, has antiapoptotic properties (Takayama et al., 1995). Although the present studies provide further support for the critical role of the molecular chaperones as an antiapoptotic system, they also indicate that this effect is likely to be a global one linking many cellular events, some of which are regulated by CHIP, rather than through individual interactions of chaperones with a particular process in the apoptotic cascade.
The validity of the central role for CHIP in modulating the cellular response to stress, as determined in our cell culture studies, is corroborated by the data derived from our physiologic studies in mice that lack CHIP. To our knowledge, this is the first report of loss-of-function of a co-chaperone in mice. That mice develop normally in the absence of CHIP is remarkable, but perhaps not surprising based on studies in yeast demonstrating that many members of the chaperone family are dispensable for normal growth and cellular maintenance under optimal growth conditions. Our consistent findings are of a failure to mount an appropriate response to physiologic stress (whether due to parturition or thermal challenge) that is accompanied by apoptotic cell death. In all likelihood the antiapoptotic effects of CHIP represent the sum activity on degradation of damaged proteins and regulation of the stress response. It is interesting to note that mice lacking HSF1 also exhibit growth retardation at birth and fail to induce heat shock protein expression appropriately, and that fibroblasts from these mice are heat-sensitive (McMillan et al., 1998; Xiao et al., 1999; Zhang et al., 2002). One difference between HSF1 (–/–) and CHIP (–/–) mice is the extent to which in utero lethality occurs in HSF1 (–/–) mice, although this appears to be a strain-sensitive phenotype so that direct comparisons of these studies may not be applicable. CHIP (–/–) cells may also be more susceptible to a single heat challenge than are HSF1 (–/–) cells, which are primarily defective in the development of thermotolerance after preconditioning (McMillan et al., 1998) [although some HSF1 (–/–) cell lines exhibit enhanced thermal sensitivity in the absence of preconditioning (Zhang et al., 2002)]. The differences noted are consistent with a model in which CHIP plays multiple roles in the stress response, regulation of HSF1 being but one aspect of this activity that is also likely to include effects on protein folding and degradation (Ballinger et al., 1999; Connell et al., 2001; Meacham et al., 2001). The phenotype of CHIP (–/–) mice also stands in contrast to reported phenotypes of mice lacking Hsp70 isoforms, which are more modest (Dix et al., 1996; Huang et al., 2001). This is probably due to functional redundancy among Hsp70 family members, and indicates that the actions of CHIP are especially crucial to buffering the organism against physiologic stress. To our knowledge, this is the first example of a temperature-sensitive mouse, which will be an important resource for addressing the role of the cellular stress response in a variety of physiologic settings.
Materials and methods
Cell culture
COS7 cells were cultured as previously described (Connell et al., 2001). HSF1 (–/–) and HSF1 (+/+) murine embryonic fibroblasts were provided by Ivor Benjamin (McMillan et al., 1998; Xiao et al., 1999) and CHIP (–/–) and CHIP (+/+) fibroblasts were cultured according to standard protocols. Recombinant adenoviruses expressing CHIP or control viruses were constructed with the Ad-Easy system and cultures were routinely infected at a multiplicity of infection of five with an infection efficiency of >98%. Western blotting and immunoprecipitations were performed as previously described (Connell et al., 2001).
Reporter gene and electrophoretic mobility gel shift assays (EMSAs)
Hsp70 promoter:reporter constructs were provided by David Latchman (Stephanou et al., 1999). Reporter constructs were co-transfected in COS7 with a β-galactosidase reporter (to control for transfection efficiency), with or without plasmids expressing CHIP, CHIP mutants or other co-chaperones as indicated. For EMSA, a radiolabeled HSE oligonucleotide was incubated with nuclear extract from cells treated as indicated. The reaction mixture was incubated at room temperature for 20 min and fractionated on a 5% native polyacrylamide gel in 0.5× TBE buffer. To determine the specificity of the DNA–protein complexes, we performed competition assays using 50-fold molar excess of the unlabeled double-stranded HSE oligonucleotide (specific inhibitor) or excess of an unrelated NFκB oligonucleotide of comparable length (non-specific inhibitor). To characterize specific DNA-binding proteins, we incubated nuclear extracts with anti-HSF1 or anti-HSF2 antibody before adding probe.
Assays of apoptosis
CHIP (+/+) and CHIP (–/–) fibroblasts were incubated at 37°C, or heated at 42°C for 30 min, followed by 6 h recovery at 37°C and heated again at 45°C for 45 min (to stimulate maximal preconditioning), or only heated at 45°C for 45 min. Cells were incubated at 37°C as indicated in the figure legends before lysing. Viability was measured with the XTT Assay (Roche). Caspase 3 activity was measured using a fluorometric assay (Roche). Cleaved caspase 3, which is the activated form of the enzyme, was detected with cleaved caspase 3 (Asp175) antibody 9661 (Cell Signaling Technology), which detects only the active form of caspase 3.
Generation of the CHIP targeting construct and mutant mice
The CHIP targeting construct was generated using the pKO Scrambler vector (Stratagene), which contains a neomycin-resistance and thymidine kinase gene for positive and negative selection. Following electroporation into a 129 SvEv cell line, selected clones were analyzed for homologous recombination by Southern blotting. Three clones were injected into C57BL/6 blastocysts, and chimeric mice were bred to C57BL/6 females to achieve germline transmission of the targeted allele.
Thermal challenge
CHIP (–/–) mice and wild-type littermates of both sexes were maintained on a 129 SvEv X C57BL/6 background. Mice were anesthetized with 1.2% avertin (0 375 mg/kg body weight by intraperitoneal injection), immobilized and covered with a blanket, and placed underneath a 250 W infrared lamp. Core rectal temperatures were raised to 42°C over 15 min and maintained at this temperature transiently (for analysis of Hsp70 expression) or for 15 min (for mortality studies). After thermal challenge, mice were volume-repleted with intraperitoneal saline, and slowly returned to isothermal conditions on a warm blanket. Mice were directly observed for 24 h after thermal challenge. No deaths were observed in mice that survived beyond 24 h after thermal challenge.
Acknowledgments
Acknowledgements
The authors thank Michael Chinkers, Ivor Benjamin, Richard Morimoto, David Latchman and Jörg Höhfeld for provision of reagents, Scott Trasti for assistance with histology, Shiloh Souza and Ken Fowler for technical assistance, and Yue Xiong for critical reading of the manuscript. Supported by grants from the National Institutes of Health (GM61728 and HL65619 to C.P. and GM56981 to D.C.). C.P. is an Established Investigator of the American Heart Association and a Burroughs Wellcome Fund Clinical Scientist in Translational Research.
References
- Aravind L. and Koonin,E.V. (2000) The U box is a modified RING finger—a common domain in ubiquitination. Curr. Biol., 10, R132–R134. [DOI] [PubMed] [Google Scholar]
- Ballinger C.A., Connell,P., Wu,Y., Hu,Z., Thompson,L.J., Yin,L.-Y. and Patterson,C. (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol., 19, 4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beere H.M. and Green,D.R. (2001) Stress management: heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol., 11, 6–10. [DOI] [PubMed] [Google Scholar]
- Bercovich B., Stancovski,I., Mayer,A., Blumenfeld,N., Laszlo,A., Schwartz,A.L. and Ciechanover,A. (1997) Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J. Biol. Chem., 272, 9002–9010. [DOI] [PubMed] [Google Scholar]
- Bharadwaj S., Ali,A. and Ovsenek,N. (1999) Multiple components of the HSP90 chaperone complex function in regulation of heat shock factor 1 in vivo. Mol. Cell. Biol., 19, 8033–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell P., Ballinger,C.A., Jiang,J., Wu,Y., Thompson,L.J., Höhfeld,J. and Patterson,C. (2001) Regulation of heat shock protein-mediated protein triage decisions by the co-chaperone CHIP. Nat. Cell Biol., 3, 93–96. [DOI] [PubMed] [Google Scholar]
- Cotto J., Fox,S. and Morimoto,R. (1997) HSF1 granules: a novel stress-induced nuclear compartment of human cells. J. Cell Sci., 110, 2925–2934. [DOI] [PubMed] [Google Scholar]
- Dix D.J., Allen,J.W., Collins,B.W., Mori,C., Nakamura,N., Poorman-Allen,P., Goulding,E.H. and Eddy,E.M. (1996) Targeted gene disruption of Hsp70-2 results in failed meiosis, germ cell apoptosis and male infertility. Proc. Natl Acad. Sci. USA, 93, 3264–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman B.C. and Yamamoto,K.R. (2002) Disassembly of transcriptional regulatory complexes by molecular chaperones. Science, 296, 2232–2235. [DOI] [PubMed] [Google Scholar]
- Gabai V.L., Meriin,A.B., Mosser,D.D., Caron,A.W., Rits,S., Shifrin,V.I. and Sherman,M.Y. (1997) Hsp70 prevents activation of stress kinases. J. Biol. Chem., 272, 18033–18037. [DOI] [PubMed] [Google Scholar]
- Guo Y., Guettouche,T., Fenna,M., Boellmann,F., Pratt,W.B., Toft,D.O., Smith,D.F. and Voellmy,R. (2001) Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J. Biol. Chem., 276, 45791–45799. [DOI] [PubMed] [Google Scholar]
- Helmbrecht K., Zeise,E. and Rensing,L. (2000) Chaperones in cell cycle regulation and mitogenic signal transduction: a review. Cell Prolif., 33, 341–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohfeld J., Cyr,D.M. and Patterson,C. (2001) From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep., 2, 885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss R., Nunnally,I., Lindquist,S., Taulien,J., Perdrizet,G. and Karl,I. (1993) Hyperthermia protects mice against the lethal effects of endotoxin. Am. J. Physiol., 265, R1447–R1457. [DOI] [PubMed] [Google Scholar]
- Huang L., Mivechi,N.F. and Moskophidis,D. (2001) Insights into regulation and function of the major stress-induced hsp70 molecular chaperone in vivo: analysis of mice with targeted gene disruption of the hsp70.1 or hsp70.3 gene. Mol. Cell. Biol., 21, 8575–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter J.J., Mestril,R., Tam,E.K., Sievers,R.E., Dillmann,W.H. and Wolfe,C.L. (1996) Overexpression of heat shock protein 72 in transgenic mice decreases infarct size in vivo. Circulation, 94, 1408–1411. [DOI] [PubMed] [Google Scholar]
- Imai Y., Soda,M., Hatakeyama,S., Akagi,T., Hashikawa,T., Nakayama,K.I. and Takahashi,R. (2002) CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease and enhances its ubiquitin ligase activity. Mol. Cell, 10, 55–67. [DOI] [PubMed] [Google Scholar]
- Jaattela M. (1999) Escaping cell death: survival proteins in cancer. Exp. Cell Res., 248, 30–43. [DOI] [PubMed] [Google Scholar]
- Jiang J., Ballinger,C., Wu,Y., Dai,Q., Cyr,D., Höhfeld,J. and Patterson,C. (2001) CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem., 276, 42938–42944. [DOI] [PubMed] [Google Scholar]
- Liu P.C. and Thiele,D.J. (1999) Modulation of human heat shock factor trimerization by the linker domain. J. Biol. Chem., 274, 17219–17225. [DOI] [PubMed] [Google Scholar]
- Luders J., Demand,J., Schonfelder,S., Frien,M., Zimmermann,R. and Höhfeld,J. (1998) Cofactor-induced modulation of the functional specificity of the molecular chaperone Hsc70. Biol. Chem., 379, 1217–1226. [DOI] [PubMed] [Google Scholar]
- Marchler G. and Wu,C. (2001) Modulation of Drosophila heat shock transcription factor activity by the molecular chaperone DROJ1. EMBO J., 20, 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan A.J. and Frydman,J. (2001) Molecular chaperones and the art of recognizing a lost cause. Nat. Cell Biol., 3, E51–E53. [DOI] [PubMed] [Google Scholar]
- McMillan D.R., Xiao,X., Shao,L., Graves,K. and Benjamin,I.J. (1998) Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem., 273, 7523–7528. [DOI] [PubMed] [Google Scholar]
- Meacham G., Patterson,C., Zhang,W. and Cyr,D.M. (2001) CHIP is a component of the Hsp70 chaperone system that targets immature CFTR to the proteasome for degradation. Nat. Cell Biol., 3, 100–105. [DOI] [PubMed] [Google Scholar]
- Morimoto R.I. (1998) Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones and negative regulators. Genes Dev., 12, 3788–3796. [DOI] [PubMed] [Google Scholar]
- Mosser D.D., Caron,A.W., Bourget,L., Denis-Larose,C. and Massie,B. (1997) Role of the human heat shock protein Hsp70 in protection against stress-induced apoptosis. Mol. Cell. Biol., 17, 5317–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S.C., Toran,E.J., Rimerman,R.A., Hjermstad,S., Smithgall,T.E. and Smith,D.F. (1996) A pathway of multi-chaperone interactions common to diverse regulatory proteins: estrogen receptor, Fes tyrosine kinase, heat shock transcription factor Hsf1 and the aryl hydrocarbon receptor. Cell Stress Chaperones, 1, 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirkkala L., Nykanen,P. and Sistonen,L. (2001) Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J., 15, 1118–1131. [DOI] [PubMed] [Google Scholar]
- Rabindran S.K., Wisniewski,J., Li,L., Li,G.C. and Wu,C. (1994) Interaction between heat shock factor and hsp70 is insufficient to suppress induction of DNA-binding activity in vivo. Mol. Cell. Biol., 14, 6552–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C., Brinker,A., Bourenkov,G., Pegoraro,S., Moroder,L., Bartunik,H., Hartl,F.U. and Moarefi,I. (2000) Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell, 101, 199–210. [DOI] [PubMed] [Google Scholar]
- Schneider C., Sepp-Lorenzino,L., Nimmesgern,E., Ouerfelli,O., Danishefsky,S., Rosen,N. and Hartl,F.U. (1996) Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc. Natl Acad. Sci. USA, 93, 14536–14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Mosser,D.D. and Morimoto,R.I. (1998) Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev., 12, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibolet S., Coll,S.R., Gilat,T. and Sohar,E. (1967) Heatstroke: its clinical picture and mechanism in 36 cases. Q. J. Med., 144, 525–548. [PubMed] [Google Scholar]
- Stephanou A., Isenberg,D.A., Nakajima,K. and Latchman,D.S. (1999) Signal transducer and activator of transcription-1 and heat shock factor-1 interact and activate the transcription of the Hsp-70 and Hsp-90beta gene promoters. J. Biol. Chem., 274, 1723–1728. [DOI] [PubMed] [Google Scholar]
- Takayama S., Sato,T., Krajewski,S., Kochel,K., Irie,S., Millan,J.A. and Reed,J.C. (1995) Cloning and functional analysis of BAG-1: a novel Bcl-2-binding protein with anti-cell death activity. Cell, 80, 279–284. [DOI] [PubMed] [Google Scholar]
- Tybulewicz V.L., Crawford,C.E., Jackson,P.K., Bronson,R.T. and Mulligan,R.C. (1991) Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell, 65, 1153–1163. [DOI] [PubMed] [Google Scholar]
- Xiao X., Zuo,X., Davis,A.A., McMillan,D.R., Curry,B.B., Richardson,J.A. and Benjamin,I.J. (1999) HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J., 18, 5943–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Marcu,M., Yuan,X., Mimnaugh,E., Patterson,C. and Neckers,L. (2002) The chaperone-dependent E3 ubiquitin ligase CHIP mediates a novel degradative pathway for c-ErbB2/Neu. Proc. Natl Acad. Sci. USA, 99, 12847–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Huang,L., Zhang,J., Moskophidis,D. and Mivechi,N.F. (2002) Targeted disruption of hsf1 leads to lack of thermotolerance and defines tissue-specific regulation for stress-inducible Hsp molecular chaperones. J. Cell. Biochem., 86, 376–393. [DOI] [PubMed] [Google Scholar]
- Zou J., Guo,Y., Guettouche,T., Smith,D.F. and Voellmy,R. (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell, 94, 471–480. [DOI] [PubMed] [Google Scholar]
- Zuo J., Rungger,D. and Voellmy,R. (1995) Multiple layers of regulation of human heat shock transcription factor 1. Mol. Cell. Biol., 15, 4319–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]