

Abstract
Induction of apoptosis in keratinocytes by UV light is a critical event in photocarcinogenesis. Although p53 is of importance in this process, evidence exists that other pathways play a role as well. Therefore, we studied whether the apoptosis-related surface molecule CD95 (Fas/APO-1) is involved. The human keratinocyte cell line HaCaT expresses CD95 and undergoes apoptosis after treatment with UV light or with the ligand of CD95 (CD95L). Incubation with a neutralizing CD95 antibody completely prevented CD95L-induced apoptosis but not UV-induced apoptosis, initially suggesting that the CD95 pathway may not be involved. However, the protease CPP32, a downstream molecule of the CD95 pathway, was activated in UV-exposed HaCaT cells, and UV-induced apoptosis was blocked by the ICE protease inhibitor zVAD, implying that at least similar downstream events are involved in CD95- and UV-induced apoptosis. Activation of CD95 results in recruitment of the Fas-associated protein with death domain (FADD) that activates ICE proteases. Immunoprecipitation of UV-exposed HaCaT cells revealed that UV light also induces recruitment of FADD to CD95. Since neutralizing anti-CD95 antibodies failed to prevent UV-induced apoptosis, this suggested that UV light directly activates CD95 independently of the ligand CD95L. Confocal laser scanning microscopy showed that UV light induced clustering of CD95 in the same fashion as CD95L. Prevention of UV-induced CD95 clustering by irradiating cells at 10°C was associated with a significantly reduced death rate. Together, these data indicate that UV light directly stimulates CD95 and thereby activates the CD95 pathway to induce apoptosis independently of the natural ligand CD95L. These findings further support the concept that UV light can affect targets at the plasma membrane, thereby even inducing apoptosis.
Apoptosis is an important and well-controlled form of cell death that occurs under a variety of physiological and pathological conditions. This process has been recognized to be of major importance for embryonic development, tissue homeostasis, neurodegeneration, autoimmune diseases, AIDS, carcinogenesis, cancer progression, and the killing of cancer cells induced by chemotherapeutic drugs (Cohen, 1991; Ameisen, 1994; Kerr et al., 1994; Friesen et al., 1996; Gehri et al., 1996; Kusiak et al., 1996; Jacobson et al., 1997). Once the apoptosis program is activated, it starts with blebbing of the membrane, followed by degradation of the chromosomal DNA by nucleases, resulting in condensation and fragmentation (Cohen, 1993). Finally, cell fragments are removed by phagocytes without causing any inflammatory reaction. Since apoptosis represents a physiological event that essentially contributes to the homeostasis of the organism, inappropriate apoptosis is involved in many disorders, including immune deficiency and autoimmune diseases, Alzheimer's disease, and various malignancies (Carson et al., 1993; Barr and Tomei, 1994; Kusiak et al., 1996). Consequently, control of apoptosis has been recognized as an important target for therapeutic intervention, making elucidation of the molecular mechanisms regulating this process of primary interest.
UV light represents one of the most important environmental factors. Besides its well-known advantages and its indispensable effects on human life, UV light, and in particular the middle wave length range (290–320 nm), called UVB, can be a hazard to human health by inducing cancer, premature skin aging, immunosuppression, inflammation, and cell death (Young, 1987; Gilchrest, 1990; Kripke, 1990; Fisher et al., 1996; Kraemer, 1997). A hallmark event of UV exposure is the occurrence of sunburn cells within the epidermis (Danno and Horio, 1987; Young, 1987). By using simple morphological criteria, these cells have been recognized for a long time as keratinocytes undergoing apoptosis. By applying more advanced techniques, it was later confirmed that UV light induces apoptosis in keratinocytes and epithelial cell lines (Martin and Cotter, 1991; Casciola-Rosen et al., 1994; Schwarz et al., 1995; Benassi et al., 1997; Gniadecki et al., 1997; Leverkus et al., 1997). Until recently, the functional role of sunburn cells was completely obscure and just regarded as a marker for severity of sun damage. Ziegler et al. (1994) currently provided evidence that, in contrast to the conventional view, sunburn cell formation may be important for preventing skin cancer. In this process, the tumor suppressor gene p53 appears to be critically involved since mice devoid of functional p53 develop almost no sunburn cells compared with control mice after irradiation with equal doses of UV light (Ziegler et al., 1994). This supports the concept that UV-damaged keratinocytes that failed to repair the damage will die as sunburn cells, thus escaping the risk of becoming malignant. Therefore, the formation of sunburn cells can be regarded as a scavenging phenomenon protecting the individual from developing UV-induced skin cancer. Consequently, keratinocytes with p53 mutations appear to be more susceptible to the tumor-promoting effects of UV light. Because of diminished p53-mediated apoptotic cell death, these cells can now survive, whereas neighboring cells carrying damaged DNA but wild-type p53 are eliminated by apoptosis (Brash et al., 1996; Kraemer, 1997). By preferentially mutating p53 (Brash et al., 1991), UV light can exert a selective pressure for the mutated, damage-resistant keratinocytes, thereby allowing these cells to clonally expand and to form actinic keratosis, the prestage of skin cancer (Ziegler et al., 1994). However, because up to 4% of keratinocytes of normal appearing sun-exposed skin cells carry p53 mutations but far less develop into actinic keratoses or cancer, the majority has to undergo squamous differentiation or apoptosis (Jonason et al., 1996; Kraemer, 1997). This implies that pathways other than p53 must be involved in UV-induced apoptosis of keratinocytes. This is also supported by the observation that the spontaneously transformed human keratinocyte cell line HaCaT, which carries p53 mutations (Lehman et al., 1993), undergoes apoptosis upon UV exposure (Schwarz et al., 1995).
CD95 (Fas/APO-1) is a death-promoting receptor that belongs to the tumor necrosis factor (TNF)1 receptor family (Trauth et al., 1989; Ito et al., 1991). Triggering of the CD95 molecule either by agonistic antibodies or by the natural ligand CD95L (FasL) induces apoptosis (Suda et al., 1993). Ligand binding induces trimerization of CD95, and the trimerized cytoplasmic region then transduces the signal by recruiting a molecule called FADD (Fas-associating protein with death domain) or MORT1 (mediator of receptor-induced toxicity), which binds to CD95 via interaction of the death domain at its COOH terminus (Boldin et al., 1995; Chinnayan et al., 1995). The NH2-terminal region of FADD is responsible for downstream signal transduction by recruitment of a protein called FLICE (FADD-like interleukin-1β–converting enzyme [ICE]) or MACH (MORT1-associated CED-3 homologue), recently designated as caspase-8 (Alnemri et al., 1996; Boldin et al., 1996; Muzio et al., 1996). The NH2 terminus of caspase-8 binds to FADD/MORT1, while its COOH-terminal region is related to the caspase-3 (CPP32) subfamily. Therefore, FLICE/MACH preferentially cleaves caspase-3 substrates such as poly(ADP)ribose polymerase (PARP) (Boldin et al., 1996) but can also cleave ICE proteases including CPP32 (Muzio et al., 1997).
The importance of the CD95/CD95L system for tissue homeostasis is best demonstrated by the mouse mutations lpr and gld, respectively. Mice carrying homozygous mutations in lpr do not express functional CD95 (Watanabe-Fukunaga et al., 1992), while mice with gld mutations lack functional CD95L (Takahashi et al., 1994). Both mice strains develop autoimmune features like splenomegaly and lymphadenopathy. CD95 is ubiquitously expressed in various tissues. CD95L was initially found on activated T lymphocytes, natural killer cells, and cells of “immune privileged organs” like testis and the anterior chamber of the eye (Bellgrau et al., 1995; Griffith et al., 1995; Friesen et al., 1996; Tanaka et al., 1996), yet the list of cells being able to express CD95L under certain conditions is steadily increasing (Hahne et al., 1996; Giordano et al., 1997).
Keratinocytes have been shown to express CD95 both in vitro and in vivo (Sayama et al., 1994; Matsue et al., 1995). Accordingly, treatment of keratinocytes with agonistic CD95 antibodies after upregulation of CD95 by interferon-γ results in apoptosis (Matsue et al., 1995; Takahashi et al., 1995). Although there is no doubt that keratinocytes express CD95L at the mRNA and protein level (Leverkus et al., 1997), it has not yet been proven that it is functionally active (Viard, I., A. Limat, M. Schröter, M. Hahne, T. Hunziker, J.-H. Saurat, J. Tschopp, and L. French. 1997. J. Invest. Dermatol. 108:569a).
Since pathways other than p53 must be involved in UV-induced apoptosis of keratinocytes, we investigated whether the CD95/CD95L system plays a role in this process. Here, we show that UV light induces activation of CPP32, PARP cleavage, and finally apoptosis in the human keratinocyte cell line HaCaT similarly to CD95L. UV-induced apoptosis of HaCaT cells, however, could neither be prevented nor reduced by neutralizing CD95 antibodies, suggesting that interaction of CD95 with its ligand CD95L may not be of major importance in UV- induced apoptosis. However, immunoprecipitation of UV-exposed HaCaT cells with an anti-CD95 antibody followed by Western blotting with an anti-FADD antibody revealed that UV light induces recruitment of FADD to CD95. This indicates that UV light activates CD95 directly and independently of the ligand CD95L. This was confirmed by confocal laser scanning microscopy showing that UV light induces clustering of the CD95 receptor similarly to CD95L. Prevention of UV-induced CD95 clustering by keeping cells at low temperatures was associated with a significantly reduced death rate. Moreover, UV-induced apoptosis was reduced in HaCaT cells transfected with a dominant negative FADD mutant. Together, these data indicate that UV light directly activates CD95 and thereby stimulates the CD95 pathway to induce apoptosis independently of the natural ligand CD95L.
Materials and Methods
Cell Culture and Irradiation
The spontaneously transformed human keratinocyte cell line HaCaT was kindly provided by Dr. Fusenig (German Cancer Research Center, Heidelberg, FRG) (Boukamp et al., 1988), and the human skin–derived squamous cell carcinoma line SCL-1 was provided by Dr. Boukamp (German Cancer Research Center) (Boukamp et al., 1988). UVB irradiation was performed as described (Köck et al., 1990). Briefly, cells were seeded in DME supplemented with 10% FCS into tissue culture dishes at a density of 2 × 105/ml and grown until subconfluency. Immediately before UV irradiation, cells were washed twice with prewarmed PBS and exposed to UV through PBS. For UV irradiation, we used a bank of four FS20 bulbs (Westinghouse Electric Corp., Pittsburgh, PA), which emit most of their energy within the UVB range (290–320 nm) with an emission peak at 313 nm. Throughout this study, a dose of 300 J/m2 was used since according to pilot experiments this dose induced apoptosis in a significant percentage of cells. Control cells were subjected to the identical procedure without being UV exposed.
Reagents
The following antibodies directed against human CD95 were used: A rabbit IgG (Santa Cruz Biotechnology Inc., Santa Cruz, CA) was used for Western blot analysis; a mouse IgG1 (ZB4; Upstate Biotechnology, Inc., Lake Placid, NY) was used for neutralization; a mouse IgG3 (Celldiagnostica, Münster, Germany) was used for immunoprecipitation; and a mouse IgM (CH-11; Immunotech, Hamburg, FRG) was used to induce apoptosis. A rabbit IgG against human FADD was obtained from Santa Cruz. Recombinant CD95L was purchased from Alexis (San Diego, CA). To enhance the efficacy of killing, CD95L was preincubated with an equal amount of an enhancer protein (Alexis) at 37°C for 30 min before addition to cells. Activation of ICE-proteases was blocked by the tripeptide z-Val-Ala-Asp-CH2F (zVAD; Enzyme Systems Products, Livermore, CA). Recombinant human TNF-α was obtained from Boehringer Mannheim (Mannheim, Germany). Monoclonal antibody 985 against the extracellular domain of TNF receptor 1 (TNFR1) and a rabbit polyclonal antibody against TNRF1-associated death domain (TRADD) were kindly provided by D. Goeddel (Tularik, San Francicso, CA) (Hsu et al., 1995). The FADD dominant negative mutant construct pcDNA3-FADD-DN was kindly provided by V. Dixit (University of Michigan, Ann Arbor, MI) (Chinnaiyan et al., 1996).
Detection of Cell Death
Unless otherwise stated, for evaluation of apoptosis, a cell death detection ELISA (Cell Death Detection ELISAPLUS; Boehringer Mannheim) was used according to the manufacturer's instructions. 16 h after treatment with UV light or recombinant CD95L, cells were detached from dishes by trypsinization and centrifuged, and cell pellets were resuspended in PBS. The principle of this test is based on the detection of mono- and oligonucleosomes in the cytoplasmic fractions of cell lysates by using biotinylated antihiston- and peroxidase-coupled anti-DNA antibodies. The enrichment of mono- and oligonucleosomes released into the cytoplasm is calculated as absorbance of sample cells/absorbance of control cells. Enrichment factor was used as a parameter of apoptosis and shown on the y-axis as mean ± SD of triplicates. An enrichment factor of 1.0 corresponded approximately to 7–10% of dead cells as determined in parallel by FACS® analysis after staining with FITC-labeled annexin V (Bender Corp., Vienna, Austria). Assays were performed at least three times, and data shown are representatives of those.
Transfection
Cells were cotransfected with 1 μg of pCMVβ-gal vector (Stratagene, Heidelberg, FRG) and 1 μg of either pcDNA3-FADD-DN or 1 μg of pcDNA3 using Lipofectamine (Life Technologies, Gaithersburg, MD). Cells were treated 24 h after transfection with either 200 ng/ml anti– human CD95 antibody (Immunotech, Hamburg, FRG) or 300 J/m2 UV light. Control cells were left untreated. 16 h later, cells were fixed in PBS buffer containing 2% paraformaldehyde, 0.2% glutaraldehyde at 4°C for 20 min, and stained for 5 h with X-gal (100 μg/ml), 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, and 1 mM MgCl2 in PBS. Positive cells were microscopically observed for apoptosis according to the method described by Bertin et al. (1997) and Hsu et al. (1995) with slight modifications. Apoptotic cells were differentiated as blue rounded cells from intact living flat cells. Four random fields were counted. Data are given as percentage of living cells calculated according to the following formula: (number of living cells per field/number of total cells per field) × 100.
Immunoprecipitation
After stimulation, cells were incubated with the cleavable cross-linker 3,3′-dithiobis[sulfosuccinimidyl-propionate] (Pierce Chemical Co., Rockford, IL) for 10 min at 4°C. The reaction was stopped by incubation in PBS containing 10 mM ammonium acetate for 5 min at 4°C. Cells were detached from dishes by use of a rubber policeman, washed twice in ice cold PBS, and lysed in lysis buffer (30 mM Tris, pH 7.5, 150 mM NaCl, 1 mM PMSF, 4 μg/ml aprotinin, 1% NP-40, 10% glycerol; all purchased from Sigma Chemical Co., St. Louis, MO) for 15 min on ice. After centrifugation at 14,000 rpm at 4°C for 15 min, an anti–human CD95 antibody or an anti–human TNFR1 antibody and protein G–Sepharose were added and reacted at 4°C overnight. The protein-antibody-beads complex was washed five times in lysis buffer, and proteins were eluted from the beads by incubation in boiling water for 5 min in the presence of 2-mercaptoethanol at a final concentration of 2.5% and centrifuged at 14,000 rpm, 4°C for 1 min. Supernatants were applied to 12% SDS-PAGE at 150 V for 2.5–3 h. Subsequently, proteins were blotted to nitrocellulose membranes. For Western blot analysis, membranes were first blocked in TBS-T solution (10 mM Tris, pH 8, 150 mM NaCl, 0.1% Tween-20 [Sigma Chemical Co.]) containing 5% ovalbumin (Sigma Chemcial Co.) at room temperature for 2 h before addition of the respective antibodies.
Western Blot Analysis
Cells were harvested by use of a rubber policeman and lysed in RIPA buffer (10 mM Tris, pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 4 μg/ml aprotinin, 1 mM sodium orthovanadate) for 15 min on ice. After centrifugation, supernatants were collected, and the protein content was measured with the Bio-Rad Protein assay kit (Bio-Rad Labs, Hercules, CA). The protein samples were subjected to SDS-PAGE, blotted to nitrocellulose membranes, and incubated with antibodies directed against CPP32 (Pharmingen, San Diego, CA) or against PARP (Pharmingen). To monitor equal loading of proteins, membranes were incubated with an antibody directed against α-tubulin (Pharmingen). Signals were detected by use of an ECL-kit (Amersham International, Buckinghamshire, UK).
Confocal Laser Scanning Microscopy
Cells were seeded into tissue culture chambers (Chamber Slide; Nunc, Naperville, IL). 24 h later, medium was changed, and cells were treated with 100 ng/ml CD95L and 10 ng/ml TNF-α or 300 J/m2 UV. Control cells were left untreated. 30 min after stimulation, cells were fixed in 4% paraformaldehyde (Sigma Chemical Co.) and washed twice with PBS for 10 min. Cells were preincubated with 2% BSA (Sigma Chemical Co.) for 30 min and incubated with an antibody directed against CD95 (ZB4) diluted in 1% BSA for 2 h at room temperature. The samples were further processed by an indirect immunofluorescence technique using FITC-conjugated goat anti–mouse antibodies (dilution 1:50; Dianova, Hamburg, FRG). For retardation of fading of the fluorochrome during laser microscopy, 1,4-diazobicyclo-(2,2,2)-octane (Merck, Darmstadt, FRG) was added to the glycerol used for mounting the preparations. The specimens were analyzed with a confocal laser scanning microscope equipped with an argon laser (model Wild Leitz-CLSM Diaplan; Leitz, Heidelberg, FRG). The confocal principle eliminates light out of focus, allows optical sectioning, and gives an improvement in resolution by a factor of 1.4 (Brakenhof et al., 1979; White et al., 1987). The cells were x/y scanned in the reflecting mode. For optical sectioning, the pinhole was closed to 60 nm, and an image averaging was performed. The images were stored on an optic disc, later processed with a highpass filter, and displayed in glow overflow pseudocolor.
Results
Anti-CD95 Antibodies Prevent CD95L-mediated but Not UV-induced Apoptosis of HaCaT Cells
To study whether the CD95 pathway is involved in UV- induced apoptosis of keratinocyte cell lines, the spontaneously transformed human keratinocyte cell line HaCaT was used since this cell line expresses CD95 (data not shown) and undergoes apoptosis after UV exposure (Schwarz et al., 1995). Treatment of HaCaT cells with 100 ng/ml recombinant CD95L caused remarkable killing, as measured by a cell death detection ELISA (Fig. 1). A similar DNA fragmentation rate was observed in HaCaT cells after exposure to 300 J/m2 UV light, confirming previously published data (Schwarz et al., 1995). CD95L-induced killing of HaCaT cells was completely prevented by coincubating the cells with a neutralizing antibody directed against CD95 (1 μg/ml) for 30 min (Fig. 1). In contrast, UV-induced killing was not at all affected by the anti-CD95 antibody, implying so far that the CD95 pathway may not be involved in UV-induced apoptosis.
Figure 1.

Anti-CD95 antibody prevents CD95L- but not UV- induced apoptosis. HaCaT cells (2 × 105/ml) were pretreated with a neutralizing anti-CD95 antibody (anti-CD95) or with an isotype control (iso). 30 min later, cells were either exposed to 300 J/m2 UV light or stimulated with 100 ng/ml recombinant CD95L. For control purposes cells were left untreated (−). 16 h later, apoptosis was examined by determining nucleosomal DNA fragmentation using an apoptosis determination kit. Rate of apoptosis is reflected by the enrichment of nucleosomes in the cytoplasm shown on the y-axis. Data presented show one representative of three independently performed experiments.
Like CD95L, UV Light Activates CPP32 Followed by Cleavage of the Death Substrate PARP
To further elucidate the mechanisms involved in UV- induced apoptosis, we investigated whether UV light induces activation of CPP32, a member of the family of ICE proteases. CPP32 was recently identified to be involved in CD95-mediated killing (Enari et al., 1996; Schlegel et al., 1996). CPP32 can only act in a functional manner when it is cleaved into its 17-kD active form (Nicholson et al., 1995). HaCaT cells were exposed to 300 J/m2 UV and 2, 4, 8, and 16 h later cell lysates were prepared for Western blotting using an antibody against CPP32. Since this antibody is directed against a domain of the CPP32 proform, it cannot recognize the processed 17-kD form, resulting in loss of the immunoreactive band in samples in which CPP32 has been activated. As shown in Fig. 2 a, a significant disappearance of CPP32 was observed in samples of UV-exposed HaCaT cells. UV-induced cleavage (activation) of CPP32 occurred as early as 2 h after irradiation, and complete processing was observed after 8 h. To exclude the possibility that the loss of the immunoreactive band is due to differences in the amounts of proteins loaded, the same samples were subjected to Western blotting against α-tubulin and showed identical intensities of signals (Fig. 2 c).
Figure 2.

Cleavage of CPP32 and PARP by UV light. HaCaT cells were irradiated with 300 J/m2 UV or left untreated. 2, 4, 8, and 16 h after stimulation proteins were extracted, and Western blot analysis was performed using antibodies directed against CPP32 (a) or PARP (b). To monitor equal loading of protein samples, Western blot using an antibody directed against α-tubulin was performed (c).
Since CPP32 cleaves the death substrate PARP (Lazebnik et al., 1994), we next examined whether UV exposure results in cleavage of PARP. As shown in Fig. 2 b, PARP was found cleaved from its intact 116-kD form into the 85-kD fragment in samples of UV-exposed cells. PARP cleavage followed the activation of CPP32 since PARP remained uncleaved until 4 h after irradiation, but cleaved 85-kD fragments appeared 8 h after irradiation. A similar cleavage pattern was also observed in HaCaT cells treated with CD95L (data not shown).
UV-induced Apoptosis Is Prevented by the ICE Inhibitor zVAD
To prove that activation of ICE-like proteases is functionally relevant in UV-induced apoptosis, the effect of the ICE family inhibitor zVAD on UV-induced apoptosis was investigated. UV-induced killing of HaCaT cells was almost completely prevented when cells were preincubated with 20 μM zVAD for 30 min (Fig. 3). For control purposes, cells were treated with CD95L in the presence or absence of zVAD, and viability was monitored for 16 h thereafter. CD95L-mediated killing was completely blocked by zVAD, confirming previous findings demonstrating the involvement of ICE-like proteases in CD95-mediated apoptosis (Enari et al., 1995; Los et al., 1995). Together, these data suggest that activation of ICE proteases is not only crucial in CD95-mediated apoptosis but also in UV-induced cell death.
Figure 3.

The ICE inhibitor zVAD inhibits both UV- and CD95L-induced cell death. HaCaT cells were either pretreated with 20 μM zVAD or left untreated. 30 min later, cells were either exposed to 300 J/m2 UV light, stimulated with 100 ng/ml recombinant CD95L, or left untreated (−). 16 h later, apoptosis was examined by determining nucleosomal DNA fragmentation using an apoptosis determination kit. Rate of apoptosis is reflected by the enrichment of nucleosomes in the cytoplasm shown on the y-axis. Data presented show one representative of three independently performed experiments.
UV Light Induces Recruitment of FADD to CD95
It has been reported that activation of CD95 requires trimerization of the receptor, which results in recruitment of the FADD protein (for review see Chinnaiyan et al., 1995; Nagata, 1997). After recruitment of FADD to CD95, the most upstream ICE-like protease, FADD-homologous ICE/CED-3 like protease (FLICE) (Boldin et al., 1996; Muzio et al., 1996) is further recruited, completing the formation of the death-inducing signaling complex (Kischkel et al., 1995). Since our data obtained so far revealed similarities between UV- and CD95L-induced cell death of HaCaT cells but also clearly showed that UV-induced cell death cannot be prevented by blocking CD95 with neutralizing antibodies, we studied whether UV exposure induces recruitment of FADD to CD95. For this purpose, cells were exposed to UV light, and 1 and 4 h later cell lysates were obtained and immunoprecipitated with an antibody directed against CD95. Western blot analysis using an antibody directed against FADD revealed that UV light rapidly induced recruitment of FADD to CD95 (Fig. 4 a). To examine the amounts of recovered proteins loaded to the gel, the same membranes were reprobed with an antibody directed against CD95 (Fig. 4 b). As a positive control, the same procedure was performed with HaCaT cells, which were treated with CD95L instead of UV light. As expected, stimulation of CD95 by the ligand also resulted in recruitment of FADD to CD95.
Figure 4.

UV light induces recruitment of FADD to CD95. HaCaT cells were either stimulated with 100 ng/ml recombinant CD95L, irradiated with 300 J/m2 UV light, or left untreated. 1 and 4 h later, proteins were extracted and immunoprecipitated with an antibody directed against CD95. After blotting to nitrocellulose membranes, Western blot analysis was performed using an antibody directed against FADD (a). To monitor loading of protein samples, the same membranes were reprobed with an anti-CD95 antibody (b).
UV Light Induces Aggregation of CD95 on HaCaT Cells
Recently, Rosette and Karin (1996) have reported that UV light can induce aggregation of cytokine receptors, including those for TNF, EGF, and interleukin-1 independently of their respective ligands. Since FADD recruitment has been shown to be a consequence of CD95 trimerization (Chinnayan et al., 1995), we next addressed whether UV light can induce CD95 receptor aggregation. Therefore, cells were either irradiated with UV light, stimulated with CD95L, or left untreated. 30 min later, cells were fixed with 4% paraformaldehyde, incubated with a murine antibody directed against CD95, and stained by an indirect immunofluorescence technique using FITC-labeled goat anti–mouse IgG for further analysis by confocal laser scanning microscopy. While untreated HaCaT cells showed an extremely weak diffuse staining of CD95 (Fig. 5 a), stimulation of cells with CD95L resulted in rapid aggregation of CD95, enabling a dense patchy staining that was primarily membrane localized (Fig. 5 b). The staining appears to be specific because stimulation of cells with TNF-α did not result in aggregation of CD95 (Fig. 5 d) but did for TNFR1, which was shown by using a TNFR1-specific antibody (data not shown). As shown in Fig. 5 c, UV exposure induced a similar aggregation pattern of CD95, which was almost indistinguishable from that induced by CD95L (Fig. 5 b). To gain further insight into the kinetics of UV-induced CD95 clustering, HaCaT cells were exposed to 300 J/m2 UV light, fixed after 0.5, 2, 6, or 10 h, stained with the anti-CD95 antibody, and subjected to confocal laser scanning microscopy. As the time sequence shows (Fig. 6), UV-induced CD95 clustering lasted up to 6 h after UV exposure and then faded, yielding after 10 h a less intense and much more diffuse, partially cytoplasmic staining pattern (Fig. 6 d). Evaluation of staining at later time points did not yield further assessable data since cells then start to die, which is associated with significant changes in cell morphology. Taken together, these data strongly suggest that UV light activates the CD95 receptor directly by inducing its aggregation.
Figure 5.
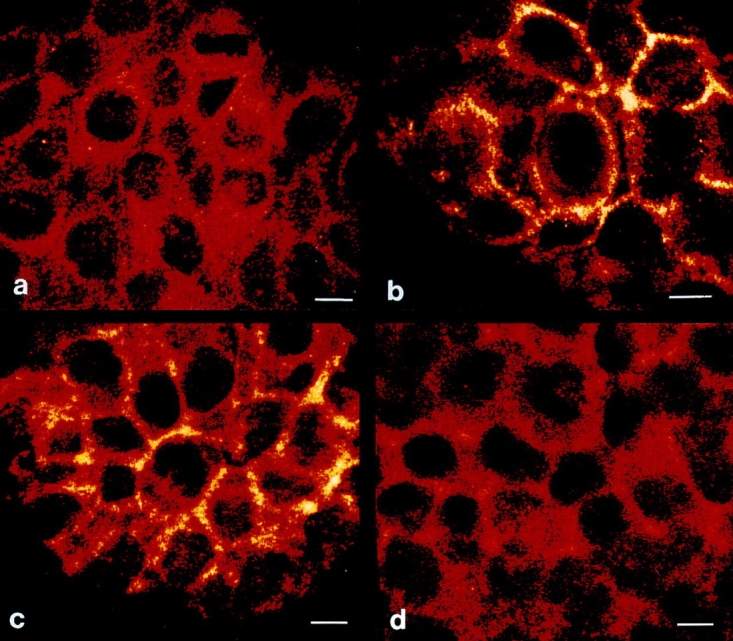
UV light induces CD95 clustering on HaCaT cells. HaCaT cells were either left untreated (a), stimulated with 100 ng/ml recombinant CD95L (b), irradiated with 300 J/m2 UV light (c), or treated with 10 ng/ml TNF-α (d). 30 min later cells were fixed, incubated with an antibody directed against CD95, incubated with an FITC-conjugated secondary antibody, and subjected to confocal laser scanning microscopy. Bars, 5 μm.
Figure 6.
Kinetics of UV- induced CD95 clustering. HaCaT cells were irradiated with 300 J/m2 UV light. Cells were fixed after 0.5 (a), 2 (b), 6 (c), or 10 h (d) of incubation with an antibody directed against CD95. They were then incubated with an FITC-conjugated secondary antibody and subjected to confocal laser scanning microscopy. Bars, 5 μm.
Reduction of UV-induced Apoptosis by Prevention of CD95 Aggregation
To test whether UV-induced clustering of CD95 is functionally relevant for the induction of apoptosis by UV light, the effect of exposing HaCaT cells to UV light at lower temperatures was examined. Rosette and Karin (1996) showed that incubating cells at 10°C prevented receptor clustering after UV irradiation and consequently inhibited JNK activation. Therefore, we surmised that if the clustering of CD95 caused by UV light is functionally relevant for the induction of apoptosis, cells exposed to UV light at low temperatures should be less susceptible to apoptosis. In contrast, keeping cells exposed to CD95L at low temperature for the same period should have no significant effect, provided that CD95L is not washed off and remains present after culturing at 37°C for the next 16 h. Therefore, HaCaT cells were kept at 10°C for 30 min and exposed to 300 J/m2 at this temperature. After 10 min, cells were incubated at 37°C for 16 h, and the apoptosis rate was determined. Compared with HaCaT cells UV- irradiated at 37°C, cells irradiated at 10°C were significantly less susceptible to UV-induced killing (Fig. 7). In contrast, no difference in the rate of apoptosis was observed when cells were exposed to recombinant CD95L under identical conditions.
Figure 7.

Prevention of CD95 aggregation reduces UV-induced apoptosis. HaCaT cells were kept at 10°C, and 30 min later cells were either exposed to 300 J/m2 UV light, stimulated with 100 ng/ ml recombinant CD95L, or left untreated (−). 10 min after stimulation, cells were incubated at 37°C. 16 h later apoptosis was examined by determining nucleosomal DNA fragmentation using an apoptosis determination kit. For control purposes, cells were kept all the time at 37°C but were otherwise treated in an identical way. Rate of apoptosis is reflected by the enrichment of nucleosomes in the cytoplasm shown on the y-axis. Data presented show one representative of three independently performed experiments.
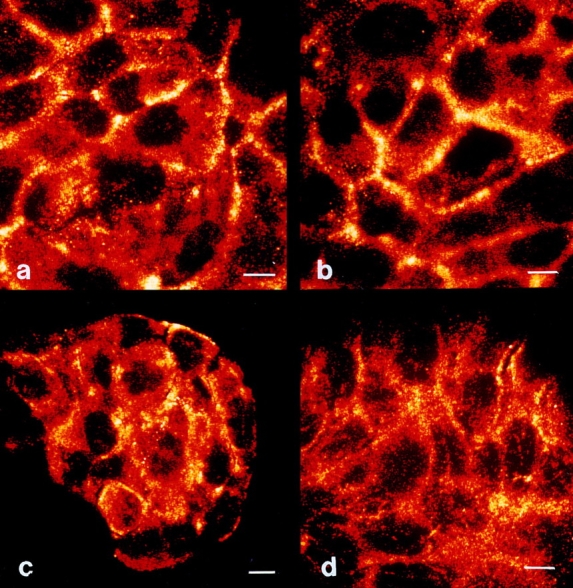
To prove that keeping cells at 10°C prevents UV-induced clustering of CD95, HaCaT cells were UV-exposed at low temperatures and kept at 37°C for 2 h, and receptor clustering was examined by confocal laser scanning microscopy as described above. In contrast to the clear clustering observed in HaCaT cells exposed to UV light at 37°C (Fig. 5 b), CD95 aggregation was significantly reduced when HaCaT cells were UV-irradiated at low temperatures (Fig. 8 a). Adding recombinant CD95L at low temperatures did not reduce clustering intensity, provided the cells were cultured at 37°C for the following 2 h in the presence of CD95L (Fig. 8 b). Together, these data show that UV light induces clustering of CD95 on HaCaT cells, which is of functional importance for UV-induced apoptosis since inhibition of clustering is associated with a reduced death rate.
Figure 8.

Low temperature prevents UV-induced clustering of CD95. HaCaT cells were kept at 10°C, and 30 min later cells were exposed to 300 J/m2 UV light (a) or stimulated with 100 ng/ml recombinant CD95L (b). 10 min after stimulation, cells were incubated at 37°C. 2 h later cells were fixed, incubated with an antibody directed against CD95, incubated with an FITC-conjugated secondary antibody, and subjected to confocal laser scanning microscopy. Bars, 5 μm.
Reduction of UV-induced Apoptosis in FADD Dominant Negative Mutant HaCaT Cells
To further prove that activation of CD95 by UV light is of functional relevance in UV-mediated apoptosis, HaCaT cells were cotransfected with pCMVβ-gal vector and with the FADD dominant negative mutant construct pcDNA3-FADD-DN or with the vector pcDNA3. 24 h after transfection, cells were treated with either 300 J/m2 UV, with an agonistic anti-CD95 antibody (200 ng/ml), or left untreated. 16 h later, cells were fixed, and viability of β-galactosidase–expressing cells was evaluated as described (Hsu et al., 1995; Bertin et al., 1997). Compared with mock-transfected HaCaT cells, FADD dominant negative mutant HaCaT cells were almost resistant to anti-CD95 antibody–mediated killing, indicating the efficacy of the FADD dominant negative mutant construct (Table I). The transfection procedure itself appeared to increase cell fragility to some extent since the death rate after 300 J/m2 UV light was enhanced in mock-transfected HaCaT cells as compared with untransfected cells. UV light was still capable of inducing killing of FADD dominant negative HaCaT cells; however, the rate of apoptosis was significantly reduced in comparison to UV-exposed mock-transfected cells. Thus, these data suggest that triggering of the CD95 receptor followed by FADD activation is functionally relevant and contributes to UV-induced apoptosis.
Table I.
UV-induced Apoptosis Is Reduced by FADD-DN Expression
| Transfectant* | Treatment‡ | Field: | Percentage of viable cells§ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||||||
| pc-DNA3-HaCaT | — | 96 | 100 | 95 | 95 | |||||||
| UV‖ | 5 | 3 | 8 | 2 | ||||||||
| anti-CD95 ¶ | 15 | 13 | 16 | 11 | ||||||||
| FADD-DN-HaCaT | — | 99 | 97 | 97 | 100 | |||||||
| UV‖ | 40 | 36 | 45 | 45 | ||||||||
| anti-CD95 ¶ | 88 | 82 | 90 | 85 | ||||||||
HaCaT cells were cotransfected with β-galactosidase and FADD-DN or pcDNA3.
HaCaT cells were treated 24 h after transfection.
Percentage of viable cells was determined according to the formula: (number of viable blue cells per field/number of total blue cells per field) × 100. Four fields were counted. Results show one representative of three independently performed experiments.
HaCaT cells were exposed to 300 J/cm2 UV light.
HaCaT cells were treated with an agonistic anti-CD95 antibody (CH-11, 200 ng/ml).
UV Light Induces CD95 Clustering in Cells Carrying Wild-Type p53
As mentioned above, HaCaT cells were selected for this study since these cells carry p53 mutations. After having shown that UV light directly activates the CD95 receptor in HaCaT cells, the question of whether the same phenomenon can be observed in cells carrying wild-type p53 emerged. For that purpose, the squamous cell carcinoma line SCL-1, which carries wild-type p53 (Boukamp, P., personal communication), was used. SCL-1 cells were irradiated with 300 J/m2 UV light and subjected to confocal laser scanning microscopy, as described above (Fig. 9). Similar to HaCaT cells, SCL-1 cells reacted with CD95 clustering to UV exposure (Fig. 9 b), and clustering was suppressed by UV irradiating SCL-1 cells at 10°C (Fig. 9 c). Moreover, to prove that CD95 clustering induced by UV light on SCL-1 cells is of functional relevance, rate of apoptosis after UV exposure at 10°C was evaluated. Although SCL-1 cells reacted both to UV light and CD59L treatment with apoptosis, SCL-1 cells appeared to be more susceptible to UV light than to CD95L, which may be explained by the fact that p53 is involved in UV- but not in CD95L-mediated apoptosis. As observed with HaCaT cells, UV irradiation of SCL-1 at 10°C was associated with a reduced death rate as compared with UV exposure at 37°C (Fig. 10). In contrast, no difference in the rate of apoptosis was observed when SCL-1 cells were exposed to recombinant CD95L under identical conditions. This indicates that CD95 clustering is also functionally relevant in p53 wild-type cells.
Figure 9.

UV light induces CD95 clustering on SCL-1 cells. SCL-1 cells were either left untreated (a) or irradiated with 300 J/m2 UV light at 37°C (b) or at 10°C (c). 30 min later, cells were fixed, incubated with an antibody directed against CD95, incubated with an FITC-conjugated secondary antibody, and subjected to confocal laser scanning microscopy. Bars, 5 μm.
Figure 10.

Prevention of CD95 aggregation reduces UV-induced apoptosis in SCL-1 cells. SCL-1 cells were kept at 10°C, and 30 min later cells were either exposed to 300 J/m2 UV light, stimulated with 100 ng/ml recombinant CD95L, or left untreated (−). 10 min after stimulation, cells were incubated at 37°C. 16 h later apoptosis was examined by determining nucleosomal DNA fragmentation using an apoptosis determination kit. For control purposes, cells were kept all the time at 37°C but were otherwise treated in an identical way. Rate of apoptosis is reflected by the enrichment of nucleosomes in the cytoplasm shown on the y-axis. Data presented show one representative of three independently performed experiments.
Discussion
Skin cancer is the most frequently observed neoplasm in Caucasians. It has a lifetime risk nearly equal to that of all other cancers combined (Kraemer, 1997). There is unanimous agreement that sunlight is the major environmental agent involved in the induction of skin cancer. Taking into account that more than 800,000 people are expected to develop skin cancer this year in the United States (Kraemer, 1997), UV light can be regarded as one of the most important if not the most important carcinogen affecting mankind to date. Nature has equipped individuals with protection strategies to counteract the potent carcinogenic effects of UV light, DNA repair being the most important one. Consequently, DNA damage caused by UV light is constantly repaired, thereby preventing carcinogenesis. Patients with the inherited disorder, xeroderma pigmentosum, have a defect in DNA nucleotide excision repair and thus are very sensitive to the effects of sunlight and exhibit a 1000-fold higher risk of developing skin cancer than the average population (Kraemer et al., 1994).
Another way for a cell with damaged DNA to escape malignant transformation is to commit suicide by activating the apoptosis program. UV exposure of healthy skin results in the regular formation of apoptotic keratinocytes, called sunburn cells (Danno and Horio, 1987; Young, 1987). According to current hypotheses, sunburn cells represent keratinocytes that are so severely damaged that they cannot repair the DNA and thus induce apoptosis as the last escape mechanism, thereby eliminating themselves in the interest of the rest of the organism (Brash et al., 1996). In this process, the tumor suppressor gene p53 appears to play an important role. UV irradiation is known to arrest some cells during the G1 phase of the cell cycle in a p53-dependent manner (Campbell et al., 1993). This arrest gives the cell the chance to repair the DNA damage. Cells that do not express functional p53 protein are unable to arrest in G1 after irradiation and proceed unchecked through DNA synthesis. This prevents cells from repairing DNA damage and thus results in a higher rate of mutation. In addition, p53 has another “guardian” role because it can induce apoptosis, thereby eliminating cells being at risk to become malignant. Therefore, UV-induced mutation of p53 enhances the risk of development of skin cancer by exerting a clonal selection pressure of mutated damage-resistant cells. However, since a considerable number of keratinocytes in UV-exposed skin carry p53 mutations (Jonason et al., 1996) but only a few develop into actinic keratoses or skin cancer, mechanisms other than p53 should be involved in the induction of UV-mediated apoptosis of keratinocytes (Kraemer, 1997). This is also supported by the fact that HaCaT cells that carry p53 mutations (Lehman et al., 1993) become apoptotic upon UV exposure (Schwarz et al., 1995). Since the decision of whether keratinocytes upon UV irradiation undergo apoptosis or not may be a crucial process in photocarcinogenesis, we were interested in the molecular mechanisms involved in UV-induced apoptosis, in particular whether the apoptosis-related surface molecule CD95 is of importance.
Exposure of HaCaT cells to a UV dose that causes a significant rate of apoptosis in the presence or absence of a neutralizing antibody directed against CD95 revealed that this antibody had no effect on UV-induced apoptosis, whereas it effectively blocked CD95L-induced apoptosis of HaCaT cells. On the first glance, these findings somehow contradict the observations made by Leverkus et al. (1997), who recently reported that addition of a neutralizing antibody directed against CD95L inhibited UV-induced apoptosis of keratinocytes to some extent. However, in this study keratinocytes had to be pretreated with interferon-γ before UV exposure to observe this effect. Moreover, although CD95L expression by keratinocytes was detected by semiquantitative PCR, Western blot, and FACS® analysis, it has not been shown whether it is functionally active. The same applies for HaCaT cells; although we can detect CD95L both at the RNA and protein level (Maeola, A., Y. Aragane, A. Schwarz, T. Luger, and T. Schwarz. 1997. J. Invest. Dermatol. 108:569a), we were not able to show that CD95L-expressing HaCaT cells can kill CD95- expressing target cells. In addition, we were not able to isolate functionally active soluble CD95L from HaCaT cell supernatants. Thus, it remains to be determined whether CD95L expressed by keratinocytes is of functional relevance. Furthermore, the UV doses applied in our study did not have a significant effect on the expression of either CD95 or CD95L in HaCaT cells. Together these findings and the failure of the neutralizing CD95 antibody to inhibit UV-induced apoptosis of HaCaT cells initially suggested that the CD95 pathway should not be relevant in UV-mediated apoptosis. However, the observation that UV irradiation of HaCaT cells activates the ICE-protease CPP32 followed by cleavage of its substrate PARP and that UV-induced apoptosis can be inhibited by the ICE family inhibitory tripeptide, zVAD, suggested similarities in the pathways involved in UV-induced and CD95-mediated apoptosis (Enari et al., 1996; reviewed in Schulze- Osthoff et al., 1996). Further insights into the mechanisms involved were obtained by addressing whether UV light directly activates the CD95 receptor. One of the earliest consequences of CD95 activation is recruitment of the signaling protein FADD (Chinnayan et al., 1995). When UV-exposed HaCaT cells were immunoprecipitated with an antibody directed against CD95, FADD protein was found to be coprecipitated. The amount of coprecipitated FADD after UV exposure was similar to the amount of FADD protein that associated with the CD95 receptor after incubation with the natural ligand CD95L. This indicates that UV exposure activates the CD95 receptor but obviously independently of CD95L.
Recently, Rosette and Karin (1996) reported that UV light and osmotic shock, respectively, can activate the JNK cascade via activation of multiple growth factor and cytokine receptors. Exposure of HeLa cells to UV light induced clustering and internalization of cell surface receptors for EGF, TNF, and interleukin-1. Since the initial event during activation of CD95 is its trimerization followed by FADD recruitment, we addressed whether UV light directly induces CD95 clustering. Therefore, HaCaT cells were exposed to UV light and stained with an antibody against CD95 followed by a second FITC-conjugated antibody. Evaluation of the staining pattern by confocal laser scanning microscopy revealed only very weak, barely discernible CD95 staining on the surface of untreated HaCaT cells. This weak staining is not due to absence of CD95 on the surface of HaCaT cells but is probably due to the sensitivity of the method used. This assumption is primarily based on the fact that Rosette and Karin observed an identically weak constitutive staining, and secondly that the expression of CD95 by HaCaT cells is clearly visible when using FACS® analysis as a detection method (data not shown). However, when UV-exposed HaCaT cells were subjected to confocal laser scanning microscopy, a marked patchy staining compatible with receptor clustering was observed. A similar staining pattern was found on HaCaT cells that were stimulated with recombinant CD95L. These data indicate that UV light triggers the CD95 pathway by directly activating the CD95 receptor. Since interaction of CD95L with the CD95 receptor is not needed for this direct activation, this explains our initial finding that addition of a neutralizing CD95 antibody had no effect on UV-induced apoptosis. Based on their findings, Rosette and Karin predicted that any receptor whose activation mechanism involves multimerization should be activatable by UV light. Our present findings strongly support this prediction.
Since HaCaT cells are known to express TNFR1 and that autocrine release of TNF-α has been shown to contribute at least partially to UV-induced apoptosis (Schwarz et al., 1995), we also studied whether UV exposure of HaCaT cells activates TNFR1. One of the early events after TNFR1 activation is recruitment of the TRADD protein (Hsu et al., 1995). Immunoprecipitation with an antibody directed against TNFR1 followed by Western blot analysis against TRADD showed that UV light, similar to the ligand TNF-α, induced TRADD recruitment in HaCaT cells (data not shown), thus confirming the observation reported by Rosette and Karin (1996). Since TRADD finally recruits FADD, this pathway may also contribute to UV-induced apoptosis. However, based on immunoprecipitation alone it is not possible to differentiate to which extent CD95 and TNFR1 activation, respectively, are involved in the apoptotic pathway. TNFR1 may also be activated at a later time point after UV exposure by TNF-α released by keratinocytes, since it was shown that UV-induced apoptosis can be partially reduced by blocking TNF-α (Schwarz et al., 1995).
How UV light leads to multimerization of cell surface receptors remains unclear, but physical perturbation of the plasma membrane or a conformational change caused by energy absorption have to be considered as likely possibilities (Rosette and Karin, 1996). To gain further insights into whether UV-induced clustering of CD95 on HaCaT cells is of functional relevance for UV-mediated apoptosis, we wondered whether blocking of the clustering is associated with a reduced death rate after UV irradiation. Exposing HaCaT cells to UV light at 10°C, which is below the transition temperature of the membrane, resulted in a significantly reduced killing rate, demonstrating that UV- induced clustering of CD95 is functionally linked to UV-mediated apoptosis. Furthermore, HaCaT cells transfected with a FADD dominant negative mutant construct were less susceptible to UV-induced apoptosis than mock-transfected cells, supporting the functional relevance of this pathway. However, it is important to mention that neither exposing HaCaT cells to UV light at low temperature nor eliminating the FADD pathway completely prevented UV-induced apoptosis. This suggests that additional pathways may be involved. An important phenomenon might be DNA damage, as suggested by Ziegler et al. (1994). It is therefore possible that two phases of UV-mediated death might exist: a first phase in which direct clustering of surface receptors, such as CD95 and TNFR1, is induced, and a second phase in which UV irradiation induces DNA damage. It is important to mention that both pathways are not mutually exclusive. However, the only direct proof that DNA damage is involved would be to enhance DNA repair and determine whether rate of apoptosis is reduced. This issue is currently being addressed in our laboratory. While DNA damage–induced apoptosis after UV exposure is suggested to be p53 dependent (Ziegler et al., 1994), CD95 activation might be independent of p53 since similar findings were obtained with p53-mutated (HaCaT) and p53 wild-type (SCL-1) cells.
Taken together, these results for the first time demonstrate that UV light directly activates the CD95 receptor and thereby contributes to the induction of apoptosis. Since UV-induced apoptosis of keratinocytes is a cancer protective mechanism, disturbed expression of CD95 may be of importance for photocarcinogenesis. Therefore, clarification of the mechanisms involved in the regulation of CD95 expression on keratinocytes may contribute to a better understanding of photocarcinogenesis. In particular, potential stimuli downregulating CD95 expression should be identified. Furthermore, the present findings strongly support the concept that UV light can affect targets at the plasma membrane (Devary et al., 1992, 1993; Sachsenmaier et al., 1994; Simon et al., 1994) and that targeting of such structures is of functional relevance.
Acknowledgments
This work was supported by grants from the European Community (EV5V-CT94-0564, PL 970659) and from the German Research Foundation (Schw 625/1-2).
Abbreviations used in this paper
- CD95L
CD95 ligand
- FADD
Fas- associated protein with death domain
- ICE
interleukin-1β–converting enzyme
- PARP
poly(ADP)ribose polymerase
- TNF
tumor necrosis factor
- TNFR
tumor necrosis factor receptor
- TRADD
TNFR1-associated death domain
Note Added in Proof
While this paper was in press, similar observations were reported by Rehemtulla et al., 1997 (J. Biol. Chem. 272:25783–25786).
Footnotes
We gratefully thank K. Große-Heitmeyer, A. Mehling, and A. Schwarz for help and discussions, and we thank J. Bückmann and P. Wissel for preparing the graphs. The authors are grateful to Drs. P. Boukamp, N. Fusenig, V. Dixit, and D. Goeddel for providing reagents. The authors are thankful to Dr. T. Tezuka (Department of Dermatology, Kinki University School of Medicine, Osaka, Japan) for his continuous and generous support.
References
- Alnemri ES, Livingston DJ, Nicholson DW, Salyesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Ameisen JC. Programmed cell death (apoptosis) and cell survival regulation: relevance to AIDS and cancer. AIDS. 1994;8:1197–1213. doi: 10.1097/00002030-199409000-00001. [DOI] [PubMed] [Google Scholar]
- Barr PJ, Tomei LD. Apoptosis and its role in human disease. Biotechnology NY. 1994;12:487–493. doi: 10.1038/nbt0594-487. [DOI] [PubMed] [Google Scholar]
- Bellgrau D, Gold D, Selawry H, Moore J, Franzusoff A, Duke RC. A role for CD95 ligand in preventing graft rejection. Nature. 1995;377:630–632. doi: 10.1038/377630a0. [DOI] [PubMed] [Google Scholar]
- Benassi L, Ottani D, Fantini F, Marconi A, Chiodino C, Gianetti A, Pincelli C. 1,25-Dihydroxyvitamin D3, transforming growth factor β1, calcium and ultraviolet B radiation induce apoptosis in cultured human keratinocytes. J Invest Dermatol. 1997;109:276–282. doi: 10.1111/1523-1747.ep12335756. [DOI] [PubMed] [Google Scholar]
- Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, Wang G-H, Senkevich TG, Alnemri ES ,. B. Moss, et al. Death effector domain-containing herpesvirus and poxvirus proteins inhibit Fas and TNFR1-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:1172–1176. doi: 10.1073/pnas.94.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D. A novel protein that interacts with the death domain of Fas/ APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Boukamp P, Tilgen W, Dzarlieva RT, Breitkreutz D, Haag D, Riehl RK, Bohnert A, Fusenig NE. Phenotypic and genotypic characteristics of a cell line from a squamous carcinoma of human skin. J Natl Cancer Inst. 1982;68:415–427. [PubMed] [Google Scholar]
- Boukamp P, Petrussevska RT, Breikreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakenhoff GL, Blom P, Barends P. Confocal scanning light microscope with high aperture immersion lens. J Microsc. 1979;117:219–222. [Google Scholar]
- Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Ponten J. A role for sunlight in skin cancer: UV- induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash DE, Ziegler A, Jonason AS, Simon JA, Kunala S, Lefell DJ. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J Invest Dermatol Symp Proc. 1996;1:136–142. [PubMed] [Google Scholar]
- Campbell C, Quinn AG, Angus B, Farr PM, Rees JL. Wavelength specific patterns of p53 induction in human skin following exposure to UV radiation. Cancer Res. 1993;53:2697–2699. [PubMed] [Google Scholar]
- Carson DA, Ribeiri JM. Apoptosis and disease. Lancet. 1993;341:1251–1254. doi: 10.1016/0140-6736(93)91154-e. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Tepper CG, Sedlin MF, O'Rourke K, Kischkel FC, Hellbardt S, Krammer PH, Peter ME, Dixit VM. FADD/ MORT1 is a common mediator of CD95(Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J Biol Chem. 1996;271:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- Cohen JJ. Programmed cell death in the immune system. Adv Immunol. 1991;50:55–85. doi: 10.1016/s0065-2776(08)60822-6. [DOI] [PubMed] [Google Scholar]
- Cohen JJ. Apoptosis. Immunol Today. 1993;14:126–130. doi: 10.1016/0167-5699(93)90214-6. [DOI] [PubMed] [Google Scholar]
- Danno K, Horio T. Sunburn cell: factors involved in its formation. Photochem Photobiol. 1987;45:683–690. doi: 10.1111/j.1751-1097.1987.tb07401.x. [DOI] [PubMed] [Google Scholar]
- Devary Y, Gottlieb RA, Smeal T, Karin M. The mammalian ultraviolet response is triggered by activation of Src tyrosine kinases. Cell. 1992;71:1081–1091. doi: 10.1016/s0092-8674(05)80058-3. [DOI] [PubMed] [Google Scholar]
- Devary Y, Rosette C, DiDonato JA, Karin M. NF-κB activation by ultraviolet light not dependent on a nuclear signal. Science. 1993;261:1442–1445. doi: 10.1126/science.8367725. [DOI] [PubMed] [Google Scholar]
- Enari M, Hug H, Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- Fisher GJ, Datta SC, Talwar HS, Wang ZQ, Varani J, Kang S, Voorhees JJ. Molecular basis of sun-induced premature skin aging and retinoid antagonism. Nature. 1996;379:335–339. doi: 10.1038/379335a0. [DOI] [PubMed] [Google Scholar]
- Friesen C, Herr I, Krammer PH, Debatin KM. Involvement of the CD95 (APO-1/Fas) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat Med. 1996;2:574–577. doi: 10.1038/nm0596-574. [DOI] [PubMed] [Google Scholar]
- Gehri R, Hahn S, Rothen M, Steuerwald M, Nuesch R, Erb P. The Fas receptor in HIV infection: expression on peripheral blood lymphocytes and role in the depletion of T cells. AIDS. 1996;10:9–16. [PubMed] [Google Scholar]
- Gilchrest BA. Actinic injury. Annu Rev Med. 1990;41:199–210. doi: 10.1146/annurev.me.41.020190.001215. [DOI] [PubMed] [Google Scholar]
- Giordano C, Stassi G, De Maria R, Todaro M, Richiusa P, Papoff G, Ruberti G, Bagnasco M, Testi R, Galluzzo A. Potential involvement of Fas and its ligand in the pathogenesis of Hashimoto's thyroiditis. Science. 1997;275:960–963. doi: 10.1126/science.275.5302.960. [DOI] [PubMed] [Google Scholar]
- Gniadecki R, Hansen M, Wulf HC. Two pathways for induction of apoptosis by ultraviolet radiation in cultured human keratinocytes. J Invest Dermatol. 1997;109:163–169. doi: 10.1111/1523-1747.ep12319216. [DOI] [PubMed] [Google Scholar]
- Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- Hahne M, Rimoldi D, Schröter M, Romero P, Schreier M, French LE, Schneider P, Bornaand T, Fontana A, Lienard D, Cerottini J-C, Tschopp J. Melanoma cell expression of Fas (APO-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- Hsu H, Xong J, Goeddel DV. The TNF receptor1-associated protein TRADD signals cell death and NF-κB activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66:233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Jonason AS, Kunala S, Price GJ, Restifo RJ, Spinelli HM, Persing JA, Lefell D, Tarone RE, Brash DE. Frequent clones of p53- mutated keratinocytes in normal human skin. Proc Natl Acad Sci USA. 1996;93:14025–14029. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JFR, Winterford CM, Harmon BV. Apoptosis: its significance in cancer and cancer therapy. Cancer. 1994;73:2013–2026. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxity-dependent APO-1 (Fas/CD95)-associated proteins (CAP) form death-inducing signaling complex (DISC) with the receptor. EMBO (Eur Mol Biol Organ) J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köck A, Schwarz T, Kirnbauer R, Urbanski A, Perry P, Ansel JC, Luger TA. Human keratinocytes are a source for tumor necrosis factor α. Evidence for synthesis and release upon stimulation with endotoxin or ultraviolet light. J Exp Med. 1990;172:1609–1614. doi: 10.1084/jem.172.6.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer KH. Sunlight and skin cancer: another link revealed. Proc Natl Acad Sci USA. 1997;94:1–14. doi: 10.1073/pnas.94.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130:1018–1021. [PubMed] [Google Scholar]
- Kripke ML. Photoimmunology. Photochem Photobiol. 1990;52:919–924. doi: 10.1111/j.1751-1097.1990.tb08703.x. [DOI] [PubMed] [Google Scholar]
- Kusiak JW, Izzo JA, Zhao B. Neurodegeneration in Alzheimer disease. Is apoptosis involved? . Mol Chem Neuropathol. 1996;28:153–162. doi: 10.1007/BF02815217. [DOI] [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Desnoyer S, Poirier GG, Earnshaw WC. Cleavage of poly (ADP-ribose) polymerase by a protease with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Lehman TA, Modali R, Boukamp P, Stanek J, Bennett WP, Welsh JA, Metcalf RA, Stampfer MR, Fusenig N, Rogan EM, Hurris CC. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis. 1993;14:833–839. doi: 10.1093/carcin/14.5.833. [DOI] [PubMed] [Google Scholar]
- Leverkus M, Yaar M, Gilchrest BA. Fas/Fas ligand interaction contributes to UV-induced apoptosis in human keratinocytes. Exp Cell Res. 1997;232:255–262. doi: 10.1006/excr.1997.3514. [DOI] [PubMed] [Google Scholar]
- Los M, van de Craen M, Penning L, Schenk H, Westendorp M, Baeuerle P, Dröge W, Krammer PH, Fiers W, Schulze-Osthoff K. Requirement of an ICE/CED-3 protease for Fas/APO-1-mediated apoptosis. Nature. 1995;375:81–83. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Cotter TG. Ultraviolet B irradiation of human leukemia HL-60 cells in vitro induces apoptosis. Int J Radiat Biol. 1991;59:1001–1016. doi: 10.1080/09553009114550891. [DOI] [PubMed] [Google Scholar]
- Matsue H, Kobayashi H, Hosokawa T, Akitaya T, Ohkawara A. Keratinocytes constitutively express the Fas antigen that mediates apoptosis in IFNγ-treated cultured keratinocytes. Arch Dermatol Res. 1995;287:315–320. doi: 10.1007/BF01105085. [DOI] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/CED-3 like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Muzio M, Salvesen GS, Dixit VM. FLICE induced apoptosis in a cell-free system. J Biol Chem. 1997;272:2952–2956. doi: 10.1074/jbc.272.5.2952. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labele M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Rosette C, Karin M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- Sachsenmaier C, Radler-Pohl A, Zinck R, Nordheim A, Herrlich P, Rahmsdorf HJ. Involvement of growth factor receptors in the mammalian UVC response. Cell. 1994;78:963–972. doi: 10.1016/0092-8674(94)90272-0. [DOI] [PubMed] [Google Scholar]
- Sayama K, Yonehara S, Watanabe Y, Miki Y. Expression of Fas antigen on keratinocytes in vivo and induction of apoptosis in cultured keratinocytes. J Invest Dermatol. 1994;103:330–334. doi: 10.1111/1523-1747.ep12394858. [DOI] [PubMed] [Google Scholar]
- Schlegel J, Peter I, Orrenius S, Miller DK, Thornberry NA, Yamin TT, Nicholson DW. CPP32/apopain is a key interleukin 1β converting protease involved in Fas-mediated apoptosis. J Biol Chem. 1996;271:1841–1844. doi: 10.1074/jbc.271.4.1841. [DOI] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Bauer MKA, Vogt M, Los M. Role of ICE-related and other proteases in Fas-mediated apoptosis. Cell Death Diff. 1996;3:177–184. [PubMed] [Google Scholar]
- Schwarz A, Bhardwaj RS, Aragane Y, Mahnke K, Riemann H, Metze D, Luger TA, Schwarz T. Ultraviolet-B-induced apoptosis of keratinocytes: evidence for partial involvement of tumor necrosis factor-α in the formation of sunburn cells. J Invest Dermatol. 1995;104:922–927. doi: 10.1111/1523-1747.ep12606202. [DOI] [PubMed] [Google Scholar]
- Simon MM, Aragane Y, Schwarz A, Luger TA, Schwarz T. UVB light induces nuclear factor κB (NFκB) activity independently from chromosomal DNA damage in cell-free cytosolic extracts. J Invest Dermatol. 1994;102:422–427. doi: 10.1111/1523-1747.ep12372194. [DOI] [PubMed] [Google Scholar]
- Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand: a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation of the Fas ligand. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Kobayashi H, Hashimoto Y, Matsuo S, Iizuka H. Interferon-γ-dependent stimulation of Fas antigen in SV40-transformed human keratinocytes: modulation of the apoptotic process by protein kinase C. J Invest Dermatol. 1995;105:810–815. doi: 10.1111/1523-1747.ep12326577. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Suda T, Haze K, Nakamura N, Sato K, Kimura F, Motoyoshi K, Mizuki M, Tagawa S, Ohga S, et al. Fas ligand in human serum. Nat Med. 1996;2:317–322. doi: 10.1038/nm0396-317. [DOI] [PubMed] [Google Scholar]
- Trauth BC, Klas C, Peters AMJ, Matzku S, Möller P, Falk W, Debatin KM, Krammer PH. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 1989;245:301–305. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
- Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- White JG, Amos WB, Fordham M. An evaluation of confocal versus conventional imaging of biological structures by fluorescence light microscopy. J Cell Biol. 1987;105:41–48. doi: 10.1083/jcb.105.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AR. The sunburn cell. Photodermatology. 1987;4:127–134. [PubMed] [Google Scholar]
- Ziegler A, Jonason JS, Leffel DW, Simon JA, Sharma HW, Kimmelman J, Remington L, Jacks T, Brash DE. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]