

Abstract
Amyotrophic lateral sclerosis (ALS) is characterized by progressive degeneration of the motor neurons in the cerebral cortex, brain stem, and spinal cord. However, the mechanisms that regulate the initiation and/or progression of motor neuron loss in this disease remain enigmatic. Cell-cycle proteins and transcriptional regulators such as cyclins, cyclin-associated kinases, the retinoblastoma gene product (pRb), and E2F-1 function during cellular proliferation, differentiation, and cell death pathways. Recent data has implicated increased expression and activation of various cell-cycle proteins in neuronal cell death. We have examined the expression and subcellular distribution of G1 to S phase cell-cycle regulators in the spinal cord, motor cortex, and sensory cortex from clinically and neuropathologically diagnosed sporadic ALS cases and age-matched controls. Our results indicate hyperphosphorylation of the retinoblastoma protein in motor neurons during ALS, concurrent with increased levels of cyclin D, and redistribution of E2F-1 into the cytoplasm of motor neurons and glia. These data suggest that G1 to S phase activation occurs during ALS and may participate in molecular mechanisms regulating motor neuron death.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease exemplified by neuronal loss in the motor cortex, brainstem, and spinal cord ventral horn. This progressive neurodegeneration results in muscle atrophy, paralysis, and death. Disease onset may occur at any age but is most common between 40 to 70 years of age. The average time interval from diagnosis to mortality is ∼4 years. 1,2 Familial ALS comprises a fraction (5 to 10%) of ALS cases and is predominantly inherited in an autosomal dominant manner and includes mutations in the SOD1 and the ALS2 genes. 3-7
The etiology of ALS is thought to be multifactorial. Factors believed to participate in motor neuron degeneration include glutamate-mediated excitotoxicity, free radical accumulation because of oxidative stress, increased intracellular calcium, mitochondrial dysfunction, cytoskeletal abnormalities, astrogliosis, and genetic mutations. 8-13 Neuronal dysfunction because of retrograde degeneration of the presynaptic axons may occur as the result of insufficient release of activity-dependent target-derived neurotrophic factors. 14 One important consequence of inappropriate trophic factor support is altered intracellular signaling to the nucleus. Signaling from the cell surface to the nucleus modulates chromatin structure and the activity of transcription factors, resulting in altered gene transcription. One potential mechanism leading to neuronal death in ALS includes altered expression of pro- and anti-apoptotic genes. 15 Another potential cell death mechanism is the inappropriate expression or activation of cell-cycle proteins. 16 The cell cycle is associated with the phase-specific expression or modification of defined sets of cell-cycle regulatory genes that regulate cellular proliferation, differentiation or entry into a quiescent state. 17 However re-entry of quiescent, terminally differentiated neurons, into the cell cycle may result in a mitotic catastrophe and cell death. 16,18,19
For entry into the cell cycle, quiescent neurons of the adult brain must first exit G0 and enter the G1 phase of the cell cycle. Multiple cell-cycle proteins regulate progression through G1, the most important being the products of retinoblastoma (pRb) tumor suppressor and E2F gene families. 20 Numerous lines of investigation have implicated pRb and E2F-1 in neuronal cell death. Studies using transgenic mouse models revealed that neuronal death in pRb knockouts was rescued by concurrent mutations in E2F-1, suggesting a role for E2F-1 in neuronal death. 21-23 In vitro studies using pharmacological agents in PC12 cells or primary neuronal cultures suggest a role for several cell-cycle elements such as cyclin-associated kinase (CDK)4/6, pRb/p107, and E2F in neuronal death evoked by insults such as β-amyloid toxicity, UV irradiation, DNA-damaging agents, trophic factor withdrawal, and depolarizing conditions. 24-35 E2F-1 participates in both caspase-dependent and caspase-independent death pathways, both of which have been postulated to function in motor neuron cell death in ALS. 36-38 We hypothesize that activation of G1 to S phase cell-cycle transcriptional regulators in motor neurons during ALS leads to altered gene expression and directly regulates cell death.
In quiescent cells, the retinoblastoma protein (pRb) remains in a hypophosphorylated state and sequesters members of the E2F gene family of transcription factors, which suppresses cell-cycle progression. 39 The E2F gene family consists of six members, which exist in a functional heterodimeric complex with DP proteins. 40 The transactivational potential of E2F is held in check because of interaction of its C-terminal transactivation domain with pRb. However activation of D-type cyclins triggers phosphorylation of pRb via cyclin-dependent kinases (CDK4/CDK6). Hyperphosphorylation of pRb (ppRb) releases and derepresses E2F. 41 Hence the activation of cyclin/cdks and inactivation of pRb directs cellular proliferation via the E2F proteins. The diversity of forming different multimeric complexes defines distinct functional roles for the different E2F members. E2F-1 may also induce cell death under the appropriate conditions. 24-32 Although cell death during ALS is believed to be apoptotic in nature, this is an area of active research and debate. 42-51 Evidence from studies on nonneuronal cells suggests that E2F-1-induced cell death is via apoptosis and can be either p53-dependent or p53-independent. 52,53 However a role for E2F-1 or other cell-cycle proteins in neurodegeneration during ALS is unknown.
In the current study we examined the phosphorylation and potential activation of G1 to S phase cell-cycle proteins in motor neurons during ALS. We report an enhanced nuclear accumulation of hyperphosphorylated pRb (ppRb) and altered localization of E2F-1 in both lower and upper motor neurons in patients with ALS. These results indicate that, similar to in vitro models of neuronal cell death, motor neurons hyperphosphorylate pRb and exhibit altered distribution of E2F-1 during ALS, suggesting motor neurons re-enter the G1 phase of the cell cycle, which may contribute to cell death mechanisms.
Materials and Methods
Source of Tissue Samples
The lumbar spinal cord and pre-/postcentral gyrus (motor/sensory cortex) region from 18 cases of clinically diagnosed sporadic ALS, and 9 non-ALS age-matched controls, were used to examine protein expression and distribution. All tissues were obtained from the University of Pittsburgh ALS tissue bank. The average age at death was 60.05 ± 12.22 years for ALS cases (range, 40 to 76 years) and was not significantly different from control cases (65.33 ± 15.37 years; range, 51 to 95 years; P = 0.34). The average postmortem interval times for ALS and control cases were 6.02 ± 3.35 hours (range, 2.5 to 14 hours) and 9.88 ± 5.68 hours (range, 5 to 20 hours), respectively. The difference in the postmortem interval time was statistically significant (P = 0.039). Some of the controls and ALS cases (indicated with an asterisk in Table 1 ▶ ) were neuropathologically diagnosed as cases with possible Alzheimer disease (Braak II or III/VI). Approval for use of human tissues was obtained from the University of Pittsburgh Interval Review Board. For immunohistochemistry, all tissues were fixed in 10% buffered formalin for 1 week and 8-μm paraffin-embedded sections were examined as follows.
Table 1.
A List of the Cases Utilized in This Study
| Cases | Age/sex | PMI (hours) | Neuropathology spinal cord | Neuropathology motor cortex |
|---|---|---|---|---|
| Control 1 | 53/F | 7 | No LMN loss or gliosis | No UMN loss or gliosis |
| Control 2 | 54/M | 6 | No LMN loss or gliosis | No UMN loss or gliosis |
| Control 3 | 62/M | 5 | No LMN loss or gliosis | No UMN loss or gliosis |
| Control 4 | 58/F | 5 | No LMN loss or gliosis | No UMN loss or gliosis |
| Control 5* | 82/F | 5 | No LMN loss or gliosis | No UMN loss or gliosis |
| Control 6* | 76/M | 13 | No LMN loss or gliosis | No UMN loss or gliosis |
| Control 7 | 57/F | 11 | No LMN loss or gliosis | No UMN loss or gliosis |
| Control 8 | 95/M | 20 | No LMN loss or gliosis | No UMN loss or gliosis |
| Control 9 | 51/F | 17 | No LMN loss or gliosis | No UMN loss or gliosis |
| ALS 1 | 71/F | 5 | Severe LMN loss and gliosis | Moderate UMN loss and gliosis |
| ALS 2 | 71/M | 3 | Severe LMN loss and gliosis | Moderate UMN and axonal loss |
| ALS 3 | 75/M | 2.5 | Severe loss of LMN and gliosis | Moderate loss of UMN and gliosis |
| ALS 4 | 43/M | 4 | Severe LMN loss, and gliosis | Severe UMN loss and gliosis |
| ALS 5 | 51/F | 5 | Severe LMN loss, moderate gliosis | Severe UMN loss and gliosis |
| ALS 6 | 67/M | 4 | Severe LMN loss and gliosis | Severe UMN loss and gliosis, severe loss of axons |
| ALS 7 | 51/M | 5 | Severe LMN loss, axonal loss, and gliosis | Moderate UMN loss and mild gliosis, mild axonal loss |
| ALS 8 | 40/M | 6 | Severe LMN and axonal loss, gliosis | Moderate UMN loss, mild gliosis |
| ALS 9 | 70/F | 3 | Severe loss of LMN and gliosis | Moderate UMN loss and gliosis |
| ALS 10* | 49/M | 14 | Severe loss of LMN and gliosis | Moderate UMN loss and gliosis |
| ALS 11 | 44/M | 3 | Severe loss of LMN, axonal loss and gliosis | Severe UMN loss, moderate gliosis, mild axonal loss |
| ALS 12 | 54/F | 6 | Severe loss of LMN and gliosis | Moderate loss of UMN and gliosis |
| ALS 13 | 57/M | 3 | Severe LMN loss, moderate gliosis | No loss of UMN and mild gliosis |
| ALS 14 | 73/F | 13 | Moderate LMN loss and gliosis | No loss of UMN and mild gliosis |
| ALS 15 | 53/M | 10 | Severe loss of LMN and gliosis | Moderate loss of UMN and gliosis |
| ALS 16 | 73/F | 11 | Severe loss of LMN and gliosis | Mild loss of UMN and gliosis |
| ALS 17* | 76/F | 6 | Severe loss of LMN and moderate gliosis | Moderate loss of UMN and gliosis |
| ALS 18* | 63/F | 5 | Severe loss of LMN and gliosis | Moderate loss of UMN and gliosis |
The age and postmortem interval (PMI) in hours (average control, 9.88 hours; ALS, 6.02 hours) are indicated for the ALS (65.33 years) and age-matched control (60.05 years) cases. MN, motor neurons; UMN, upper motor neurons, asterisk next to cases indicates possible Alzheimer’s disease.
Antibodies
Monoclonal antibodies were used to detect hypophosphorylated pRb (Pharmingen, La Jolla, CA), E2F-1 (clones KH95 and C-20; Santa Cruz Biotechnology, Santa Cruz, CA), and cyclin D1 (Santa Cruz Biotechnology). Polyclonal antibodies were used to detect hyperphosphorylated pRb (ser-795, NEN Biolabs, Boston, MA), active form of CDK4 (Santa Cruz Biotechnology), actin (Chemicon, Temecula, CA), and glial fibrillary acidic protein (GFAP) (DAKO, Carpinteria, CA). Western gels were probed at dilutions of 1:750 (pRb, ppRb, E2F-1), 1:200 (cyclin D1, CDK4), or 1:15000 (actin). For immunohistochemistry, these antibodies were used at dilutions of 1:150 (pRb, ppRb, E2F-1) and 1:200 (CDK4, cyclin D1, GFAP).
Immunohistochemistry
Paraffin-embedded tissue sections were microwave treated for 4 minutes at full power followed by 7 minutes at 40% power in 1× citra antigen retrieval (Biogenex, San Ramon, CA), cooled to room temperature for 90 minutes, and then incubated in 3% H2O2 and 0.25% Triton X-100 in phosphate-buffered saline (PBS) for 30 minutes. The sections were then blocked in 5% milk/PBS for 1 hour. Primary antibodies were added in 1× PBS and incubated overnight at 4°C. After four 15-minute washes in PBS, sections were incubated in biotinylated goat anti-rabbit or anti-mouse IgG secondary antibody (1:1000 dilution; Southern Biotechnology Labs., Birmingham, AL) for 1.5 hours. The signal was further amplified using biotinylated tyramide according to the manufacturer’s protocol (TSA Biotin System, NEN Biolabs). On washing, the sections were incubated in streptavidin-horseradish peroxidase (1:1000 dilution) for 1 hour and the reaction product visualized using 3-amino-9-ethylcarbazole (AEC) (for 3 to 5 minutes) (Biogenex, San Ramon, CA). This reaction results in a red end product and all sections were then counterstained with hematoxylin.
Protein Extraction
Spinal cord and motor cortical frozen tissue samples from controls cases and ALS cases (Table 1) ▶ were used for immunoblotting and DNA-binding assays. For total cell lysates, tissue samples were homogenized using polytron homogenizer (PGC Scientific, Gaithersburg, MD) set at 15,000 rpm for 45 seconds. It was performed in lysis buffer containing 25 mmol/L HEPES (pH 7.4), 50 mmol/L NaCl, protease inhibitor cocktail II (Sigma Chemical Co., St. Louis, MO), and 1% Triton X-100. The homogenized product was spun at 14,000 rpm in a cold microfuge and the supernatant saved as the total cell lysate. Nuclear and postnuclear extracts were extracted as described previously. 54 Briefly, protein extracts were prepared by detergent lysis on ice (0.1% Nonidet P-40, 10 mmol/L Tris, pH 8.0, 10 mmol/L MgCl2, 15 mmol/L NaCl, 0.5 mmol/L phenylmethyl sulfonyl fluoride, 2 μg/ml pepstatin A, and 1 μg/ml leupeptin). The nuclei were collected by low-speed centrifugation at 800 × g for 5 minutes. The supernatant was saved as the postnuclear supernatant and the pellet containing the nuclei was further extracted with high-salt buffer (0.42 mol/L NaCl, 20 mmol/L HEPES, pH 7.9, 20% glycerol, 0.5 mmol/L phenylmethyl sulfonyl fluoride, 2 μg/ml pepstatin A, and 1 μg/ml leupeptin) on ice for 10 minutes. Residual insoluble material was removed by centrifugation at 14,000 × g for 5 minutes. The resulting supernatant fraction was collected and termed the nuclear extract. Protein concentrations were determined by the Bio-Rad protein assay (Bio-Rad, Richmond, CA).
Immunoblotting
Total cell lysates, nuclear extracts and postnuclear supernatants were fractionated by electrophoresis on an 8, 10, or 12% sodium dodecyl sulfate-polyacrylamide gels. The proteins were transferred to polyvinylidene difluoride nylon membranes (NEN Biolabs) and blocked in 5% nonfat milk/1× PBS or 0.5% bovine serum albumin/0.15% glycine in 1× PBS overnight at 4°C. The blots were probed individually with the antibodies and concentrations as aforementioned, overnight at 4°C in 0.5% milk/PBS. The blots were washed three times in PBS/0.1% Tween-20 for 15 minutes. Isotype-specific horseradish peroxidase-conjugated secondary antibodies (Chemicon) specific for each primary antibody were added for 2 hours at room temperature. The secondary antibodies were washed extensively in PBS/0.1% Tween-20 (three times for 20 minutes). The final reaction products were visualized using enhanced chemiluminescence (Pierce, Rockford, IL) and the band intensities were within the linear range of detection. The density of bands was measured using the NIH Image software version 1.58 (National Institutes of Health, Atlanta, GA). Actin was used to normalize protein levels within each sample.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extracts (20 to 25 μg) were preincubated with salmon sperm DNA (120 ng) as a nonspecific competitor in 12 to 15 μl of EMSA buffer (20% glycerol, 150 mmol/L KCl) before addition of 32P-labeled oligo to reduce nonspecific DNA-protein interactions. Wild-type and mutant oligonucleotides were synthesized and gel purified (Oligos ETC.). The sequences were: wild type E2F-1 5′-ATTTAAGTTTCGCGCCCTTTCTCAA-3′; mutant E2F-1 5′-ATTTAAGTTTCGATCCCTTTCTCAA-3′. For competition reactions, unlabeled E2F-1 competitor (3, 30, and 100 ng) or unlabeled unrelated competitor (5′-GATCATTCAGGTCATGACCTGA-3′; 100 and 300 ng) oligos were preincubated with the protein for 5 minutes on ice before addition of labeled probe. For positive controls, nuclear extracts were prepared from NIH3T3 cells. The reaction mixture was loaded onto a 6% nondenaturing polyacrylamide gel and electrophoresed at 100 V in 1× Tris-borate-ethylenediaminetetraacetic acid. The polyacrylamide gel was removed from the apparatus, dried, and exposed to autoradiography film. The density of the complexes was measured using the NIH Image software version 1.58 (National Institutes of Health).
Statistical Analysis
Comparisons between any two groups of data were done using the single-factorial analysis of variance. A P value of ≤0.05 was considered statistically significant. Numerical data were expressed as means ± the SD with n = number of experiments or cases.
Results
Based on the reported activation of cell-cycle proteins in neurodegenerative diseases such as Alzheimer’s disease, 24,55-58 we investigated the expression and distribution of G1 to S phase cell-cycle proteins in human postmortem tissues from ALS and age-matched control cases. The G1 to S phase cell-cycle transition is necessary for cell-cycle progression and regulated by the activation of cyclin/cyclin-dependent kinases that hyperphosphorylate pRb, thus de-repressing the transactivational ability of E2F. 20,39-41 Therefore, the cell-cycle proteins analyzed in this study include cyclin D1, CDK4, ppRb, and E2F-1. Because ALS affects the lower motor neurons in the spinal cord and upper motor neurons (Betz cells) in the motor cortex, we used tissue samples from both CNS regions to determine subcellular localization of cell-cycle proteins by immunohistochemistry. Regions primarily unaffected during ALS, namely the dorsal horn of the spinal cord and the sensory cortex, were used as internal controls.
Increased Immunoreactivity of G1 to S Phase Cell-Cycle Proteins in ALS Spinal Cord Motor Neurons
Sections of lumbar spinal cord and pre-/postcentral gyri were immunostained using commercially available antibodies (see Materials and Methods). For light microscopy, the antigen-antibody complex was visualized with AEC and counterstained with hematoxylin (see Materials and Methods). Cyclin D1, a D-type cyclin functional in the hyperphosphorylation of pRb and G1 to S phase transition of the cell cycle, exhibited increased cytoplasmic distribution in the spinal motor neurons of ALS patients (Figure 1, A and B) ▶ . Sections were also stained for the active form of CDK4. Although active CDK4 immunoreactivity was primarily negligible in the ventral horn of control cases (Figure 1D) ▶ , increased and punctate CDK4 immunoreactivity was apparent in the cytoplasm and occasionally in the nucleus of ventral horn motor neurons of ALS patients (Figure 1E) ▶ . CDK4 and cyclin D1 immunoreactivities were negligible in the dorsal horn sensory neurons and surrounding glia in the ALS patients (Figure 1, C and F) ▶ .
Figure 1.
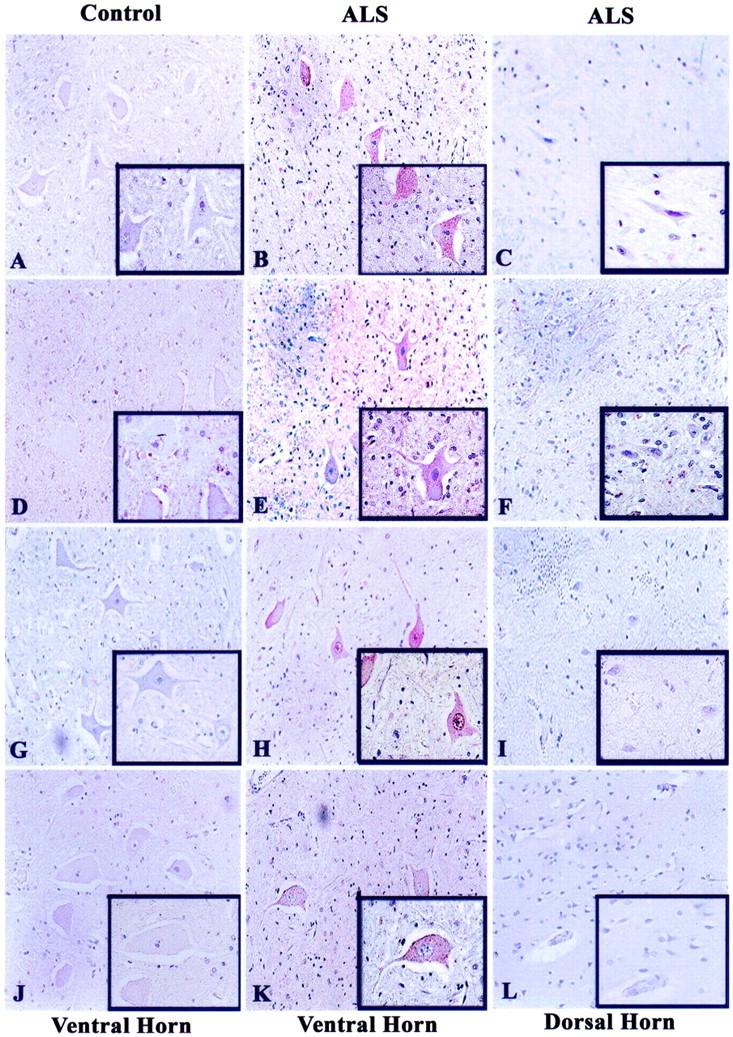
Immunohistochemical analysis of G1 to S phase regulators in human spinal cord tissues. Lumbar spinal cord sections from 10 ALS and 5 nonneurological disease controls were immunostained for cyclin D1 (A–C), CDK4 (D–F), hyperphosphorylated Rb (ppRb, G–I), and E2F-1 (J–L). AEC was used to stain the antigens of interest (red) and each section was counterstained with hematoxylin. In relation to Table 2 ▶ , panels represent cases C2 (A, J), C1 (D, G), ALS 4 (B, C), ALS 11 (E, F), ALS 5 (H, I), and ALS 8 (K, L). All insets are of the same cases as the lower magnification. Original magnifications: ×200 (A to L); ×400 (insets in A to L).
One downstream target of active cyclin D/CDK4 complex is the retinoblastoma protein (pRb), with cyclin D-specific phosphorylation of serine at position 795. Therefore, we examined the phosphorylation state of pRb in ALS using a phospho-specific anti-pRb antibody to serine-795 that has been shown to detect increased pRb phosphorylation in Alzheimer’s disease. 24,25,55 We detected abundant nuclear and punctate cytoplasmic staining of Ser-795 phosphorylated pRb (ppRb) in the motor neurons of ALS spinal cord but not in the age-matched control tissues (Figure 1, G and H) ▶ . When morphologically distinct motor neurons from multiple lumbar spinal cord sections of 10 ALS cases and 5 control cases were counted (total of 450 motor neurons per condition) the percentage of ppRb-positive lower motoneurons in ALS (85.5%) was significantly greater than controls (2.8%). The retinoblastoma protein regulates the transactivation activity of the E2F family of transcription factors. We noted punctate E2F-1 immunoreactivity specific to the cytoplasm of ALS spinal motoneurons but not in control motor neurons (Figure 1, J and K) ▶ . The percentage of E2F-1-positive motor neurons in ALS cases was significantly greater (80.8%) than in control cases (4.5%). There were negligible levels of nuclear E2F-1 irrespective of the disease state. These results were reproduced using a second E2F-1 antibody that recognizes a distinct epitope not encompassing the pRb-binding domain (data not shown). In contrast to the ALS ventral horn motor neurons, the dorsal horn sensory neurons were ppRb- and E2F-1-negative (Figure 1, I and L) ▶ .
Cellular Distribution of Cell-Cycle Proteins in Motor Cortex
We next examined the expression patterns of cell-cycle proteins in the motor and sensory cortex (pre- and postcentral gyrus) of control and ALS patients. All ALS cases except one (ALS 10, Table 1 ▶ ) exhibited substantial loss of Betz cells in the motor cortex and the presence of gliosis was used to identify the motor cortex when all Betz cells were absent. Although cyclin D1 expression was primarily cytoplasmic in any remaining large pyramidal neurons (Betz cells) of the motor cortex of ALS patients (Figure 2, A and B) ▶ , CDK4 immunoreactivity was both nuclear and cytoplasmic in these neurons (Figure 2, D and E) ▶ . There was variation in the level of CDK4 immunoreactivity within the motor cortex of ALS patients (Table 2) ▶ , although this did not correlate to any reported clinical information for the patients. The cortical neurons in the postcentral gyrus of these patients exhibited little immunoreactivity for cyclin D1 or active CDK4 (Figure 2, C and F) ▶ . Hyperphosphorylated pRb (ppRb) was detected predominantly in the nucleus of many cortical neurons in the precentral gyrus of ALS patients but not in control cases (Figure 2, G and H) ▶ . The reactivity of ppRb in the cortical sensory neurons was low or negligible (Figure 2I) ▶ . E2F-1 immunoreactivity was observed in the cytoplasm of neurons in the ALS motor cortex with no protein detected within control tissues (Figure 2, J and K) ▶ . E2F-1 immunoreactivity was absent in the postcentral gyrus (Figure 2L) ▶ . It is interesting to note that one ALS case lacking loss of upper motor neurons (ALS case 13, Table 1 ▶ ) exhibited little immunoreactivity for these G1 to S phase cell-cycle regulators (Table 2) ▶ .
Figure 2.
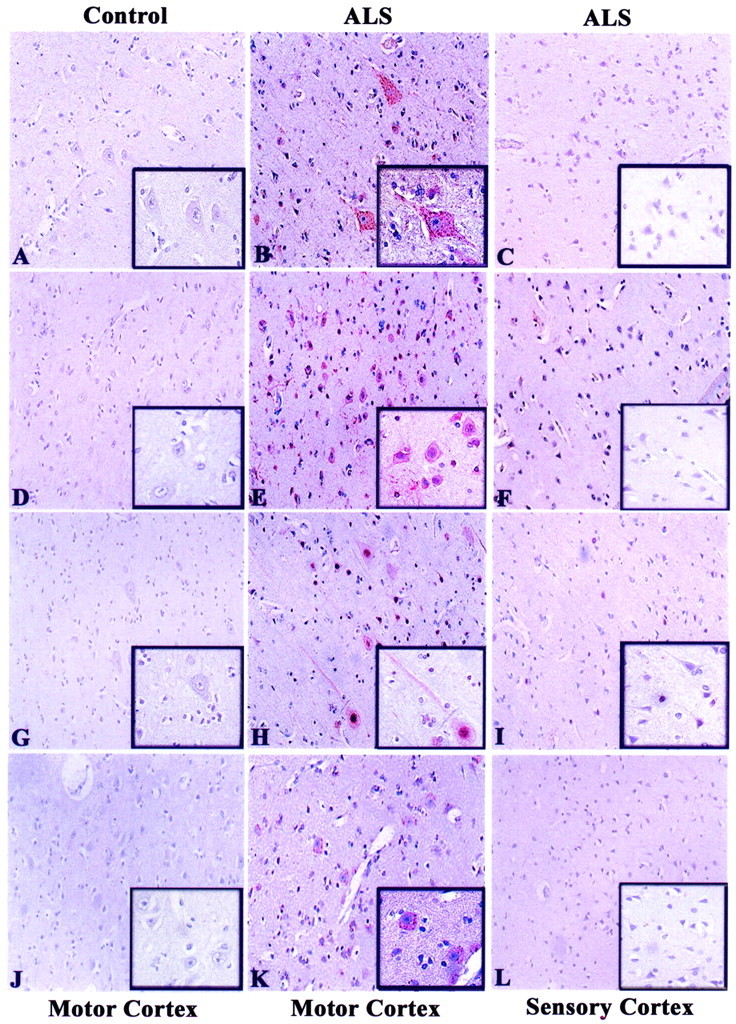
Immunohistochemical analysis of G1 to S phase regulators in human motor cortex. Precentral and postcentral gyrus from 10 ALS and 5 nonneurological disease controls were immunostained for cyclin D1 (A–C), CDK4 (D–F), hyperphosphorylated Rb (ppRb, G–I), and E2F-1 (J–L). AEC was used to stain the antigens of interest (red) and each section counterstained with hematoxylin. In relation to Table 2 ▶ , panels represent cases C1 (A, D, G, J); ALS 1 (B, C, H, I); ALS 4 (E, F, K, L). All insets are of the same cases as the lower magnification. Original magnifications: ×200 (A to L); ×400 (insets in A to L).
Table 2.
A Qualitative Analysis of the Immunohistochemical Data from Spinal Cord (SC) and Motor Cortex (MC) Tissues
| Cases | Cyclin D1 | CDK4 | ppRb | E2F-1 | ||||
|---|---|---|---|---|---|---|---|---|
| SC | MC | SC | MC | SC | MC | SC | MC | |
| C 1 | ND | Low | Low | Low | ND | Low | ND | ND |
| C 2 | ND | ND | ND | ND | Low | ND | ND | ND |
| C 3 | ND | Low | ND | ND | ND | ND | ND | ND |
| C 8 | ND | ND | ND | ND | ND | ND | ND | ND |
| C 9 | ND | ND | ND | ND | ND | ND | ND | ND |
| ALS 1 | High | Moderate | High | Low | High | High | Moderate | Low |
| ALS 2 | Moderate | Moderate | High | High | High | High | Moderate | Moderate |
| ALS 4 | High | High | Moderate | High | High | High | High | High |
| ALS 5 | High | Moderate | High | Low | High | High | High | Low |
| ALS 7 | High | Moderate | High | Low | High | High | Moderate | Low |
| ALS 8 | High | Moderate | High | High | High | High | High | High |
| ALS 9 | Moderate | Low | High | Moderate | High | High | High | Moderate |
| ALS 11 | Moderate | Moderate | Moderate | High | Moderate | High | High | Moderate |
| ALS 12 | Moderate | Moderate | Moderate | Moderate | Moderate | Low | Moderate | Moderate |
| ALS 13 | Moderate | Low | Moderate | Low | Moderate | Low | Moderate | Low |
Immunoreactivity scoring for motor neurons in controls and ALS cases (case numbers are the same as in Table 1 ▶ ). The levels of expression are indicated as low, moderate, and high with ND denoting nondetectable levels of the protein.
Distribution of Cell-Cycle Proteins in White Matter of ALS Spinal Cord and Motor Cortex
Subsequently we investigated the presence of these proteins in cells with a glial morphology. We observed moderate immunoreactivity for cyclin D1 and CDK4 in cells with morphological characteristics of astrocytes in the white matter of ALS lumbar spinal cord (data not shown). Hyperphosphorylated pRb (ppRb) immunoreactivity appeared in the nucleus of cells in the spinal cord white matter of ALS patients but not in control cases (Figure 3A) ▶ . Furthermore, glial cells with a morphology of activated astrocytes in the white matter of ALS lumbar spinal cord tissues was E2F-1 immunoreactive (Figure 3B) ▶ . Within the motor cortex, ppRb immunoreactivity was present in the cytoplasm of cells in the white matter of ALS patients but not in control cases (Figure 3C) ▶ . Using consecutive sections, we demonstrated the presence of GFAP-positive astrocytes in the white matter that were ppRb- and E2F-1-positive (asterisk in Figure 3; C to E ▶ ). Additional GFAP-labeled astrocytes were E2F-1-positive but ppRb-negative (arrowheads in Figure 3, D and E ▶ ).
Figure 3.
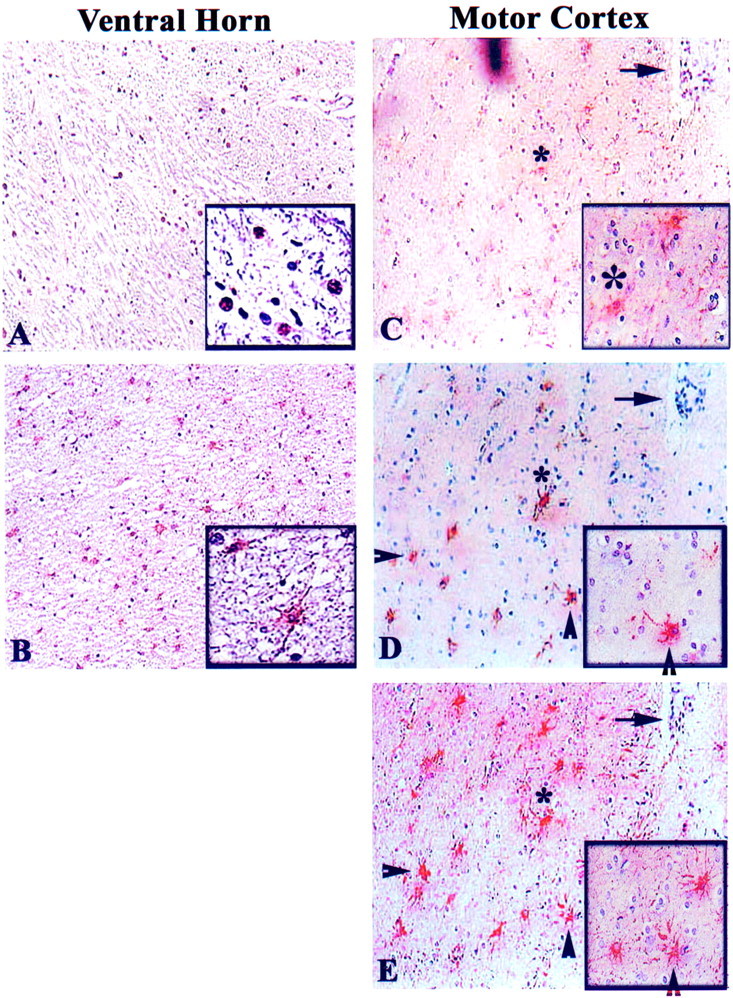
Immunohistochemical analysis of ppRb and E2F-1 in glia in the white matter of ALS spinal cord and motor cortex. Hyperphosphorylated Rb (ppRb) was in the nucleus of cells in the white matter of ventral horn of ALS spinal cord (A), whereas E2F-1 was cytoplasmic in cells that have an astrocytic morphology in ALS ventral horn white matter (B). However, both ppRb and E2F-1 appeared in the cytoplasm of cells in the motor cortex of ALS patients (C, D). C, D, and E: ppRb, E2F-1, and glial fibrillary acidic protein (GFAP) (glial marker) staining in consecutive sections, with the arrow marking a blood vessel as landmark. The asterisk indicates a ppRb+/GFAP+/E2F-1+ cell, and arrowheads indicate E2F-1+/GFAP+ cells. AEC was used to stain the antigens of interest (red) and each section counterstained with hematoxylin. In relation to Table 2 ▶ , panels represent cases ALS 8 (A), ALS 9 (B), ALS 4 (C, D, E). Original magnifications: ×200 (A to E); ×400 (insets in A to E).
Analysis of Protein Levels by Immunoblot
To determine whether altered immunostaining correlates to increased protein levels in control and ALS spinal cords, we performed immunoblot analysis using total cell lysates from lumbar spinal cord tissues for 18 ALS and 9 control cases. We show representative data from 10 ALS and 6 control cases (Figure 4) ▶ . The level of ppRb was significantly increased in the spinal cord of ALS patients, although the predominant pRb family member phosphorylated is p130. (Figure 4, A and B) ▶ . There was also an increase in phosphorylated Rb in nuclear extracts whereas it was undetected in postnuclear supernatants (data not shown). We failed to detect altered levels of total pRb in nuclear extracts from control or ALS spinal cord, suggesting that phospho-pRb results from phosphorylation of pre-existing pRb (data not shown). Cyclin D1, CDK4, and E2F-1 expression exhibited increased levels in the total cell lysates (Figure 4, A and B) ▶ . Increased levels of cyclin D1, ppRb, and E2F-1 in ALS extracts were statistically significant (P ≤ 0.05). E2F-1 was increased specifically in soluble postnuclear supernatants but undetectable in the nuclear fraction from the spinal cord of ALS patients (data not shown). All protein levels were normalized to levels of actin and quantitated to demonstrate statistical significance of these findings (Figure 4) ▶ .
Figure 4.

Protein levels in total cell extracts from human spinal cord and motor cortex. Protein extracts from lumbar spinal cord (A) and motor cortex (B) tissues of 10 ALS (ALS 4 to 7 and 13 to 18; Table 1 ▶ ) and 6 nonneurological age-matched controls (C1, C4 to C7, and C9; Table 1 ▶ ) were prepared as described in the Material and Methods. Protein (150 μg) from each extract were loaded on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and the resulting blots probed with antibodies specific to each protein. Immunoblots were repeated twice and all data quantified using NIH 1.58 software densitometry normalized to levels of actin in each sample. A: Immunoblots of lumbar spinal cord total cellular extracts with the following lane assignments: P, positive control (ppRb); lanes 1 to 6, control cases; lanes 7 to 16, ALS cases; B and C are densitometric quantified representations of total cellular extracts from spinal cord and motor cortex, respectively. Black bars are control cases (n = 6) and the gray bars are ALS cases (n = 10). ND indicates nondetectable levels. Statistical analysis was performed using single-factor analysis of variance and asterisks indicate P ≤ 0.05. In B, the P values for cyclin D1, ppRb, and E2F-1 were 0.046, 0.034, and 0.044, respectively. In C, the P value for cyclin D1 was 0.04.
We further examined protein expression in the motor cortex by immunoblot analysis. There was a decrease in levels of cyclin D1, CDK4, and E2F-1 in total cellular extracts (Figure 4C) ▶ . This decrease was statistically significant for cyclin D1 (P ≤ 0.05). Hyperphosphorylated pRb (ppRb) was undetectable in the total cell extracts. However, we did notice a significant accumulation of ppRb in nuclear extracts of ALS patients but not in the postnuclear supernatants (data not shown). Also, increased levels of cyclin D1 and E2F-1 were present in postnuclear supernatants (data not shown). There was a significant twofold increase in the levels of active CDK4 in the nuclear fraction by immunoblot correlating with nuclear CDK4 immunoreactivity as noticed in many of the ALS cases (data not shown).
DNA-Binding Activity of E2F-1
We next examined the DNA-binding activity of E2F-1 in control and ALS patients as a measure of E2F-1 functional activity. To evaluate DNA-binding activity, gel mobility shift assays (EMSA) were performed using nuclear extracts from spinal cord and motor cortex, with extracts from proliferating NIH3T3 cells as positive control. [γ-32P]-labeled double-stranded oligonucleotides containing the consensus E2F-1-binding site were incubated with 25 μg of 3T3 cells, spinal cord, or motor cortex nuclear extracts. Retardation in protein mobility because of DNA-protein complexes was visualized by autoradiography. We first demonstrated that the very sensitive EMSA could detect nuclear E2F-1 that was below the limits of detection by immunoblot. The spinal and cortical nuclear extracts contained E2F-1 protein that bound the recognized E2F-1-binding element with specificity as demonstrated by competition with excess wild-type cold oligos but not with excess mutant or unrelated oligos (Figure 5A) ▶ . In Figure 5A ▶ , the slowest migrating complex is competed away by excess unlabeled oligos but not by mutant or unrelated oligos and were interpreted to contain E2F-1. We next used nuclear extracts from proliferative 3T3 cell nuclear extracts as positive control (lane 2 in left panel of Figure 5B ▶ ) and competed the E2F-1 complex with increasing levels of excess cold oligo (Figure 5B ▶ , lanes 3 and 4). We next examined nuclear extracts of control and ALS spinal cord and motor cortex for E2F-1 DNA-binding activity (Figure 5B) ▶ . Densitometric measurement of the protein:DNA complex indicates no significant change in the intensity of DNA binding in spinal cord (P = 0.189) or motor cortex (P = 0.724) of ALS patients when compared to age-matched controls (Figure 5, B and C) ▶ .
Figure 5.
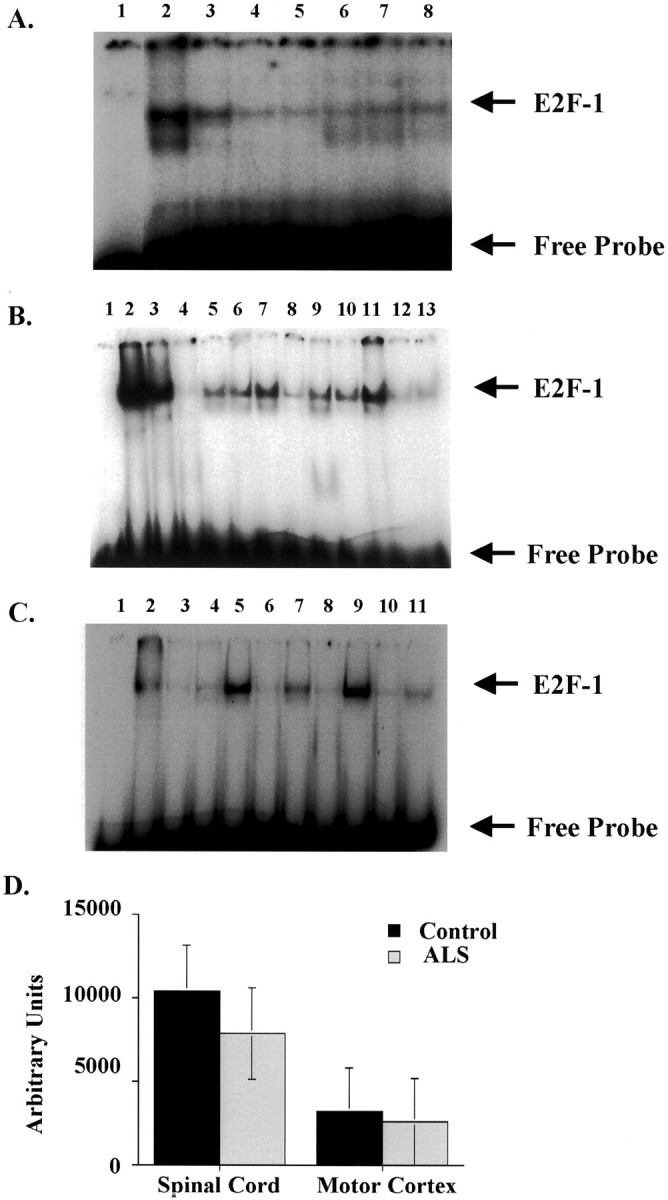
E2F-1 DNA-binding activity is unaltered in ALS. Nuclear extracts from lumbar spinal cord and motor cortex tissues of six ALS (cases 3, 4, 5, 9, 12, and 13) and three nonneurological age-matched controls (cases 1, 2, and 3) were used for EMSA. A: Competition EMSA using radiolabeled E2F-1 oligo and spinal cord nuclear extracts (25 μg) indicating a concentration-dependent competition (lanes 3 to 5) when excess cold oligos were used in the binding reaction. The complexes remain unaltered when excess mutant oligos (lanes 6 to 8) were used in the binding reactions. Lane assignments were as follows: lane 1, free probe alone; lane 2, hot probe; lanes 3 to 5, excess cold oligos (3 ng, 30 ng, and 100 ng, respectively); lanes 6 to 8, excess mutant oligos (30 ng, 100 ng, and 300 ng, respectively). B: EMSA using radiolabeled E2F-1 oligo and spinal cord nuclear extracts. Spinal cord gel lane assignments: lane 1, free probe alone; lane 2, positive control (3T3 cells); lane 3, positive control with 3 ng cold oligo; lane 4, positive control with 100 ng cold oligo; lanes 5 to 7, control cases (C1 to C3 from Table 1 ▶ ); lanes 8 to 13, ALS cases (ALS 3, 4, 5, 9, 12, and 13 from Table 1 ▶ ). C: EMSA using radiolabeled E2F-1 oligo and motor cortex nuclear extracts. Motor cortex gel lane assignments: lane 1, free probe alone; lane 2, positive control (3T3 cells); lanes 3 to 5, control cases; lanes 6 to 11, ALS cases (cases 3, 4, 5, 9, 12, and 13 from Table 1 ▶ ). The E2F-1:DNA complex and free probes are indicated to the left of the figure. D: Densitometric measurement of the E2F-1: DNA complex using NIH 1.58 software indicates unaltered levels of DNA-binding activity. Controls are denoted in black bars (n = 3) and ALS cases in stippled bars (n = 6). The P values were 0.189 for spinal cord extracts and 0.724 for motor cortex using single-factor analysis of variance with a 95% confidence interval.
Discussion
A delicate balance of signals regulates cellular homeostasis. Activation of cell-cycle proteins via extracellular signals such as excitotoxins present during neurodegenerative diseases can be toxic to postmitotic neurons as shown in a number of model systems. We initiated this study to explore improper cell-cycle activation and altered nuclear events as a possible mechanism in sporadic ALS (SALS). Aside from our previously published brief report, 55 this is the first in-depth study examining a role for the G1 to S phase cell-cycle proteins in pathogenesis of ALS. We present evidence for nuclear accumulation of hyperphosphorylated pRb (ppRb) with a concurrent increase in cytoplasmic E2F-1 immunoreactivity suggesting a role for aberrant activation of G1 to S phase regulators.
Increased levels of cyclin D1 and its associated kinase, CDK4, in motor neurons of the affected region in ALS patients suggest aberrant reactivation of the cell cycle. The activation of G1 to S phase cyclins results in hyperphosphorylation and inactivation of the retinoblastoma proteins (ppRb). Accumulation of ppRb and increased E2F-1 in ALS motor neurons and glia as shown by both immunohistochemistry and immunoblot confirms re-entry into the G1 phase of the cell cycle. We used 150 μg of tissue extracts for our immunoblot analysis because control experiments demonstrated that this amount was within the linear range of detection for each protein. As a positive control for the ppRb blots we used phosphorylated and nonphosphorylated Rb-C fusion protein that migrates at an apparent molecular weight of 70 kd and was recognized by the phospho-Rb (Ser-795) antibody. Extracts from 3T3 cells were used as positive controls for detection of cyclin D1, CDK4, and E2F-1. Immunoblot analysis for hyperphosphorylated pRb revealed multiple bands representing multiple members of the pRb gene family, although p130 was the predominant species. Furthermore, a 70-kd band was observed in both control and ALS cases with no discernible differences, which might in effect be a product of pRb degradation. The dissociation from E2F-1 makes the ubiquitin site on ppRb available such that ppRb is targeted for ubiquitin-mediated degradation. 39-41 ppRb was not detected in postnuclear supernatants because of very low protein abundance despite the fact that we observed some cytoplasmic ppRb by immunohistochemistry. This likely reflects the fact that cytosolic proteins from one cell type would not likely be detectable after tissue homogenization.
One of the intriguing results of our study was immunohistochemical data that indicates an altered localization of the E2F-1 transcription factor. Such alterations have been observed in studies on Alzheimer’s disease and SIV-E tissues. 55,57,59 The E2F gene family comprises six members sharing homology in the Rb-binding domains. 39-41 These E2F isoforms complex with the Rb family of proteins at different and defined periods of the cell cycle controlling gene expression within the G1 phase. The different binding states can translate to differences in subcellular localization of these proteins. 60 The redistribution of E2F-1 during ALS may result from the formation of alternative protein:protein complexes containing E2F-1 or the retention of newly synthesized E2F-1 in the cytoplasm. Alternatively E2F-1 protein contained in the nucleus may not be recognized by the monoclonal antibody raised against the Rb-binding epitope and used throughout this study. To discern this, a second anti-E2F-1 antibody was used whose antigenic determinant site does not encompass the Rb-binding domain. Identical results were obtained with this antibody. However the presence of E2F-1 DNA binding by EMSA indicates that E2F-1 protein resides in nuclei but at insufficient levels to be detected by immunohistochemistry or immunoblot analysis. Another explanation for the cytosolic accumulation of E2F-1 is reduced protein turnover. The presence of ubiquitin-positive protein aggregates in affected motor neurons suggests impaired proteasome function, which may lead to increased cytosolic levels of proteins such as E2F-1. 61,62 The role of cytoplasmic E2F-1 in motor neuron cell death warrants further investigations within well-defined in vitro model systems.
To examine the functional state of E2F-1 in the nucleus, gel shift assays were performed to determine DNA-binding activity of E2F-1. The DNA-binding activity of E2F-1 in the spinal cord and motor cortex did not show a significant difference between controls and ALS cases. This suggests that any changes in E2F-1 transactivational activity may be transient or that loss of nuclear E2F-1 from motor neurons is compensated by E2F-1 activation in glial cells present in the tissue extracts. Supershift analysis using available E2F-1 antibodies was not successful with our extracts, even from 3T3 cells that have detectable E2F-1 by immunoblot.
We acknowledge that protein extraction from the spinal cord and motor cortex tissue present a caveat. The presence of glial cells in the tissues will contribute to our immunoblotting and/or DNA-binding results. However it is not uncommon for activated glial cells to have a toxic effect on neurons through the release of cytokines and chemokines, which affect the neuronal milieu. 63 In fact, increased E2F-1 immunoreactivity was found in the white matter of ALS spinal cord suggesting that cell-cycle proteins in microglia and astrocytes may play a role in ALS. The question of cell type heterogeneity may be resolved in future studies by performing microdissection of the ventral spinal cord and assessing mRNA and proteins by quantitative real-time polymerase chain reaction and proteomics on a per cell basis.
E2F-1 derepression by pRb hyperphosphorylation leads to increased expression of downstream targets such as p53, p73, p14ARF, and Apaf-1, which are involved in cell death pathways. 50,51,64-69 Although there are reports indicating an increased expression of p53 in both spinal cord and motor cortex 70 further studies are required to examine the levels of other proteins regulated by E2F-1. The expression of p53 may also result in the direct activation of proapoptotic genes via p53 mediated gene expression. 71 Synergism between loss of pRb and activation of E2F-1 has been shown to contribute to p53-induced apoptosis. 50,51 In addition, accumulated DNA damage through chromatin remodeling and derepression of E2F-1 may contribute to p53-mediated apoptosis. This entails a more detailed analysis of the functional role of p53- and E2F-1-regulated gene products in ALS.
Our findings of altered subcellular distribution of the transcription factor, E2F-1, in the affected motor neurons (lower and upper) is consistent with the hypothesis that E2F-1 relocalization may trigger indirect cell death-signaling mechanisms. This event deviates from the classical model of E2F-1-mediated activation of gene expression. Interaction of E2F-1 with members of the TRAF family of adaptor proteins that mediate intracellular signaling initiates alternative death cascades. 72 The presence of TRAF2 or TRAF6 proteins in the cytoplasm of motor neurons sequesters the death receptors, aiding in survival of the neuron. 73,74 However, E2F-1 located in the cytoplasm may induce complex formation with TRAF proteins to impede their function, thus releasing death receptors that could now induce cell death via the p75NTR pathway or other death receptor pathways. 72-74 There is indeed precedence to the interactions of E2F-1 with the TRAF proteins in other cell types. 72 This hypothesis warrants further investigation.
During ALS, excess excitotoxins, accumulation of oxidative free radicals, or damage to DNA in motor neurons may cause an aberrant activation of G1 to S phase cell-cycle proteins. Although conditions may not permit DNA replication and completion of the cell cycle, cell-cycle proteins may increase the vulnerability of motor neurons to further toxic insults and induce expression of genes that function in death pathways. Improper activation of cell-cycle proteins will alter the function of DNA-binding proteins that can affect overall chromatin structure and allow for further DNA damage via oxidative injury exacerbated by various endonucleases. Support for this hypothesis comes from the recent studies that have uncovered a second gene, called Alsin, implicated in familial ALS. 6-8 This gene shares sequence homology to a putative G-protein termed RCC1 (regulator of chromosome condensation) that acts on RAN protein, which is involved in nuclear import and export. Mutation in a protein that is homologous to a regulator of chromatin structure may make the cell more susceptible to DNA damage.
Our data supports a model in which motor neurons are stimulated to enter the G1 phase of cell cycle during ALS, which may directly regulate motor neuron cell death (Figure 6) ▶ . Hyperphosphorylation of pRb via cyclin D/CDK4/6 can induce expression of E2F-regulated genes such as Bax, p53, and Apaf-1. These gene products can participate in multiple cell death pathways related to DNA damage and mitochondrial dysfunction. In addition, accumulation of E2F-1 in the cytoplasm can induce a transcription-independent form of cell death via death receptors. Both caspase-mediated and death receptor-mediated cell death pathways have been implicated in ALS and animal models of ALS. 36-38 This model suggests that activation of G1 to S phase cell-cycle proteins may directly induce cell death without progression into downstream phases of the cell cycle. Future studies will further define the intracellular signaling pathways involved in cell-cycle activation and explore the functional role of these proteins in motor neuron cell death.
Figure 6.

Model of motor neuron cell death induced by activation of cell-cycle proteins. In an adult postmitotic neuron reduced levels of cyclin D-CDK4 results in repression of gene expression by the interaction of pRb with E2F-1. Also, TRAF proteins complex with death receptors (DR) such as p75NTR blocking cell death. However, on extracellular signals, increased levels of cyclin D-CDK4 result in hyperphosphorylation of pRb (ppRb) with release of E2F-1. Transcriptional activity of E2F-1 results in initiating death signals via induction of p14ARF, p53, Bax, Apaf-1, and cytochrome C (Cyt C), apoptotic regulators that would in turn activate proapoptotic proteins such as caspase-9 and caspase-3. Alternatively, E2F-1 in the cytoplasm may interact with TNF receptor-associated factors (TRAF) and permit death receptor-mediated cell death. Activation of these pathways may sensitize the neuron to further toxic insults that pushes the susceptible neuron toward death.
Acknowledgments
We thank Ms. Jonette Werley for processing of tissues and paraffin-embedded slides.
Footnotes
Address reprint requests to Robert Bowser, Department of Pathology, Division of Neuropathology, University of Pittsburgh School of Medicine, BST S-420, 3500 Terrace St., Pittsburgh, PA 15261. E-mail: bowser@np.awing.upmc.edu.
Supported by a research grant from the ALS Association; the ALS tissue bank is supported by the Mario Lemieux Foundation and the Western Pennsylvania Chapter of the ALS Association.
References
- 1.Chou SM: Pathology of motor system disorder. Leigh PN Swash M eds. Motor Neuron Disease, Biology and Management. 1995:pp 53-92 Springer-Verlag, London
- 2.Eisen A, Weber M: The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 2001, 24:564-573 [DOI] [PubMed] [Google Scholar]
- 3.Rosen DR, Siddique TS, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH, Jr: Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362:59-62 [DOI] [PubMed] [Google Scholar]
- 4.Rosen DR, Bowling AC, Patterson D, Usdin TB, Sapp P, Mezey E, McKenna-Yasek D, O’Regan JP, Rahman Z, Ferrante RJ, Brownstein MJ, Kowall NW, Beal MF, Horvitz HR, Brown RH, Jr: A frequent ala 4 to val superoxide dismutase-1 mutation is associated with a rapidly progressive familial amyotrophic lateral sclerosis. Hum Mol Genet 1994, 3:981-987 [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, Hung WY, Ouahchi K, Yan J, Azim AC, Cole N, Gascon G, Yagmour A, Ben-Hamida M, Pericak-Vance M, Hentati F, Siddique T: The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 2001, 29:160-165 [DOI] [PubMed] [Google Scholar]
- 6.Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, Miyamoto N, Showguchi-Miyata J, Okada Y, Singaraja R, Figlewicz DA, Kwiatkowski T, Hosler BA, Sagie T, Skaug J, Nasir J, Brown RH, Jr, Scherer SW, Rouleau GA, Hayden MR, Ikeda JE: A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 2001, 29:166-173 [DOI] [PubMed] [Google Scholar]
- 7.Kunst CB, Messer L, Gordon J, Haines J, Patterson D: Genetic mapping of a mouse modifier gene that can prevent ALS onset. Genomics 2000, 70:181-189 [DOI] [PubMed] [Google Scholar]
- 8.Cleveland DW, Rothstein JD: From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2001, 2:806-819 [DOI] [PubMed] [Google Scholar]
- 9.Kruman II, Pedersen WA, Springer JE, Mattson MP: ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp Neurol 1999, 160:28-39 [DOI] [PubMed] [Google Scholar]
- 10.Shaw PJ, Egget CJ: Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J Neurol 2000, 247(Suppl 1):I/17-I/27 [DOI] [PubMed] [Google Scholar]
- 11.Tu P-H, Gurney ME, Julien J-P, Lee VM-Y, Trojanowski JQ: Oxidative stress, mutant SOD-1 and neurofilament pathology in transgenic mouse models of motor neuron disease. Lab Invest 1997, 76:441-456 [PubMed] [Google Scholar]
- 12.Kong J, Xu Z: Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci 1998, 18:3241-3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee MK, Marszalek JR, Cleveland DW: A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron 1994, 13:975-988 [DOI] [PubMed] [Google Scholar]
- 14.Martin JE: Neurotrophic factors and neurodegeneration. Leigh PN Swash M eds. Motor Neuron Disease, Biology and Management. 1995:pp 241-258 Springer-Verlag, London
- 15.Mu X, He J, Anderson DW, Trojanowski JQ, Springer JE: Altered expression of bcl-2 and bax mRNA in amyotrophic lateral sclerosis spinal cord motor neurons. Ann Neurol 1996, 40:379-386 [DOI] [PubMed] [Google Scholar]
- 16.Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, Memo M: Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci 2001, 24:25-31 [DOI] [PubMed] [Google Scholar]
- 17.Ross ME: Cell division and the nervous system: regulating the cycle from neural differentiation to death. Trends Neurosci 1996, 19:62-68 [DOI] [PubMed] [Google Scholar]
- 18.Freeman RS: The cell cycle and neuronal cell death. Koliatsos VE Ratan RR eds. Cell Death and Diseases of the Nervous System. 1999:pp 103-119 Humana Press Inc., New Jersey
- 19.Heintz N: Cell death and the cell cycle: a relationship between transformation and neurodegeneration? Trends Biochem Sci 1993, 18:157-159 [DOI] [PubMed] [Google Scholar]
- 20.Weinberg RA: The retinoblastoma protein and cell cycle control. Cell 1995, 81:323-330 [DOI] [PubMed] [Google Scholar]
- 21.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA: Effects of an Rb mutation in the mouse. Nature 1992, 359:295-300 [DOI] [PubMed] [Google Scholar]
- 22.Lee EY-HP, Chang C-Y, Hu N, Wang Y-CJ, Lai C-C, Herrup K, Lee W-H, Bradley A: Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 1992, 359:288-294 [DOI] [PubMed] [Google Scholar]
- 23.Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T: Mutation of E2F-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell 1998, 2:293-304 [DOI] [PubMed] [Google Scholar]
- 24.Jordan-Sciutto K, Rhodes J, Bowser R: Altered subcellular distribution of transcriptional regulators in response to Aβ peptide and during Alzheimer disease. Mech Ageing Dev 2001, 123:11-20 [DOI] [PubMed] [Google Scholar]
- 25.Jordan-Sciutto KL, Malaiyandi LM, Bowser R: Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J Neuropathol Exp Neurol 2002, 61:358-367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park DS, Farinelli SE, Greene LA: Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem 1996, 271:8161-8169 [DOI] [PubMed] [Google Scholar]
- 27.Park DS, Morris EJ, Greene LA, Geller HM: G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci 1997, 17:1256-1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park DS, Levine B, Ferrari G, Greene LA: Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci 1997, 17:8975-8983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA: Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol 1998, 143:457-467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, Greene LA: Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation, and oxidative stress J Neurosci 1998, 18:830-840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park DS, Morris EJ, Bremner R, Keramaris E, Padmanabhan J, Rosenbaum M, Shelanski ML, Geller HM, Greene LA: Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci 2000, 20:3104-3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Hare MJ, Hou ST, Morris EJ, Cregan SP, Xu Q, Slack RS, Park DS: Induction and modulation of cerebellar granule neuron death by E2F-1. J Biol Chem 2000, 275:25358-25364 [DOI] [PubMed] [Google Scholar]
- 33.Padmanabhan J, Park DS, Greene LA, Shelanski ML: Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci 1999, 19:8747-8756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS: Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F-DP, in beta-amyloid-induced neuronal death. J Biol Chem 1999, 274:19011-19016 [DOI] [PubMed] [Google Scholar]
- 35.Giovanni A, Keramaris E, Morris EJ, Hou ST, O’Hare M, Dyson N, Robertson GS, Slack RS, Park DS: E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J Biol Chem 2000, 275:11553-11560 [DOI] [PubMed] [Google Scholar]
- 36.Ilzecka J, Stelmasiak Z, Dobosz B: Interleukin-1beta converting enzyme/caspase-1 (ICE/caspase-1) and soluble APO-1/Fas/CD 95 receptor in amyotrophic lateral sclerosis patients. Acta Neurol Scand 2001, 103:255-258 [PubMed] [Google Scholar]
- 37.Pasinelli P, Houseweart MK, Brown RH, Jr, Cleveland DW: Caspase-1 and -3 are sequentially activated in motor neuron death in Cu, Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 2000, 97:13901-13906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yi FH, Lautrette C, Vermot-Desroches C, Bordessoule D, Couratier P, Wijdenes J, Preud’homme JL, Jauberteau MO: In vitro induction of neuronal apoptosis by anti-Fas antibody-containing sera from amyotrophic lateral sclerosis patients. J Neuroimmunol 2000, 109:211-220 [DOI] [PubMed] [Google Scholar]
- 39.Dyson N: The regulation of E2F by pRb-family proteins. Genes Dev 1998, 12:2245-2262 [DOI] [PubMed] [Google Scholar]
- 40.Black AR, Azizkhan-Clifford J: Regulation of E2F: a family of transcriptional factors involved in proliferation control. Gene 1999, 237:281-302 [DOI] [PubMed] [Google Scholar]
- 41.Chellappan SP, Heibert S, Mudryj M, Horowitz JM, Nevins JR: The E2F transcription factor is a cellular target for the RB protein. Cell 1991, 65:1053-1061 [DOI] [PubMed] [Google Scholar]
- 42.Thomas LB, Gates DJ, Richfield EK, O’Brien TF, Schweitzer JB, Steindler DA: DNA end labeling (TUNEL) in Huntington’s disease and other neuropathological conditions. Exp Neurol 1995, 133:265-272 [DOI] [PubMed] [Google Scholar]
- 43.Li WP, Chan WY, Lai HW, Yew DT: Terminal dUTP nick end labeling (TUNEL) positive cells in the different regions of the brain in normal aging and Alzheimer patients. J Mol Neurosci 1997, 8:75-82 [DOI] [PubMed] [Google Scholar]
- 44.Martin LJ: Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol 1999, 58:459-471 [DOI] [PubMed] [Google Scholar]
- 45.Bar-Peled O, Knudson M, Korsmeyer SJ, Rothstein JD: Motor neuron degeneration is attenuated in bax-deficient neurons in vitro. J Neurosci Res 1999, 55:542-556 [DOI] [PubMed] [Google Scholar]
- 46.Timsit S, Rivera S, Ouaghi P, Guischard F, Tremblay E, Ben-Ari Y, Khrestchatisky M: Increased cyclin D1 in vulnerable neurons in the hippocampus after ischaemia and epilepsy: a modulator of in vivo programmed cell death? Eur J Neurosci 1999, 11:263-278 [DOI] [PubMed] [Google Scholar]
- 47.Dobashi Y, Shoji M, Noguchi T, Kondo E, Katayama K, Kameya T: A novel apoptotic cascade mediated by CDK4 in rat pheochromocytoma PC12 cells. Biochem Biophys Res Commun 1999, 260:806-812 [DOI] [PubMed] [Google Scholar]
- 48.Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM: Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science 2000, 288:335-339 [DOI] [PubMed] [Google Scholar]
- 49.Vukosavic S, Stefanis L, Jackson-Lewis V, Guégan C, Romero N, Chen C, Dubois-Dauphin M, Przedborski S: Delaying caspase activation by bcl-2: a clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 2000, 20:9119-9125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trinh E, Boutillier AL, Loeffler JP: Regulation of the retinoblastoma-dependent Mdm2 and E2F-1 signaling pathways during neuronal apoptosis. Mol Cell Neurosci 2001, 17:342-353 [DOI] [PubMed] [Google Scholar]
- 51.Sathasivam S, Ince PG, Shaw PJ: Apoptosis in amyotrophic lateral sclerosis: a review of the evidence. Neuropathol Appl Neurobiol 2001, 27:257-274 [DOI] [PubMed] [Google Scholar]
- 52.Macleod KF, Hu Y, Jacks T: Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J 1996, 15:6178-6188 [PMC free article] [PubMed] [Google Scholar]
- 53.Phillips AC, Bates S, Ryan KM, Helin K, Vousden KH: Induction of DNA synthesis and apoptosis are separable functions of E2F-1. Genes Dev 1997, 11:1853-1863 [DOI] [PubMed] [Google Scholar]
- 54.Jordan-Sciutto K, Dragich JM, Bowser R: DNA binding activity of the fetal Alz-50 clone1 (FAC1) protein is enhanced by phosphorylation. Biochem Biophys Res Comm 1999, 260:785-789 [DOI] [PubMed] [Google Scholar]
- 55.Ranganathan S, Scudiere S, Bowser R: Hyperphosphorylation of the retinoblastoma gene product and altered subcellular distribution of E2F-1 in Alzheimer disease and amyotrophic lateral sclerosis. J Alzheimer Dis 2001, 3:377-385 [DOI] [PubMed] [Google Scholar]
- 56.Yang Y, Geldmacher DS, Herrup K: DNA replication precedes neuronal cell death in Alzheimer disease. J Neurosci 2001, 21:2661-2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vincent I, Jicha G, Rosado M, Dickson DW: Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer disease brain. J Neurosci 1997, 17:3588-3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McShea A, Harris P, Webster K, Wahl A, Smith M: Abnormal expression of the cell cycle regulators p16 and cdk4 in Alzheimer disease. Am J Pathol 1997, 150:1933-1939 [PMC free article] [PubMed] [Google Scholar]
- 59.Jordan-Sciutto K, Wang G, Murphy-Corb M, Wiley C: Induction of cell cycle regulators in simian immunodeficiency virus encephalitis. Am J Pathol 2000, 157:497-507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verona R, Moberg K, Estes S, Starz M, Vernon JP, Lees JA: E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Mol Cell Biol 1997, 17:7268-7282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lowe J: New pathological findings in amyotrophic lateral sclerosis. J Neurol Sci 1994, 124:38-51 [DOI] [PubMed] [Google Scholar]
- 62.Migheli A, Attanasio A, Schiffer D: Ubiquitin and neurofilament expression in anterior horn cells in amyotrophic lateral sclerosis: possible clues to the pathogenesis. Neuropathol Appl Neurobiol 1994, 20:282-289 [DOI] [PubMed] [Google Scholar]
- 63.He BP, Wen W, Strong MJ: Activated microglia (BV-2) facilitation of TNF-α-mediated motor neuron death in vitro. J Neuroimmunol 2002, 128:31-38 [DOI] [PubMed] [Google Scholar]
- 64.Qin X, Livingston DM, Kaelin WGJ, Adams PD: Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA 1994, 91:10918-10922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stiewe T, Putzer BM: Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat Genet 2000, 26:464-469 [DOI] [PubMed] [Google Scholar]
- 66.Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W, Flores ER, Tsai KY, Jacks T, Vousden KH, Kaelin WG, Jr: Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 2000, 407:645-648 [DOI] [PubMed] [Google Scholar]
- 67.Stewart B, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH: p14ARF links the tumor suppressors RB and p53. Nature 1998, 395:124-125 [DOI] [PubMed] [Google Scholar]
- 68.Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H, Helin K: Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol 2001, 3:552-558 [DOI] [PubMed] [Google Scholar]
- 69.Luciakova K, Barath P, Li R, Zaid A, Nelson BD: Activity of the human cytochrome c1 promoter is modulated by E2F. Biochem J 2000, 351:251-256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martin LJ: P53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol Dis 2000, 7:613-622 [DOI] [PubMed] [Google Scholar]
- 71.Miyashita T, Reed JC: Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80:293-299 [DOI] [PubMed] [Google Scholar]
- 72.Phillips AC, Ernst MK, Bates S, Rice NR, Vousden KH: E2F-1 potentiates cell death by blocking antiapoptotic signaling pathways. Mol Cell 1999, 4:771-781 [DOI] [PubMed] [Google Scholar]
- 73.Khursigara G, Orlinick JR, Chao MV: Association of the p75 neurotrophin receptor with TRAF6. J Biol Chem 1999, 274:2597-2600 [DOI] [PubMed] [Google Scholar]
- 74.Ye X, Mehlen P, Rabizadeh S, VanArsdale T, Zhang H, Shin H, Wang JJ, Leo E, Zapata J, Hauser CA, Reed JC, Bredesen DE: TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J Biol Chem 1999, 274:30202-30208 [DOI] [PubMed] [Google Scholar]