

Abstract
We survey computational approaches that tackle membrane protein structure and function prediction. While describing the main ideas that have led to the development of the most relevant and novel methods, we also discuss pitfalls, provide practical hints and highlight the challenges that remain. The methods covered include: sequence alignment, motif search, functional residue identification, transmembrane segment and protein topology predictions, homology and ab initio modeling. Overall, predictions of functional and structural features of membrane proteins are improving, although progress is hampered by the limited amount of high-resolution experimental information available. While predictions of transmembrane segments and protein topology rank among the most accurate methods in computational biology, more attention and effort will be required in the future to ameliorate database search, homology and ab initio modeling.
Keywords: membrane proteins, protein structure prediction, protein function prediction, alignments, transmembrane segment prediction, homology modeling, ab initio modeling
Introduction
The fold space of membrane proteins is relatively small...
Integral membrane proteins (IMPs) are polypeptide chains embedded into biological membranes. In this review, we focus exclusively on polytopic membrane proteins, i.e. proteins that span the membrane at least once. We will not consider monotopic membrane proteins, which despite being anchored to the membrane do not cross it (anchors include amphipathic helices and intra-membranous molecules covalently attached to the protein).
Biological membranes are phospholipid bilayers. The polar heads of the phospholipids face the aqueous solution on both sides of the membrane, while the lipid tails form a thick (∼30 Å) hydrophobic core. The sequence and structure of IMPs reflects an extreme effort of adaptation to this environment. Since the insertion of polar groups in the hydrophobic core of the membrane is energetically unfavorable, IMPs have evolved to minimize the interaction of such groups with the lipid tails. Inside the membrane, IMP chains form long regular secondary structure elements that cross the entire bilayer, i.e are transmembrane (TM) segments. In this way they satisfy the hydrogen-bond potential of the backbone amide and carbonyl groups. These secondary structure elements can either be TM helices (TMH) or TM beta strands (TMB); they are connected by regions that generally extend beyond the membrane core, into the aqueous solution. The assembly of TM elements is strongly influenced by the bilayer. Alpha helices form bundles, with each helix strongly oriented towards the normal to the membrane plane (1). Beta strands are arranged in barrel-like structures, with the barrel axis normal to the membrane plane. While helical IMPs are practically ubiquitous (only being absent in the outer membrane of Gram-negative bacteria), beta-barrel IMPs are only found in the outer membrane of Gram-negative bacteria, mitochondria and chloroplasts. Proteins with both integral TMHs and TMBs have not been observed. Predicting the location of TM segments in the protein sequence turns out to be a relatively easy task, due to the strong compositional biases imposed by the bilayer onto the amino acid sequence. In fact, in order to cross the membrane, TMHs need to be at least 15 residues long and composed predominantly of hydrophobic amino acids. TMBs are generally longer than 10 residues and are comprised of alternating hydrophobic and polar amino acids (hydrophobic side chains face the lipids while polar residues face the water- or protein-filled interior of the barrel). Also, regions connecting TM segments that are not translocated across the bilayer (“inside” or cytoplasmic regions) are enriched in positively charged amino acids, following the so-called ‘positive-inside rule’ (2, 3). This simplifies the prediction of the way in which TM segments cross the membrane (inside-out or outside-in).
In conclusion, in comparison to water-soluble proteins, IMP chains are able to sample only a limited number of folds (4). This reduced conformational space is easier to search through traditional sampling algorithms (e.g. molecular dynamics and Monte Carlo). The number, location and cross-membrane direction of TM segments can be predicted rather accurately (Methods); it is, therefore, reasonable to believe that 3D-structure prediction for IMPs is within reach of computational methods.
... but membrane proteins still exhibit remarkable structural variability
Notwithstanding the previously described constraints and biases, presently no method can accurately predict the 3D-structure of any IMP from sequence alone. On one hand, this is due to the very limited number of high-resolution structures available, giving us a myopic view of the IMP structural space; on the other, IMP architectures may not be as simple as initially thought. Indeed, as more experimental structures have become available, IMPs have revealed an unexpected structural diversity. Constraints on TMH length and tilt angles are not as strict as previously hypothesized (5). Also, analysis of known structures in the Protein Data Bank (6) (PDB) indicated that about 50% of TMHs contain non-canonical elements (i.e. kinks, 310-helix and π-helix turns) (7) and 5% of all TMHs cross the membrane only partially, i.e. form half TMHs (8). Stable loop regions have been found inside the hydrophobic core of the membrane (9, 10). In some cases, membrane domains contain crevices and cavities that can accommodate numerous water molecules and other ligands, thus forming a more diverse environment with which the protein can interact. There is also evidence that TMH topology is not solely determined by the protein sequence, but depends on complex interactions with the translocon, the machinery responsible for inserting TMHs into the membrane (11). Consequently, topology may depend on the organism in which the protein is expressed. Finally, functional IMPs are very often the result of the assembly of several chains, posing the problem of finding the correct protein-protein docking solution, a notoriously difficult task, despite recent progress (12), e.g., receptor binding and activity in G-protein coupled receptors have been reported to be linked to oligomerization (13 FEBS Journal 2005). Thus, the IMP structural space, although smaller than that sampled by water-soluble proteins, is still intricate enough to make computational structure prediction a very challenging task.
Two main prediction tasks: identify and characterize
In this paper we review computational methods that can be applied to study IMPs. Some of these approaches have been developed specifically for IMPs, others have been adapted or simply transferred from the techniques originally devised for water-soluble proteins. Computational methods have been developed to address two different goals: to identify novel IMPs or to characterize the functional and structural features of a protein experimentally known to be inside the membrane.
The first task (identification of IMPs) can be solved by sequence or profile alignment techniques that detect relatives of known IMPs. Alternatively, we can search motif databases (e.g. PROSITE (14), PRINTS (15)) to identify functional motifs that are characteristic of IMPs and use these motifs to extend the database searches, or we can use prediction methods that locate TM segments and see whether or not the protein under study is compatible with membrane insertion. Note that these approaches produce different and often complementary information. For example, Ruta et al. (16) used database searches and sequence analysis to detect a previously uncharacterized voltage-gated potassium channel in the archae bacterium Aeropyrum pernix and later confirmed the prediction experimentally. On the other hand, prediction of TM segments has been widely used for estimating the fraction of membrane proteins in various genomes and kingdoms of life (Fig. 3, inset).
Fig. 3.

Genomics and Structural Genomics of helical IMPs.
The second task (functional and structural characterization) can also be achieved by different means, depending on the type of information that is available (Fig. 1). If the high-resolution structure of a protein (template) homologous to the target is known, we can use homology modeling to obtain a prediction for the target structure. The quality of the resulting model typically depends on the sequence similarity between template and target (17). Once built, the model can prove instrumental for studying the function of the protein. If low-resolution structural information (e.g. from cryo-electron microscopy or mutation analysis) is available either for the target or for a homologue, we can analyze sequence conservation and co-variation to identify functional residues, or, in combination with molecular modeling, to provide a better guess of the target structure. When no experimental structural information is available, we can apply de novo and ab initio methods. De novo methods predict TM segments and topology through knowledge-based approaches, while ab initio methods attempt to model structural features from first principles. Both de novo and ab initio methods may help in finding optimal packing through the application of energy-based scoring functions. The less is known experimentally, the lower is the expected accuracy of the models, with homology modeling being, when applicable, by far the most reliable modeling technique.
Fig. 1. Predicting structure and function for a protein experimentally known to be an IMP.
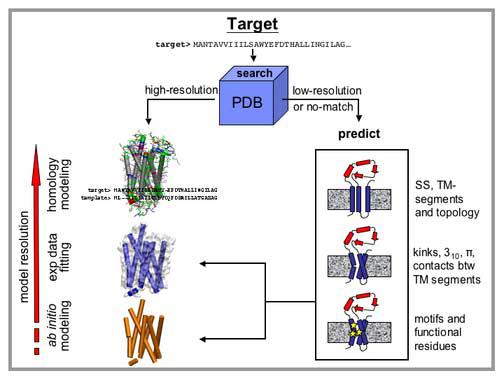
First, the target sequence will be searched against the PDB (6), looking for homologs of known 3D structure. If this search returns at least one good match (template) with a high-resolution structure, it will be possible to apply homology modeling techniques to obtain a model for the target proteins whose resolution will in general depend on the similarity with the template. If the search is either unsuccessful or returns a low-resolution structure, further analysis will instead be needed. Prediction of TM segments, kinks, functional residues and motifs can all help in elucidating the target structural and functional features, either in combination with low-resolution structural information (e.g. from cryo-electron microscopy) or with ab initio modeling techniques. When predicting function it will be useful to search not only the PDB but also other databases, such as SWISS-PROT (147), Interpro (148) and Pfam (50). Note that database searches are performed through alignments methods using either substitution matrices or HMMs.
Goals and standards of this review
While we present empirical observations and theoretical background when needed to understand computational approaches, our main aim is to highlight the state-of-the-art methods that readers can rely upon for the study of IMPs. As far as the quality of the predictions is concerned, it is not always possible to report independent estimates of performance. In fact, despite the important progress witnessed in the last few years in membrane protein structure determination (18), the small number of high-resolution structures available has so far prevented experiments of the type devised for the assessment of prediction methods for water-soluble proteins, such as CASP (19), CAFASP (20), EVA (21) and LIVEBENCH (22). Thus, when no independent assessment data are available, we refrain from reporting self-assessed performance measures, while attempting to point out the strengths and weaknesses of the methods. Finally, since in many respects computational analysis of IMPs may still be considered in its infancy, new and more effective methods are likely to emerge in the very near future. Throughout the paper, we will try to point to those techniques that are likely to break new ground and lead to novel tools or increased performance.
Methods
Databases
Several databases have been built as comprehensive repositories of IMP sequences or structures and for the purpose of helping computational biologists to develop and test their prediction methods (Table 1). PDB_TM (23) and Stephen White’s database (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html) contain all IMPs of known structure, with links to the PDB (6) and PubMed (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=PubMed) entries. MPtopo (24) additionally includes a list of proteins of unknown 3D structure, the topology of which has been experimentally annotated through low-resolution techniques such as gene fusion or proteolytic degradation. The reliability of such annotations is typically much lower than that of annotations from 3D structures (25). The PDB_TM and OPM databases (26) contain predictions for the orientation of IMPs of known structure relative to the hydrophobic core of the membrane. The web server TMDET (a companion to PDB_TM) (27) can also be used to predict the membrane orientation of model structures, e.g. models obtained by homology. The Transport Classification Database (TCDB) (28) is a comprehensive classification of membrane transporters (analogous to the Enzyme Classification system for enzymes) that incorporates both functional and phylogenetic information. It classifies about 3,000 transporter sequences into more than 550 families. Interestingly, the TCDB website also reports a list of transporters that have been associated with disease. Finally, there are several databases that collect information on specific IMP families, such as G-protein coupled receptors (GPCR) (the database is GPCRDB (29)) and potassium channels (KchannelDB, see ‘usage notice’ at http://www.receptors.org/KCN/htmls/consortium.html), or structural classes, such as porins (PRNDS (30)).
Table 1.
Membrane proteins databases.
| Database | Description/URL |
|---|---|
| GPCRDB (29), KchannelDB and others | Several receptor databases http://www.receptors.org |
| OPM (26) | Database reporting predictions for the orientation of IMPs within the membrane http://opm.phar.umich.edu/ |
| PDB_TM (94) | Database of known membrane protein structures http://pdbtm.enzim.hu/ |
| MPtopo (24) | Database of experimentally-determined protein topologies http://blanco.biomol.uci.edu/mptopo/ |
| Stephen White’s database | Database of known membrane protein structures http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html |
| PRNDS (30) | Database of porins http://gene.tn.nic.in/PRNDS |
| TCDB (28) | Transport classification database http://www.tcdb.org/ |
| TMDET (27) | Web server for predicting the orientation of a query membrane protein structure http://www.enzim.hu/TMDET |
*Methods in this and in the following Tables are listed in alphabetical order.
Sequence alignments, motifs and functional residues
Sequence alignment methods are ubiquitous tools for the prediction of structure and function; they are primarily used to identify related sequences via database searches and to detect template structures needed for the construction of homology models (17). Different applications may require different alignment approaches since so far no single method combines the sensitivity and efficiency necessary for a database search with the accuracy required for the construction of a homology model. Thus, we first consider developments that relate to all types of alignment approaches, and then address separately methods for database searching and for the purpose of building homology models.
A general consideration: substitution matrices
Pair-wise sequence alignment methods generate many possible matches between two protein sequences. Matches are typically scored as a sum over the probabilities for each observed amino-acid substitution (match), as defined in the so-called substitution matrix, and the highest-scoring alignment is reported. Thus, when selecting a sequence alignment method there are two major considerations: the substitution matrix and the algorithm for optimizing the scores. For water-soluble proteins, the two most widely used substitution matrices are PAM (31) and BLOSUM (32). However, since the amino-acid composition of TM domains differs from that of water-soluble proteins (33), several groups have introduced amino-acid substitution matrices specific to helical IMPs, including JTT (34), PHAT (35) and SLIM (36). Of these, SLIM was reported to have the highest accuracy for detecting remote relationships between sequences in the manually-curated GPCR database (36). To date, however, the membrane protein-specific matrices have not been independently assessed on large-scale IMPs data sets, and as such are not recommended for general use.
Database searching for detection of sequence relations
For the detection of related sequences in databases, novel methods have been developed that use the signal from the long hydrophobic stretches of TMHs, as well as more complex approaches (Table 2). These methods involve: matching sequences with an equivalent number of predicted TMHs (37-39); per-residue alignment of hydropathy profiles (40); matching patterns of peaks and troughs extracted from hydropathy profiles (41) or of TM topology (GPCRHMM) (42); matching of functional motifs (PRED-GPCR) (43) or of amino acid composition (42); and favoring matches of predicted TM residues within a standard PSI-BLAST search (TM-PSI) (44). For these approaches, success was often measured by the identification of novel family members not found by sequence alone, although TM-PSI, PRED-GPCR and GPCRHMM were assessed for their ability to detect GPCR sequence relationships (42-44). No systematic comparisons of all these methods exist making it impossible to name the most effective approach. However, matching numbers of TMHs is likely to be the least sensitive method, whereas the family-specific methods may prove to be the most sensitive. Hydropathy-profile alignments may be useful for classification of novel protein families (45, 46), and are reported to be appropriate for detecting close homologies (41). Hydropathy-profile pattern matching, on the other hand, which may be more sensitive for remote-homology detection, has the disadvantage that it requires manual selection of representative patterns (41). TM-PSI is probably the most general and easily-automated method, but it requires that a TMH prediction is made for all sequences in the database before a search can proceed, and is not currently available through a web server. It will be interesting to compare these methods with general sequence profile-based or Hidden Markov Models (HMM) methods used for water-soluble proteins (47), which are not limited to one fold type or family.
Table 2.
Sequence alignment programs, family HMM profiles and motifs databases.*
| Method | Description/URL |
|---|---|
| BLAST or PSI-BLAST (149) | Database searching http://www.ncbi.nlm.nih.gov/BLAST/ |
| ClustalW (150) | Multiple sequence alignment http://www.ebi.ac.uk/clustalw/ |
| GPCRHMM (42) | Identification and classification of GPCRs http://gpcrhmm.cgb.ki.se |
| HMAP (57) | Profile-to-profile alignment including secondary and tertiary structure information. http://trantor.bioc.columbia.edu/hmap/ |
| Hydropathy profile alignment | Direct alignment of hydropathy profiles for database search http://bioinformatics.weizmann.ac.il/hydroph/hydroph.html |
| Hydropathy pattern matching | Conversion of representative profile into pattern of peaks/troughs for database search |
| PHAT search (35) | http://blocks.fhcrc.org/sift/PHAT_submission.html |
| PSI-BLAST with compositional bias (149) | Database searching allowing for differences in composition in membrane proteins. http://www.ncbi.nlm.nih.gov/blast/ |
| PFAM (50) | Database of protein families including HMMs and sequence alignments http://www.sanger.ac.uk/Software/Pfam/ |
| PRED-GPCR (43) | Identification and classification of GPCRs http://bioinformatics.biol.uoa.gr/PRED-GPCR |
| PRINTS (15) | Database of protein family fingerprints (motifs) http://umber.sbs.man.ac.uk/dbbrowser/PRINTS/ |
| PROSITE (14) | Database of protein families including patterns and profiles http://www.expasy.ch/prosite/ |
| T-Coffee (56) | Advanced multiple sequence alignment http://www.igs.cnrs-mrs.fr/Tcoffee/tcoffee_cgi/index.cgi |
| TM-PSI (44) | Matching of predicted TM regions from TMHMM within PSI-BLAST profiles for database search |
Code for other methods mentioned in the text is usually available from the authors
In contrast to the significant number of novel methods developed for database searching of helical IMPs, the analysis of beta-barrel IMPs has generally focused on assigning function through the prediction of the number of TMBs (see TM segment prediction section, below), or has relied on standard BLAST searches for the detection of related sequences.
A recent development in database searching is the adjustment of the scoring in BLAST to reflect the composition of the sequences (48, 49), which theoretically should improve results for membrane protein searches. However, this method has not yet been assessed specifically on membrane proteins, and thus should be used with caution. Note also that during BLAST or PSI-BLAST searches, it is recommended not to apply the “low-complexity filter”, since this is likely to remove hydrophobic regions from the target sequence (44) (see also http://ca.expasy.org/tools/blast/blast_help.html).
Detecting relations through motifs and patterns
When database searching fails to find homologs, it is still possible to retrieve useful information about the target by searching databases that contain motifs or patterns relating to specific protein families. Such databases, for example PFAM (50), PROSITE (14), and PRINTS (15) (Table 2), usually cover water-soluble proteins as well as IMPs. Although two proteins that share a common motif may not have common ancestry, a match can often provide very useful information about the function of the target.
Sequence alignments
The accuracy of the alignment between two related membrane protein sequences is another important consideration (Table 2). The bipartite alignment method for helical IMPs uses a TM substitution matrix in the (predicted) membrane regions of a sequence, and a standard matrix in the remaining regions (e.g. STAM) (35, 51, 52). This can be achieved by separating out the predicted TMHs from other residues, and by aligning them independently and finally reassembling the full sequence (e.g. (52, 53)). Often, known functionally-important motifs are matched in order to guide the alignment (54, 55). Alternatively, one can simply adopt a standard alignment algorithm but apply substitution probabilities from different matrices according to whether the position is predicted to be in the membrane or not. Using this approach with the PHAT matrix however does not provide an improvement over the BLOSUM matrix [Forrest, Tang and Honig, personal communication]. Of all these methods, STAM (52) is possibly the most user-friendly, since it incorporates a graphical user interface, and allows manual adjustment of the definitions of the ends of the TM segments. Most of these methods are not currently widely available, and, in the absence of more thorough benchmarks, are not recommended for the casual user. Advanced multiple-sequence alignment and profile-to-profile alignment methods, however, have been shown to work well for water-soluble proteins, and can in fact be used for membrane proteins [Forrest and Honig, personal communication]. Multiple-sequence alignment methods such as T-Coffee (56) are also very accurate at high sequence identities (> 40%), despite using a single generic substitution matrix. At lower sequence identities, the use of profile-based methods, such as HMAP (57), which incorporate structural information, can improve the alignment accuracy significantly. For a review of the best methods for water-soluble proteins, and their availabilities, see (58).
Marking functional residues
The residues that are most relevant for the structure and function of a protein are not always recognizable as part of a global sequence similarity pattern or as motifs of neighboring conserved positions on the sequence. Sometimes conservation patterns are irregular and sparse; in other cases, the relevant signal is not the conservation but rather the co-variation of two or more sequence positions. Specific methods have been developed to deal with these situations and they can be very useful for predicting protein structural and functional features, either in combination with the previously described approaches, or in cases where these approaches are not applicable. Unfortunately, most of these methods have not yet been implemented as web servers.
Amino acid conservation at a specific position within a family (i.e. an ensemble of proteins sharing a common ancestor) is considered to be an indication of strong evolutionary pressure at that site. Hence, conserved residues, easily identified through multiple sequence alignments (Table 2), represent potential targets for structural and functional studies. Also, within a family it is often possible to identify several subfamilies that exhibit different functional characteristics (for example, binding different ligands or substrates). When this is the case, functional variability is usually encoded in subfamily-specific conservation at particular sites (dubbed tree determinants). In two recent papers the Valencia and Lichtarge groups reviewed these methods and attempted an evaluation of their performance in predicting functionally and structurally important residues (59, 60). Another approach that aims at the identification of functionally relevant residues is co-variation analysis. In this case, it is assumed that proteins can accommodate a potentially deleterious mutation at a given position if this is compensated by concerted mutations at other sites. Co-variation is usually considered to be an indication of spatial proximity (61 Proteins 1994) and can hence be used for predicting intra-protein contacts (61 Proteins 1994, 62) or protein-protein interactions (63).
In general, all methods based on sequence conservation within families and subfamilies or co-variation can hardly distinguish between different evolutionary constraints. For this reason, interpretation of a specific signal often relies on some additional experimental information. In the realm of membrane proteins, conservation and co-variation studies performed on GPCRs have taken advantage of the high-resolution structure of rhodopsin (64) to identify putative oligomerization interfaces and G-protein interaction sites (65); knowledge of the crystal structure of a voltage-gated potassium channel from Aeropyrum pernix (66) prompted the use of correlated mutation analysis to identify gating-related residues (67); recently, conservation and co-variation analysis has been applied in combination with molecular modeling to the gap junction channel, starting from a low-resolution cryo-electron microscopy structure (68).
Prediction of TM segments
There are two goals of methods that predict TM segments. The first, whole-protein prediction, identifies IMPs among a set of protein sequences for which structures and membrane/non-membrane localization are unknown. The second is per-residue prediction, which labels the residues in a protein according to whether they span the membrane or not. Whole-protein predictions are frequently used in structural genomics projects when defining the target lists, with the objective to either exclude or to specifically select IMPs. In contrast, per-residue predictions are routinely used by experimentalists, e.g. as a guide for engineering mutants.
Early approaches
Early predictions of TM segments for helical IMPs generally used the following four-step procedure (Fig. 2): 1. Derive a ‘propensity scale’, a set of 20 numbers corresponding to properties or statistics of the 20 amino acids when found in TM regions. 2. Generate a plot of propensity values along the query sequence. 3. Smooth the plot by taking the average propensity value in a window of N residues and plot the average at the center of the window (i.e. a sliding window average). 4. Identify TM stretches on the smoothed plot using some propensity threshold.
Fig. 2. Timeline of TMH prediction methods.
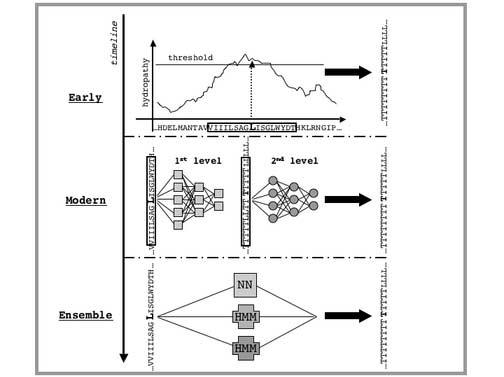
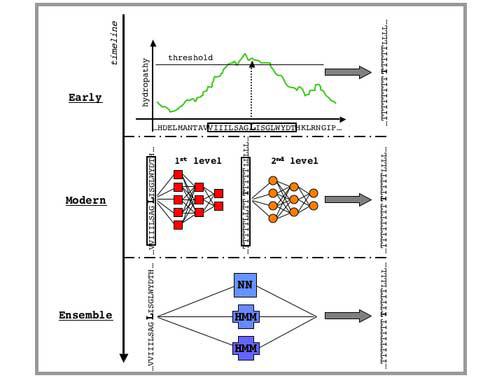
Early methods used per-residues hydropathy scales and a window around each residue to produce a smoothed sequence profile. A threshold, usually manually-adjusted, was then introduced to predict TMHs. Modern approaches use machine-learning algorithms such as NNs (illustrated), HMMs or SVMs to predict each residue in e.g. two states, TM or non-TM. In this figure, we schematically show a common NN architecture used for predicting TMHs. As discussed in more detail in the text, there are two NN levels: the first (also called sequence-to-structure NN) produces a per-residue score (analogous to the per-residue value from hydropathy scales); the second (structure-to-structure) takes the output of the first NN and smoothes its values by taking into account the first level predictions for the neighboring residues (somehow similar to the window used in early approaches). Finally, ensemble approaches combine several different methods (in this example, one NN and two HMMs) to produce a consensus prediction.
Early studies derived propensity scales from biophysical or chemical measurements such as water/vapor transfer free energy. A well-known early example was the ‘hydropathy index’ of Kyte and Doolittle (69). As more experimental structures have become available, propensity scales have been expanded to reflect more than the original two (TM/non-TM) structural states. Careful inspection of 3D structures has led to scales derived from ‘snorkeling preferences’ (polar amino acids enriched at TMH N-terminal ends, due to chain-direction dependent rotamer preferences) (70), helical hairpins (71), or residue orientation in helix bundles (72).
Modern approaches
Many popular modern approaches (Fig. 2), based on Hidden Markov Models (HMMs) or neural networks (NNs), are related to sliding-window hydropathy plot methods. The most successful NN approaches use the concept of combining two consecutive NNs, first introduced by PHDhtm (73, 74), to predict TMHs (Fig. 2). The first (dubbed ‘sequence-to-structure’ network) receives a window of the amino acid sequence as input and outputs a score representing the propensity of the central residue in that window to be within the membrane. When sliding the window over the entire sequence, the NN produces a succession of propensities (analogous to the propensity plot mentioned above). The second NN (dubbed ‘structure-to-structure’ network) smoothes the propensities generated from the first one. These NNs are capable of capturing subtle patterns, such as amphipathicity in helices, or TMH length preferences, which may evade simple hydropathy plot methods.
HMMs are more specialized models. Like hydropathy plots and NN-based methods, they capture both ‘sequence-to-structure’ and ‘structure-to-structure’ relationships. The difference is that in HMMs the two steps are merged into one integrated model. This has advantages and disadvantages, and HMMs are often complementary to NNs. For example, HMMs can take advantage of global patterns in the structure, such as the repeated strand-periplasmic loop-strand-extracellular loop pattern in beta-barrel IMPs. NNs have no such “global view” of proteins. However, NNs can recognize local sequence patterns involving several residues, which HMMs cannot. The best performing HMM- and NN-based methods will be discussed in a later section (see also Tables 3 and 4).
Table 3.
Recommended TMH prediction methods.*
| Method | URL | Service |
|---|---|---|
| DAS-TMfilter (82) | http://mendel.imp.ac.at/sat/DAS/DAS.html | PR2, WP |
| HMMTOP 2.0 (86) | http://www.enzim.hu/hmmtop/ | PR3 |
| MEMSAT3 (151) | http://bioinf.cs.ucl.ac.uk/psipred/ | PR3 |
| MINNOU (152) | http://minnou.cchmc.org/ | PR2 |
| OrienTM (153) | http://o2.biol.uoa.gr/orienTM/ | PR3 |
| PHDhtm (87) | http://rostlab.org/predictprotein/submit_adv.html | PR3, WP |
| Phobius (101) | http://phobius.cgb.ki.se/ | PR3 + SP |
| SOSUI (83) | http://sosui.proteome.bio.tuat.ac.jp/sosuiframe0.html | PR2 |
| Split4 (88) | http://split.pmfst.hr/split/4/ | PR2 |
| THUMBU | http://phyyz4.med.buffalo.edu/service.htm | PR2, WP |
| TMAP (90) | http://bioinfo.limbo.ifm.liu.se/tmap/index.html | PR3 |
| TMHMM (84) | http://www.cbs.dtu.dk/services/TMHMM/, http://www.sbc.su.se/~erikgr/tmhmm/index.html | PR3, WP, Database |
| TOP-PRED (154) | http://bioweb.pasteur.fr/seqanal/interfaces/toppred.html | PR3 |
The methods in this Table are recommended either because they stood out in independent assessments or because otherwise interesting. PR2: two-state, TM/non-TM per-residue prediction. PR3: three-state TM/inside/outside per-residue prediction, i.e. implicitly a topology prediction. SP: N-terminal signal peptide prediction. WP: suitable for whole-proteome screening. Results are returned in seconds or minutes by most servers.
Table 4.
Recommended TMB prediction methods*
| Method | URL | Service |
|---|---|---|
| B2TMR, HMMB2TMR (95) | http://gpcr.biocomp.unibo.it | PR3 |
| BOMP (155) | http://www.bioinfo.no/tools/bomp | WP |
| PRED-TMBB (91) | http://bioinformatics2.biol.uoa.gr/PRED-TMBB | PR3 |
| PROFtmb (93) | http://rostlab.org/services/proftmb | PR3, WP |
| TMB-HUNT (156, 157) | http://www.bioinformatics.leeds.ac.uk/ | WP |
The methods in this Table are recommended either because they stood out in independent assessments or because otherwise interesting. See Table 3 for legend.
More recently, Support Vector Machines (SVM) have also been applied to this problem. An SVM is a general algorithm used to classify patterns into two groups (known as the ‘binary classification problem’). Yuan et. al. (75) use an SVM for per-residue prediction of helical IMPs with a sliding window, similar to ‘sequence-to-structure’ NN or early propensity value methods. SVMs (like NNs) are capable of learning complex relationships among the amino acids comprised in the local windows with which they are trained.
Ultimately, selection and preparation of training data affects performance of each model, and it is unclear whether one kind of model is clearly better than others.
Ensemble methods
An ensemble method consists of taking the output of individual predictors and combining them by a weighted vote (Fig. 2). This idea has been extensively tested in statistics, and it is known that, if the predictors have roughly equal accuracy and loosely correlated errors, the ensemble will yield a better prediction than each individual method (76). The majority vote tends to cancel the errors, choosing the correct prediction among the individuals (77) and thus combining the advantages of the different methods. Martelli and Casadio combined a NN and two different HMM predictors for predicting helical IMPs (78). Taylor and coworkers (79) combined five methods for predicting helical IMP topology. Nilsson and coworkers also used five topology prediction methods to predict partial membrane topologies (80). Bagos et al. (81) created an ensemble predictor for beta-barrel IMPs from several available methods.
Detecting membrane proteins in genomes (whole-protein predictions)
While some methods provide whole-protein predictions, fewer are suitable for whole-proteome screening, either because they have not been specifically evaluated for this task, or because the websites do not accept multiple sequence submissions, making screening impractical. The whole-protein prediction methods are invariably applicable to only one of the two membrane protein fold types (helical or beta-barrel IMPs, Tables 3 and 4). One important observation is that most methods using only hydrophobicity indices incorrectly identify TMHs in over 50% of all proteins (25). In contrast, the very best methods confuse fewer than 2-4% of the globular proteins for IMPs (25). Particularly successful are: DAS-TMFilter (82), SOSUI (83), TMHMM (84), and PHDhtm (25, 74). Note that these error rates assume predictions based on the mature protein sequence, i.e. without signal and transit peptides. Error rates are significantly higher than the values quoted if we include the confusion between signal peptides and TMHs (25, 85).
Assessing TMH predictions
Most TMH prediction methods use the evolutionary information from multiple alignments either directly or indirectly. For these methods, poor predictions often result from incorrect or uninformative (no homologs found) alignments. Some servers flag such predictions as less reliable; others do not. Another common error is to predict TMHs in globular proteins or in place of signal peptides. Both signal peptides, which comprise a hydrophobic region, and long hydrophobic stretches in globular proteins, may be easily mistaken for TMHs. The simplest defense against the mis-prediction of water-soluble proteins is only consider predictions with multiple TMHs. N-terminal signal peptides can also be excluded from predictions by a similar rule, i.e. by excluding cases where a single TMH is predicted to be within a given number of residues from the N-terminus. Because of a variety of reasons, few servers implement such rules. Another pitfall that many prediction methods have succumbed to involves setting hard limits for the minimum and maximum lengths of TMHs. When few high-resolution structures were known, low-resolution experiments found TMHs to range between 17 and 25 residues in length (25). Many methods used this information to divide what would otherwise have been predicted to be, for example, a 36-residue helix into two 18-residue helices, since it was clear that a 36-residue helix fell outside of the experimentally observed range. However, as high-resolution structures became more numerous and diverse, it became clear that membrane helices are in fact extremely diverse, and range from 10 (half-TMHs) (8) to 40 residues in length (25). As a result, the prediction of long TMHs as well as of half helices is generally inaccurate. In addition to servers enforcing strict limits on the lengths of TMHs, some servers predict all TMHs to be exactly the same length (e.g. 20 residues). This may be a way of effectively “hedging ones bets” (you don’t terribly overshoot or undershoot the boundary) at the TM region boundaries and scoring better on a per-residue basis (see Fig. 4 in (8)).
Methods that predict TM segments have been subjected to several assessment studies and they have been evaluated on both high-resolution and low-resolution datasets. While high-resolution data, including crystal and NMR structures, provide structural details at an atomic level, low-resolution experiments, mainly of C-terminal fusions with indicator proteins and antibody-binding studies, only identify which portions of an IMP are inside and outside of the membrane (85). Unfortunately, low-resolution experiments have been suggested to be no more accurate than the best prediction methods (25). Two recent studies have examined the accuracy of TMH prediction (8, 25). These studies used a wide variety of measures to gauge performance, including prediction accuracy measured on a per-residue, per-segment (a segment is correctly predicted if most of its residues are correctly predicted), and per-protein basis (a protein is correctly predicted if all of its TM segments are identified, see previous paragraph), as well as the accuracy of segment boundary and topology prediction. Methods were further evaluated on their ability to avoid false positive predictions of TMHs in signal peptide regions and in globular proteins. No single method was able to perform best according to all of the various measures. As a result, these studies did not designate a best method, but rather reported which methods consistently ranked very high across all of the measures. The top performing programs reported in the earlier study (25) were HMMTOP2 (86) and PHDhtm (87), while SPLIT4 (88), TMHMM2 (89), HMMTOP2 and TMAP (90) topped the rankings in the more recent study (8). The difference between the two lists can largely be attributed to the inclusion of more recent methods in the later study. When looking at these results, it has to be considered that although the assessments were performed under controlled conditions, the paucity of the available data forced the assessors to use the very same structures on which the methods had been trained. In other words, it is often difficult to guess from these data how the methods would perform on previously unseen examples. In this view, the fact that HMMTOP2 appears in both lists of top predictors gives some indication that it may perform well on proteins it has not seen in training. Note that the ensemble methods, most of them reporting higher accuracies than individual methods, have not yet been independently assessed.
Assessing TMB predictions
To our knowledge, Bagos et. al. (81) have provided the only independent evaluation of TMB predictors. They evaluated four HMM-based, four NN-based and two SVM-based methods in terms of several statistics focused on per-residue, per-segment, and topology prediction accuracy. Two issues affect accuracy assessment for TMBs. First, many methods were originally evaluated on the sequences taken from PDB structures. Beta-barrel IMPs, needing to be translocated to the outer membrane of Gram-negative bacteria and organelles, and in contrast with most helical IMPs, invariably carry a cleavable signal peptide. However, sequences extracted from the PDB are almost always mature, signal-peptide cleaved forms of the original sequence, and in some cases, are missing N- or C-terminal domains. Bagos et. al. found that when signal peptides were present in the query protein sequence, NN- and SVM-based methods in particular tended to mistake it for a TMB, while HMMs did not. Thus, practically speaking, removing known signal peptides from query sequences before submitting them to a TMB predictor will yield slightly improved predictions. The second important issue is that beta-strands in beta-barrel IMPs often extend well past the membrane, mostly on the extracellular side. Some methods are designed to predict the TM regions of the strands, such as PRED-TMBB (91, 92), while others predict the full extent of the beta-strands, such as PROFtmb (93). Bagos et al. assessed the ability of the methods to predict the location of the TM regions of beta-strands, as determined by PDB_TM (94). With the above factors as caveats, the study made one major conclusion: the top three methods namely, HMM-B2TMR (95), ProfTMB (93) and PRED-TMBB (91), are HMM-based and far outperform NN- and SVM-based methods in predicting topology and number of strands correctly.
Topology prediction for helical IMPs
Topology prediction identifies non-TM regions as either non-translocated (inside) or translocated (outside). Membrane insertion and topogenesis of helical IMPs is conserved between bacteria and eukaryotes, and the mechanism is complex and actively researched (reviewed in (96-98)). Future insights into these mechanisms will be invaluable for improving topology prediction. Helical IMPs generally traverse the membrane in unbroken helices, creating an alternating translocated/nontranslocated pattern of extra-membrane loops. Thus, when the exact number of TMHs is known and assuming that the protein contains only integral TMHs (i.e. no half-TMHs), the topology is uniquely determined by the location of the TM segments and of the N-terminus (that is, either inside or outside).
Current predictors rely on two topogenic signals in the protein sequence: the distribution of positively charged residues in extra-membrane loops, known as the “positive-inside rule” (2) and the existence of N-terminal signals, cleaved and uncleaved. The “positive-inside rule”, introduced by von Heijne and Gavel (99), reflects the observation that Lys and Arg are enriched in nontranslocated loops and depleted in translocated loops (although the bias is strong only for loops shorter than 60 residues). Later, Andersson and von Heijne demonstrated in the inner membrane of E. coli that ATPase sec was required for translocation of loops in a fashion linearly dependent on loop length and the number of positive charges, hypothesizing that the dependence was due to the requirement of several rounds of ATP hydrolysis to translocate positive charges against the membrane potential (100). More recently, the basis of the “positive-inside rule” has been proposed to be the interaction with the translocon itself (97). While many methods use a simplified version of the “positive-inside rule” that ignores the lengths of the loops, TMHMM (89) explicitly models short and long loops using separate HMM architecture modules, thus accounting for the length dependence. The other important topogenic signal is the N-terminal signal. Phobius (101) is the first topology model to combine signal peptide cleavage site detection with TMH prediction in an integrated probabilistic model, which seeks to compensate for mis-predictions of cleavable signal peptides. It also makes optimal use of the fact that a cleaved signal corresponds to a topology with the N-terminus outside. Recently, Bernsel and von Heijne (102) improved topology predictions of helical IMPs using searches of SMART (103) database families to identify water-soluble proteins that have compartment specific localization and are homologous to the IMP non-TM domains.
Prediction of kinks and other non-helix conformations in TMHs
A significant fraction of the TMHs exhibits kinks and other deviations from standard alpha-helical conformations. These features are likely to play an important role in determining the structural and functional diversity of proteins with the same topology (104). Therefore, they are an important target for computational predictions, particularly as a complement to homology modeling (see below). Recently, Bowie and coworkers (104) proposed a simple algorithm to predict kinks in TMHs. The method consists of producing a multiple sequence alignment of homologs of the target protein, and identifying the positions at which more than 10% of the residues are prolines. While self-assessed performances are high, the authors do not specify the number and evolutionary-distance distribution of homologues that are required in order to produce a robust prediction. Another method, based on sequence pattern descriptors, predicts kinks as well as other non-canonical helical conformations (310 and π turns) (105). Although the method is reported to perform well in identifying non-alpha conformation in TMHs of known structure, it is unclear how it would perform when evaluated on previously unseen proteins, since pattern recognition methods can be very accurate but are often unable to generalize well.
3D-structure prediction
Homology modeling and fold recognition for membrane proteins
Homology modeling (or comparative modeling) is the construction of a model for the structure of a target protein using an experimentally known 3D structure of a related protein as a template. That is, once the template has been selected, and an alignment between template and target is generated (see Alignments), the non-conserved side chains are replaced, and insertions (regions with no template structure) are modeled as ‘loop’ regions by de novo or ab initio techniques. Of the many modeling programs available, those that produce the highest quality models (i.e. the best stereochemistry) are SegMod/ENCAD (106) and Modeller (107), whereas Nest (108) makes models that are the most similar to the native structure of the target (109) (Table 5). However, none of the commonly-used homology modeling programs have been modified for membrane proteins (except for the SWISS-MODEL (110) 7TM interface, see ab initio methods): thus, care must be taken to ensure that the hydrophobic protein-lipid interface region of the protein does not incorrectly contain polar side chains, which may occur depending on the constraints imposed by the sequence alignment and the template structure. Side chain prediction algorithms such as SCWRL (111) or SCAP (112), which are certainly likely to improve the accuracy of the model within the core of the protein, may suffer from the same problem at the lipid-interacting surface.
Table 5.
Homology modeling, loop prediction and side-chain optimization methods.*
| Method | Description/URL |
|---|---|
| Modeller 7v7 (107) | Homology modeling, alignment, loop prediction etc http://salilab.org/modeller/ |
| SegMod/ENCAD (106) | Homology modeling http://csb.stanford.edu/levitt/segmod/ |
| Jackal | Homology modeling (Nest), side chain prediction (SCAP) and loop prediction (Loopy) http://trantor.bioc.columbia.edu/ |
| PLOP (113) | Homology modeling, side chain and loop prediction http://francisco.compbio.ucsf.edu/~jacobson/ |
| RAPPER (114) | Loop prediction http://raven.bioc.cam.ac.uk/ |
| SCWRL 3.0 (111) | Side chain prediction http://dunbrack.fccc.edu/SCWRL3.php |
A selection of available tools for the construction and manipulation of homology models
Note that, as for globular proteins, the accuracy of a homology model is strongly dependent on the identity between the two sequences, even if the ‘correct’ alignment is obtained [Forrest and Honig, personal communication]. Specifically, below 30% sequence identity, the accuracy of the model decreases rapidly since the difference in structure between the template and the true native structure is higher. Furthermore, there is significant variation between the extra-membranous loop regions of related IMPs, so that these regions of a homology model are likely to be particularly unreliable. On the other hand, de novo/ab initio predictions for loop regions shorter than 12 residues tend to be reliable (113, 114), if constructed using reasonable physical-chemical force fields that incorporate descriptions of the aqueous solution rather than a vacuum environment (115, 116). Existing loop-prediction programs include Loopy (117), RAPPER (114) and PLOP (113) (Table 5); of these, PLOP is reportedly the most accurate, although it requires the most computational time, whereas Loopy is the fastest. Other strategies have been reported for loop modeling in GPCRs, including simulated annealing with Monte Carlo (116) and coarse-grained backbone dihedral sampling (118), although larger benchmarks are required to confirm their general applicability. To date, no methods have solved the significant problem of predicting multiple interacting loops simultaneously.
Apart from short loops, attempts to ‘refine’ an entire homology model to try to make it more native-like, and less like the template, are presently not recommended, neither for membrane proteins nor for water-soluble proteins, since no method has been shown to consistently improve the accuracy of homology models {Tress, 2005 #10237}. An exception may be when significant amounts of experimental information are available to provide constraints for refinement, such as for GPCRs (54).
Ab initio and de novo modeling of membrane proteins
When little or no experimental information about a protein or its homologues is available, it is necessary to resort to ab initio or de novo approaches. These approaches include prediction of interactions between pairs of TM segments as well as of full 3D-structure modeling. While such predictions for water-soluble proteins can sometimes be used to obtain indications about 3D structure (119), methods developed for IMPs are still of limited practical relevance and, to our knowledge, none are presently available as web servers. Nonetheless, this is an area that is expected to grow considerably in the years ahead and we will therefore briefly review it in order to give the reader a glimpse of things to come.
Predicting helix assembly is of crucial interest in IMP structure prediction for at least two reasons: firstly, the internal packing of helical IMPs is mostly determined by interactions between TMHs, secondly, oligomerization of helical IMPs appears to be mediated by inter-helical interactions between the different monomers (e.g. in GPCRs (120, 121 Saperstein DA, Palczewski K, Engel A. J Biol Chem 2003)). A handful of motifs have been identified that are often observed in TMH-TMH interfaces. In the GxxxG motif, two glycine residues are found on the same side of the alpha helix. Due to their small size, they facilitate interaction with other helices, by maximizing van der Waals contacts between a larger number of residues and by establishing inter-helical hydrogen bonds (122). The GxxxG motif was first identified as a principal effector of glycophorin A dimerization (123, 124) only to be later discovered in internal interfaces and other oligomers (125, 126). Note, however, that recent studies dispute the relevance of this pattern for alpha helix dimerization (127, 128). Analogous motifs involving small residues, such as alanine and serine, and longer motifs referred to as glycine zippers (129) are thought to play a similar stabilizing role. More generally, helix-helix interfaces appear to be enriched in small amino acids, both in membrane proteins (130) and in globular proteins (131, 132). The simple “small residues go inside” rule was implemented with reasonable success in a TMH-TMH interaction prediction algorithm by Fleishman and Ben-Tal (133). So far, the method has been tested on a carefully selected set of helix pairs, and it is not immediately applicable to multiple-TMH bundles. Polar residues are also often located at helix-helix interfaces. Although rare in TMHs, they seem to have a role in driving helix association (134).
Several methods have been developed for predicting internal helix assembly or oligomerization of helical IMPs (135). Most have been tested on very few structures, with the glycophorin A homodimer often serving as a model system. Ponder and coworkers (136) used a potential smoothing and search algorithm to predict the structure of the glycophorin A dimer. Jones and coworkers (137) implemented a knowledge-based method, FILM, which uses the energy function of a previous globular protein fragment-assembly program (FRAGFOLD) modified by the introduction of a potential term that mimics the presence of the membrane bilayer. For the moment, the method can only predict the structure of small, less than 100 amino acid-long proteins (i.e. proteins for which the conformational space to search is small). Bowie and coworkers (138) predicted the structure of IMP oligomers using knowledge of the oligomer symmetry. They use a simple softened van der Waals potential and Monte Carlo minimization to pack ideal alpha helices. The method has been applied to homo-oligomers constituted of monomers with only one TMH. Methods for predicting 7-TMH IMPs, proteins that alone account for about 50% of all drug targets, have exploited a combination of computational modeling and experimental constraints, the latter ranging from mutation data to low-resolution cryo-electron microscopy data (139). However, most of these methods have not been extended to model helical IMPs with a different number of TM segments. At the border of homology modeling and ab initio/de novo, is the SWISS-MODEL (110) 7TM interface (http://swissmodel.ncifcrf.gov/cgi-bin/sm-gpcr.cgi), explicitly designed to assist in the modeling of 7-TMH proteins. SWISS-MODEL 7TM allows homology modeling based on experimental and computationally designed templates, with the ‘computational templates’ generated by taking into account constraints from low-resolution experimental data. The user needs only to provide information about the location of the TMHs in the query sequence and to select the most appropriate template from those available.
Very few groups addressed the ab initio/de novo modeling of beta-barrel IMPs. The task may be easier than with helical IMPs, since in all known beta-barrel IMP structures adjacent TMBs are found to be in contact. Hence, once TMBs have been correctly located in the protein sequence, the only goal is to identify the contacting residues. Jackups and Liang (140) recently published a method that predicts the contacting residues on paired strands. While the reported accuracy is not very high, this seems to be a first important step toward modeling of beta-barrel IMP structures.
Genomics and Structural Genomics of helical IMPs
The explosion of genome sequencing projects since the last decade has enabled systematic computational study of IMPs within the genome. Studies using several different methods generally agreed that in most genomes, about 20-30% of proteins have at least one TMH (89, 141-144). An early study on 14 genomes found that larger genomes contained a higher fraction of membrane proteins than smaller genomes (143), which led to the hypothesis that more complex multi-cellular organisms may need more membrane proteins to carry out inter-cellular communications. Several more recent studies based on more genomes and more complete sequence data failed to reproduce the same results (89, 142, 144). Detailed topology prediction, although not as accurate as the simple membrane vs. non-membrane classification, suggested that within an individual genome, proteins with fewer TMHs are more abundant than proteins with more helices. Proteins with seven TMHs seem to be much more abundant in higher eukaryotes, presumably due to the expansion of the GPCR family, and proteins with six or twelve TMHs are overrepresented in bacteria, reflecting the abundance of small molecule transporters (142). It has also been observed that there is an interesting correlation between the protein length and the number of TMHs. That is, most helical IMPs either have many TMHs with short connecting loops or have only 1-2 helices with large extra-membranous domains (142, 143). Recently, it was shown that topology prediction improves significantly when the periplasmic or cytoplasmic location of the C-terminus is known (145). By constraining a topology prediction algorithm (TMHMM (84)) with large-scale experimental data on locations of the C-termini, high-quality topology models for more than 600 membrane proteins in E. coli were constructed (146), providing an elegant example of the power of combining both computational and experimental approaches.
Conclusions
Predicting the structure and function of integral membrane proteins appears to be an easier task than predictions for water-soluble globular proteins. However, our knowledge and understanding of membrane protein structural features has so far been hampered by the limited experimental high-resolution information available. While recently solved structures have shown that membrane proteins are more diverse than initially thought, we are probably still unable to see the full complexity of the problem.
We have presented a survey of the state-of-the-art computational methods developed for predicting structural and functional features in membrane proteins. In the past, for the reasons mentioned, progress in many areas has been slow. However, some approaches have matured and reached high levels of performance and reliability. Transmembrane helix, transmembrane strand and protein topology predictions are among those. Alignment methods specific to membrane proteins are now being developed; however, many questions, such as which substitution matrices are more appropriate for use in transmembrane regions, remain unsolved. Homology modeling has been widely used on membrane proteins in the attempt to fill the gap between the number of known sequences and structures. Still, in most of the cases, models are built using programs originally developed for water-soluble proteins. As the number of available templates increases, is seems likely that new methods will emerge, which are tailored specifically to membrane proteins. Finally, de novo and ab initio structure prediction of membrane proteins is still in its infancy and more and better methods will have to be developed in the future.
Structural biologists solve more membrane protein structures today than ever before. Structural genomics projects targeting integral membrane proteins are now under way. If experimentalists continue to expand our knowledge of the membrane protein world, it will be possible for computational biologists to develop new and better methods, that may eventually confirm that, yes, membrane protein structure is easy to predict.
Acknowledgements
Thanks to Hans-Erik G. Aronson (Columbia) for computer assistance; thanks to Gunnar von Heijne, Henrik Nielsen, Jannick Bendtsen and Lukas Käll for very generous email clarification and discussion. This work was supported by the grant RO1-LM07329-01 from the National Library of Medicine (NLM). Last, not least, thanks to all those who deposit their experimental data in public databases, and to those who maintain these databases.
Abbreviations used
- 3D
three-dimensional
- GPCR
G-Protein Coupled Receptor
- HMM
Hidden Markov Model
- IMP
Integral Membrane Protein
- NN
Neural Network
- PDB
Protein Data Bank
- SVM
Support Vector Machine
- TM
transmembrane
- TMH
transmembrane helix
- TMB
transmembrane beta-strand
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bowie JU. J Mol Biol. 1997;272:780–9. doi: 10.1006/jmbi.1997.1279. [DOI] [PubMed] [Google Scholar]
- 2.von Heijne G, Gavel Y. Eur J Biochem. 1988;174:671–8. doi: 10.1111/j.1432-1033.1988.tb14150.x. [DOI] [PubMed] [Google Scholar]
- 3.Nilsson J, Persson B, von Heijne G. Proteins. 2005;60:606–16. doi: 10.1002/prot.20583. [DOI] [PubMed] [Google Scholar]
- 4.Bowie JU. Protein Sci. 1999;8:2711–9. doi: 10.1110/ps.8.12.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gimpelev M, Forrest LR, Murray D, Honig B. Biophys J. 2004;87:4075–86. doi: 10.1529/biophysj.104.049288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, Fagan P, Marvin J, Padilla D, Ravichandran V, Schneider B, Thanki N, Weissig H, Westbrook JD, Zardecki C. Acta Crystallogr D Biol Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 7.Riek RP, Rigoutsos I, Novotny J, Graham RM. J Mol Biol. 2001;306:349–62. doi: 10.1006/jmbi.2000.4402. [DOI] [PubMed] [Google Scholar]
- 8.Cuthbertson JM, Doyle DA, Sansom MS. Protein Eng Des Sel. 2005;18:295–308. doi: 10.1093/protein/gzi032. [DOI] [PubMed] [Google Scholar]
- 9.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Cahit BT, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 10.Fu D, Libson A, Miercke LJ, Weitzman C, Nollert P, Krucinski J, Stroud RM. Science. 2000;290:481–6. doi: 10.1126/science.290.5491.481. [DOI] [PubMed] [Google Scholar]
- 11.Goder V, Junne T, Spiess M. Mol Biol Cell. 2004;15:1470–8. doi: 10.1091/mbc.E03-08-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mendez R, Leplae R, Lensink MF, Wodak SJ. Proteins. 2005;60:150–69. doi: 10.1002/prot.20551. [DOI] [PubMed] [Google Scholar]
- 13.Maggio R, Novi F, Scarselli M, Corsini GU. Febs J. 2005;272:2939–46. doi: 10.1111/j.1742-4658.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- 14.Hulo N, Bairoch A, Bulliard V, Cerutti L, De Castro E, Langendijk-Genevaux PS, Pagni M, Sigrist CJ. Nucleic Acids Res. 2006;34:D227–30. doi: 10.1093/nar/gkj063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Attwood TK, Bradley P, Flower DR, Gaulton A, Maudling N, Mitchell AL, Moulton G, Nordle A, Paine K, Taylor P, Uddin A, Zygouri C. Nucleic Acids Res. 2003;31:400–2. doi: 10.1093/nar/gkg030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruta V, Jiang Y, Lee A, Chen J, MacKinnon R. Nature. 2003;422:180–5. doi: 10.1038/nature01473. [DOI] [PubMed] [Google Scholar]
- 17.Petrey D, Honig B. Mol Cell. 2005;20:811–9. doi: 10.1016/j.molcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 18.White SH. Protein Sci. 2004;13:1948–9. doi: 10.1110/ps.04712004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moult J. Curr Opin Struct Biol. 2005;15:285–9. doi: 10.1016/j.sbi.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 20.Bourne PE. Methods Biochem Anal. 2003;44:501–7. [PubMed] [Google Scholar]
- 21.Koh IY, Eyrich VA, Marti-Renom MA, Przybylski D, Madhusudhan MS, Eswar N, Grana O, Pazos F, Valencia A, Sali A, Rost B. Nucleic Acids Res. 2003;31:3311–5. doi: 10.1093/nar/gkg619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rychlewski L, Fischer D. Protein Sci. 2005;14:240–5. doi: 10.1110/ps.04888805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tusnady GE, Dosztanyi Z, Simon I. Bioinformatics. 2004;20:2964–72. doi: 10.1093/bioinformatics/bth340. [DOI] [PubMed] [Google Scholar]
- 24.Jayasinghe S, Hristova K, White SH. Protein Sci. 2001;10:455–8. doi: 10.1110/ps.43501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen CP, Kernytsky A, Rost B. Protein Sci. 2002;11:2774–91. doi: 10.1110/ps.0214502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. Bioinformatics. 2006;22:623–5. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 27.Tusnady GE, Dosztanyi Z, Simon I. Bioinformatics. 2005;21:1276–7. doi: 10.1093/bioinformatics/bti121. [DOI] [PubMed] [Google Scholar]
- 28.Saier MH, Jr., Tran CV, Barabote RD. Nucleic Acids Res. 2006;34:D181–6. doi: 10.1093/nar/gkj001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horn F, Bettler E, Oliveira L, Campagne F, Cohen FE, Vriend G. Nucleic Acids Res. 2003;31:294–7. doi: 10.1093/nar/gkg103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katta AV, Marikkannu R, Basaiawmoit RV, Krishnaswamy S. In Silico Biol. 2004;4:549–61. [PubMed] [Google Scholar]
- 31.Dayhoff MO, Schwartz RM, Orcutt BC. In: Atlas of protein sequence and structure. Dayhoff MO, editor. Vol. 5. 1978. pp. 345–52. [Google Scholar]
- 32.Henikoff S, Henikoff JG. Proc. Natl. Acad. Sci. USA. Vol. 89. 1992. pp. 10915–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cserzo M, Wallin E, Simon I, von Heijne G, Elofsson A. Prot. Eng. 1997;10:673–76. doi: 10.1093/protein/10.6.673. [DOI] [PubMed] [Google Scholar]
- 34.Jones DT, Taylor WR, Thornton JM. FEBS Lett. 1994;339:269–75. doi: 10.1016/0014-5793(94)80429-x. [DOI] [PubMed] [Google Scholar]
- 35.Ng PC, Henikoff JG, Henikoff S. Bioinformatics. 2000;16:760–66. doi: 10.1093/bioinformatics/16.9.760. [DOI] [PubMed] [Google Scholar]
- 36.Muller T, Rahmann S, Rehmsmeier M. Bioinformatics. 2001;17:S182–89. doi: 10.1093/bioinformatics/17.suppl_1.s182. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Engelman DM, Gerstein M. Genome Biology. 2002;3 doi: 10.1186/gb-2002-3-10-research0054., research0054.1-54.12.
- 38.Gao Q, Chess A. Genomics. 1999;60:31–39. doi: 10.1006/geno.1999.5894. [DOI] [PubMed] [Google Scholar]
- 39.Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S. FEBS Letters. 2002;520:97–101. doi: 10.1016/s0014-5793(02)02775-8. [DOI] [PubMed] [Google Scholar]
- 40.Lolkema JS, Slotboom DJ. Mol. Membr. Biol. 1998;15:33–42. doi: 10.3109/09687689809027516. [DOI] [PubMed] [Google Scholar]
- 41.Clements JD, Martin RE. Eur. J. Biochem. 2002;269:2101–07. doi: 10.1046/j.1432-1033.2002.02859.x. [DOI] [PubMed] [Google Scholar]
- 42.Wistrand M, Kall L, Sonnhammer ELL. Protein Sci. 2006;15 doi: 10.1110/ps.051745906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papasaikas PK, Bagos PG, Litou ZI, Promponas VJ, Hamodrakas SJ. Nucl. Acids Res. 2004;32:W380–82. doi: 10.1093/nar/gkh431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hedman M, Deloof H, Von Heijne G, Elofsson A. Protein Sci. 2002;11:652–58. doi: 10.1110/ps.39402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lolkema JS, Slotboom DJ. J. Mol. Biol. 2003;327:901–09. doi: 10.1016/s0022-2836(03)00214-6. [DOI] [PubMed] [Google Scholar]
- 46.Lolkema JS, Slotboom DJ. Molec. Memb. Biol. 2005;22:177–89. doi: 10.1080/09687860500063324. [DOI] [PubMed] [Google Scholar]
- 47.Marti-Renom MA, Madhusudhan MS, Sali A. Protein Sci. 2004;13:1071–87. doi: 10.1110/ps.03379804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu Y-K, Wootton JC, Altschul SF. Proc. Natl. Acad. Sci. 2003;100:15688–93. doi: 10.1073/pnas.2533904100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schaffer AA, Yu Y-K. FEBS Journal. 2005;272:5101–09. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR. Nucleic Acids Res. 2004;32:D138–41. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones DT, Taylor WR, Thornton JM. Biochemistry. 1994;33:3038–49. doi: 10.1021/bi00176a037. [DOI] [PubMed] [Google Scholar]
- 52.Shafrir Y, Guy HR. Bioinformatics. 2004;20:758–69. doi: 10.1093/bioinformatics/btg482. [DOI] [PubMed] [Google Scholar]
- 53.Bissantz C, Logean A, Rognan D. J. Chem. Inf. Comput. Sci. 2004;44:1162–76. doi: 10.1021/ci034181a. [DOI] [PubMed] [Google Scholar]
- 54.Fanelli F, De Benedetti PG. Chem Rev. 2005;105:3297–351. doi: 10.1021/cr000095n. [DOI] [PubMed] [Google Scholar]
- 55.Giorgetti A, Carloni P. Curr. Opin. Chem. Biol. 2003;7:150–56. doi: 10.1016/s1367-5931(02)00012-1. [DOI] [PubMed] [Google Scholar]
- 56.Notredame C, Higgins DG, Heringa J. J. Mol. Biol. 2000;302:205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 57.Tang CL, Xie L, Koh IYY, Posy S, Alexov E, Honig B. J. Mol. Biol. 2003;334:1043–62. doi: 10.1016/j.jmb.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 58.Ginalski K, Grishin NV, Godzik A, Rychlewski L. Nucl. Acids Res. 2005;33:1874–91. doi: 10.1093/nar/gki327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.del Sol Mesa A, Pazos F, Valencia A. J Mol Biol. 2003;326:1289–302. doi: 10.1016/s0022-2836(02)01451-1. [DOI] [PubMed] [Google Scholar]
- 60.Mihalek I, Res I, Lichtarge O. J Mol Biol. 2004;336:1265–82. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 61.Gobel U, Sander C, Schneider R, Valencia A. Proteins. 1994;18:309–17. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- 62.Pazos F, Helmer-Citterich M, Ausiello G, Valencia A. J Mol Biol. 1997;271:511–23. doi: 10.1006/jmbi.1997.1198. [DOI] [PubMed] [Google Scholar]
- 63.Pazos F, Valencia A. Proteins. 2002;47:219–27. doi: 10.1002/prot.10074. [DOI] [PubMed] [Google Scholar]
- 64.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 65.Filizola M, Weinstein H. Febs J. 2005;272:2926–38. doi: 10.1111/j.1742-4658.2005.04730.x. [DOI] [PubMed] [Google Scholar]
- 66.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 67.Fleishman SJ, Yifrach O, Ben-Tal N. J Mol Biol. 2004;340:307–18. doi: 10.1016/j.jmb.2004.04.064. [DOI] [PubMed] [Google Scholar]
- 68.Fleishman SJ, Unger VM, Yeager M, Ben-Tal N. Mol Cell. 2004;15:879–88. doi: 10.1016/j.molcel.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 69.Kyte J, Doolittle RF. J. Mol. Biol. 1982;157:105–32. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 70.Chamberlain AK, Lee Y, Kim S, Bowie JU. J. Mol. Biol. 2004;339:471–79. doi: 10.1016/j.jmb.2004.03.072. [DOI] [PubMed] [Google Scholar]
- 71.Monne M, Hermansson M, von Heijne G. J Mol Biol. 1999;288:141–5. doi: 10.1006/jmbi.1999.2657. [DOI] [PubMed] [Google Scholar]
- 72.Pilpel Y, Ben-Tal N, Lancet D. J. Mol. Biol. 1999;294:921–35. doi: 10.1006/jmbi.1999.3257. [DOI] [PubMed] [Google Scholar]
- 73.Rost B, Casadio R, Fariselli P, Sander C. Protein Science. 1995;4:521–33. doi: 10.1002/pro.5560040318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rost B. Methods Enzymol. 1996;266:525–39. doi: 10.1016/s0076-6879(96)66033-9. [DOI] [PubMed] [Google Scholar]
- 75.Yuan Z, Mattick JS, Teasdale RD. J Comput Chem. 2004;25:632–6. doi: 10.1002/jcc.10411. [DOI] [PubMed] [Google Scholar]
- 76.Hansen LK, Salamon P. IEEE Transactions on pattern analysis and machine intelligence. 1990;12:993–1001. [Google Scholar]
- 77.Dietterich TG. Lecture Notes in Computer Science. 2000;1857:1–15. [Google Scholar]
- 78.Martelli PL, Fariselli P, Casadio R. Bioinformatics. 2003;19(Suppl 1):i205–11. doi: 10.1093/bioinformatics/btg1027. [DOI] [PubMed] [Google Scholar]
- 79.Taylor PD, Attwood TK, Flower DR. Nucleic Acids Res. 2003;31:3698–700. doi: 10.1093/nar/gkg554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nilsson I, Persson B, von Heijne G. FEBS Lett. 2000;486:267–69. doi: 10.1016/s0014-5793(00)02321-8. [DOI] [PubMed] [Google Scholar]
- 81.Bagos PG, Liakopoulos T, Hamodrakas S. BMC Bioinformatics. 2005;6:7. doi: 10.1186/1471-2105-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cserzo M, Eisenhaber F, Eisenhaber B, Simon I. Bioinformatics. 2004;20:136–37. doi: 10.1093/bioinformatics/btg394. [DOI] [PubMed] [Google Scholar]
- 83.Hirokawa T, Boon-Chieng S, Mitaku S. Bioinformatics. 1998;14:378–79. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 84.Sonnhammer ELL, von Heijne G, Krogh A. ISMB-98. AAAI Press; 1998. [Google Scholar]
- 85.Moller S, Croning MDR, Apweiler R. Bioinformatics. 2001;17:646–53. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
- 86.Tusnady GE, Simon I. Bioinformatics. 2001;17:849–50. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 87.Rost B, Fariselli P, Casadio R. Protein Sci. 1996;5:1704–18. doi: 10.1002/pro.5560050824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Juretic D, Zoranic L, Zucic D. J Chem Inf Comput Sci. 2002;42:620–32. doi: 10.1021/ci010263s. [DOI] [PubMed] [Google Scholar]
- 89.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. J Mol Biol. 2001;305:567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 90.Persson B, Argos P. J. Prot. Chem. 1997;16:453–57. doi: 10.1023/a:1026353225758. [DOI] [PubMed] [Google Scholar]
- 91.Bagos PG, Liakopoulos TD, Spyropoulos IC, Hamodrakas SJ. Nucl. Acids Res. 2004;32:W400–04. doi: 10.1093/nar/gkh417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bagos PG, Liakopoulos TD, Spyropoulos IC, Hamodrakas SJ. BMC Bioinformatics. 2004;5:29. doi: 10.1186/1471-2105-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bigelow HR, Petrey DS, Liu J, Przybylski D, Rost B. Nucl. Acids Res. 2004;32:2566–77. doi: 10.1093/nar/gkh580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tusnady GE, Dosztanyi Z, Simon I. Nucleic Acids Res. 2005;33:D275–8. doi: 10.1093/nar/gki002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Martelli PL, Fariselli P, Krogh A, Casadio R. Bioinformatics. 2002;18(Suppl 1):S46–53. doi: 10.1093/bioinformatics/18.suppl_1.s46. [DOI] [PubMed] [Google Scholar]
- 96.Collinson I. Biochem Soc Trans. 2005;33:1225–30. doi: 10.1042/BST0331225. [DOI] [PubMed] [Google Scholar]
- 97.de Gier JW, Luirink J. Mol Microbiol. 2001;40:314–22. doi: 10.1046/j.1365-2958.2001.02392.x. [DOI] [PubMed] [Google Scholar]
- 98.White SH, von Heijne G. Curr Opin Struct Biol. 2005;15:378–86. doi: 10.1016/j.sbi.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 99.von Heijne G, Gavel Y. Eur. J. Biochem. 1988;174:671–78. doi: 10.1111/j.1432-1033.1988.tb14150.x. [DOI] [PubMed] [Google Scholar]
- 100.Andersson H, von Heijne G. FEBS Lett. 1994;347:169–72. doi: 10.1016/0014-5793(94)00530-3. [DOI] [PubMed] [Google Scholar]
- 101.Kall L, Krogh A, Sonnhammer EL. J Mol Biol. 2004;338:1027–36. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 102.Bernsel A, Von Heijne G. Protein Sci. 2005;14:1723–28. doi: 10.1110/ps.051395305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schultz J, Milpetz F, Bork P, Ponting CP. Proc Natl Acad Sci U S A. 1998;95:5857–64. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yohannan S, Faham S, Yang D, Whitelegge JP, Bowie JU. PNAS. 2004;101:959–63. doi: 10.1073/pnas.0306077101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rigoutsos I, Riek P, Graham RM, Novotny J. Nucl. Acids Res. 2003;31:4625–31. doi: 10.1093/nar/gkg639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Levitt M. J. Mol. Biol. 1992;226:507–33. doi: 10.1016/0022-2836(92)90964-l. [DOI] [PubMed] [Google Scholar]
- 107.Sali A, Blundell TL. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 108.Petrey D, Xiang ZX, Tang CL, Xie L, Gimpelev M, Mitros T, Soto CS, Goldsmith-Fischman S, Kernytsky A, Schlessinger A, Koh IYY, Alexov E, Honig B. Proteins. 2003;53:430–35. doi: 10.1002/prot.10550. [DOI] [PubMed] [Google Scholar]
- 109.Wallner B, Elofsson A. Protein Sci. 2005;14:1315–27. doi: 10.1110/ps.041253405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 111.Canutescu AA, Shelenkov AA, Dunbrack RL. Protein Sci. 2003;12:2001–14. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xiang ZX, Honig B. J. Mol. Biol. 2001;311:421–30. doi: 10.1006/jmbi.2001.4865. [DOI] [PubMed] [Google Scholar]
- 113.Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. Proteins. 2004;55:351–67. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 114.de Bakker PIW, DePristo MA, Burke DF, Blundell TL. Proteins. 2003;51:21–40. doi: 10.1002/prot.10235. [DOI] [PubMed] [Google Scholar]
- 115.Forrest LR, Woolf TB. Proteins: Struct. Funct. Genet. 2003;52:492–509. doi: 10.1002/prot.10404. [DOI] [PubMed] [Google Scholar]
- 116.Mehler EL, Periole X, Hassan SA, Weinstein H. J. Comp. Aid. Mol. Des. 2002;16:841–53. doi: 10.1023/a:1023845015343. [DOI] [PubMed] [Google Scholar]
- 117.Xiang Z, Soto CS, Honig B. Proc. Natl. Acad. Sci. 2002;99:7432–37. doi: 10.1073/pnas.102179699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nikiforovich GV, Marshall GR. Biophys. J. 2005;89:3780–89. doi: 10.1529/biophysj.105.070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vincent JJ, Tai CH, Sathyanarayana BK, Lee B. Proteins. 2005;61(Suppl 7):67–83. doi: 10.1002/prot.20722. [DOI] [PubMed] [Google Scholar]
- 120.Carrillo JJ, Lopez-Gimenez JF, Milligan G. Mol Pharmacol. 2004;66:1123–37. doi: 10.1124/mol.104.001586. [DOI] [PubMed] [Google Scholar]
- 121.Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A. J Biol Chem. 2003;278:21655–62. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Senes A, Gerstein M, Engelman DM. J Mol Biol. 2000;296:921–36. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- 123.Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Biochemistry. 1992;31:12719–25. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 124.MacKenzie KR, Prestegard JH, Engelman DM. Science. 1997;276:131–3. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 125.Russ WP, Engelman DM. J Mol Biol. 2000;296:911–9. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 126.Melnyk RA, Partridge AW, Deber CM. J Mol Biol. 2002;315:63–72. doi: 10.1006/jmbi.2001.5214. [DOI] [PubMed] [Google Scholar]
- 127.Doura AK, Fleming KG. J Mol Biol. 2004;343:1487–97. doi: 10.1016/j.jmb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 128.Kobus FJ, Fleming KG. Biochemistry. 2005;44:1464–70. doi: 10.1021/bi048076l. [DOI] [PubMed] [Google Scholar]
- 129.Kim S, Jeon TJ, Oberai A, Yang D, Schmidt JJ, Bowie JU. Proc Natl Acad Sci U S A. 2005;102:14278–83. doi: 10.1073/pnas.0501234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jiang S, Vakser IA. Proteins. 2000;40:429–35. doi: 10.1002/1097-0134(20000815)40:3<429::aid-prot80>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 131.Jiang S, Vakser IA. Protein Sci. 2004;13:1426–9. doi: 10.1110/ps.03505804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gimpelev M, Forrest LR, Honig B. Biophys. J. 2004;87:4075–86. doi: 10.1529/biophysj.104.049288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fleishman SJ, Ben-Tal N. J. Mol. Biol. 2002;321:363–78. doi: 10.1016/s0022-2836(02)00590-9. [DOI] [PubMed] [Google Scholar]
- 134.DeGrado WF, Gratkowski H, Lear JD. Protein Sci. 2003;12:647–65. doi: 10.1110/ps.0236503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fleishman SJ, Unger VM, Ben-Tal N. Trends Biochem Sci. 2006;31:106–13. doi: 10.1016/j.tibs.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 136.Pappu RV, Marshall GR, Ponder JW. Nat Struct Biol. Vol. 6. 1999. pp. 50–5. [DOI] [PubMed] [Google Scholar]
- 137.Pellegrini-Calace M, Carotti A, Jones DT. Proteins. 2003;50:537–45. doi: 10.1002/prot.10304. [DOI] [PubMed] [Google Scholar]
- 138.Kim S, Chamberlain AK, Bowie JU. J Mol Biol. 2003;329:831–40. doi: 10.1016/s0022-2836(03)00521-7. [DOI] [PubMed] [Google Scholar]
- 139.Fanelli F, DeBenedetti PG. Chem. Rev. 2005 [Google Scholar]
- 140.Jackups J, Ronald, Liang J. Journal of Molecular Biology. 2005;354:979–93. doi: 10.1016/j.jmb.2005.09.094. [DOI] [PubMed] [Google Scholar]
- 141.Kahsay RY, Gao G, Liao L. Bioinformatics. 2005;21:1853–58. doi: 10.1093/bioinformatics/bti303. [DOI] [PubMed] [Google Scholar]
- 142.Liu J, Rost B. Protein Sci. 2001;10:1970–9. doi: 10.1110/ps.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wallin E, von Heijne G. Protein Sci. 1998;7:1029–38. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Knight CG, Kassen R, Hebestreit H, Rainey PB. Proc Natl Acad Sci U S A. 2004;101:8390–5. doi: 10.1073/pnas.0307270101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Melen K, Krogh A, von Heijne G. J. Mol. Biol. 2003;327:735–44. doi: 10.1016/s0022-2836(03)00182-7. [DOI] [PubMed] [Google Scholar]
- 146.Daley DO, Rapp M, Granseth E, Melen K, Drew D, von Heijne G. Science. 2005;308:1321–23. doi: 10.1126/science.1109730. [DOI] [PubMed] [Google Scholar]
- 147.Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I, Pilbout S, Schneider M. Nucleic Acids Res. 2003;31:365–70. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bradley P, Bork P, Bucher P, Cerutti L, Copley R, Courcelle E, Das U, Durbin R, Fleischmann W, Gough J, Haft D, Harte N, Hulo N, Kahn D, Kanapin A, Krestyaninova M, Lonsdale D, Lopez R, Letunic I, Madera M, Maslen J, McDowall J, Mitchell A, Nikolskaya AN, Orchard S, Pagni M, Ponting CP, Quevillon E, Selengut J, Sigrist CJ, Silventoinen V, Studholme DJ, Vaughan R, Wu CH. Nucleic Acids Res. 2005;33:D201–5. doi: 10.1093/nar/gki106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucl. Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McGuffin LJ, Bryson K, Jones DT. Bioinformatics. 2000;16:404–5. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 152.Cao B, Porollo A, Adamczak R, Jarrell M, Meller J. Bioinformatics. 2006;22:303–9. doi: 10.1093/bioinformatics/bti784. [DOI] [PubMed] [Google Scholar]
- 153.Liakopoulos TD, Pasquier C, Hamodrakas SJ. Protein Eng. 2001;14:387–90. doi: 10.1093/protein/14.6.387. [DOI] [PubMed] [Google Scholar]
- 154.von Heijne G. J. Mol. Biol. 1992;225:487–94. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 155.Berven FS, Flikka K, Jensen HB, Eidhammer I. Nucleic Acids Res. 2004;32:W394–9. doi: 10.1093/nar/gkh351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Garrow AG, Agnew A, Westhead DR. Nucleic Acids Res. 2005;33:W188–92. doi: 10.1093/nar/gki384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Garrow AG, Agnew A, Westhead DR. BMC Bioinformatics. 2005;6:56. doi: 10.1186/1471-2105-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]